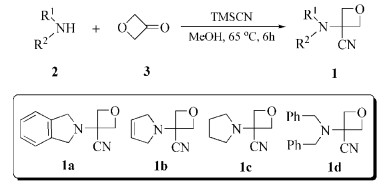
氧杂环丁烷是一类结构特殊的杂环化合物,存在于许多活性天然产物[1 3 及上市药物中[4 6 。正是由于其独特的化学结构,氧杂环丁烷衍生物表现出广泛生物活性,一方面,作为良好的氢键供体,氧杂环丁烷易于和药物靶标形成氢键相互作用;另一方面,氧杂环丁烷结构在碱性及弱酸性环境中较稳定,具有良好的成药性基础[7 。研究发现,氧杂环丁烷母核结构可以作为偕二甲基、羰基、吗啉基等活性基团的生物电子等排体,通过基团替代,将氧杂环丁烷引入药物分子结构中,不仅可以增强分子的刚性,还可以提高其水溶性以及代谢稳定性等[8 10 。因此,近年来,基于氧杂环丁烷结构作为生物电子等排体在药物分子设计合成中的应用研究得到了药物化学工作者的广泛关注[11 13 。目前,关于氧杂环丁烷衍生物的合成,主要有以下四种方法:(1)Williamson分子内醚合成法[14 ;(2)通过硫叶立德一锅法合成[15 ;(3)Paternó-Büchi环加成法合成[16 ;(4)以氧杂环丁烷衍生物为原料合成[17 18 。其中,以氧杂环丁烷衍生物为原料直接衍生合成是目前应用最多的方法。作为一种最常见的氧杂环丁烷衍生物,氧杂环丁-3-酮结构上的羰基经过简单衍生化即可得到3-取代氧杂环丁烷,该方法具有原料廉价易得、合成工艺简单等优点[9 19 。
为了能开发一种简便有效的合成3-仲氨基氧杂环丁烷-3-腈衍生物的方法,本文报道一种三组分“一锅法”合成策略,分别以四种仲胺、氧杂环丁-3-酮和三甲基氰硅烷(TMSCN)为原料,无需催化剂,一步合成得到目标化合物(1a~1d ),并对目标化合物在有机合成中的应用进行了初步研究。该反应工艺具有原料廉价易得、反应时间短、收率较高等优点。为3-仲氨基取代氧杂环丁烷-3-腈化合物的合成提供了一种简便可行的工艺方法。
图式 1
1.
实验部分
1.1
仪器与试剂
AV400型核磁共振谱仪(DMSO-d 6 或CDCl3 为溶剂,TMS为内标,德国Bruker公司);Ultima Global Spectrometer型质谱仪(ESI源,美国Waters公司)。
氧杂环丁-3-酮、异吲哚啉、2, 5-二氢吡咯盐酸盐、四氢吡咯、二苄胺、三甲基氰硅烷(TMSCN)(分析纯,阿达玛斯试剂有限公司,使用前需重蒸处理);柱层析硅胶(300~400目,青岛海洋化工厂);其他所用试剂均为市售分析纯。
1.2
3-仲氨基氧杂环丁烷-3-腈化合物(1a~1d)的合成方法
无水无氧条件下,在25mL三口瓶中依次加入仲胺2(40.0mmol)、无水甲醇15mL、氧杂环丁-3-酮1.44g(20.0mmol)和重蒸的TMSCN 4.96g(50.0mmol),室温反应1h后继续回流反应6h。TLC监测反应结束后,反应体系中加入20mL饱和NaHCO3 水溶液稀释、乙酸乙酯萃取(3×30mL)、有机相经无水硫酸钠干燥后减压旋干,粗品经硅胶柱层析分离纯化(石油醚/乙酸乙酯体积比20:1~5:1),得到目标化合物(1a~1d )。
3-(异吲哚啉-2-基)氧杂环丁烷-3-腈(1a ):白色固体,收率78.3%。1 H NMR (400MHz,DMSO-d 6 )δ :7.36~7.23(m,4H),4.86 (d,J =6.8Hz,2H),4.72 (d,J =6.8Hz,2H),4.00 (s,4H);ESI-MS,m/z: 201.16 [M+H]+ 。
3-(2, 5-二氢-1H -吡咯-1-基)氧杂环丁烷-3-腈(1b ):淡黄色固体,收率79.4%。1 H NMR(DMSO-d 6 ,400MHz)δ :5.54~5.50 (m,2H),4.84 (d,J =6.8Hz,2H),4.62 (d,J =6.8Hz,2H),3.42~3.33 (m,4H)。ESI-MS,m /z :151.12 [M+H]+ 。
3-(吡咯烷-1-基)氧杂环丁烷-3-腈(1c ):淡黄色固体,收率81.3%。1 H NMR (400MHz,DMSO-d 6 ) δ :4.86 (d,J =6.8Hz,2H),4.66 (d,J =6.8Hz,2H),2.67~2.58 (m,4H),1.79~1.70 (m,4H);ESI-MS,m /z :153.14 [M+H]+ 。
3-(二苄氨基)氧杂环丁烷-3-腈(1d ):淡黄色固体,收率86.6%。1 H NMR (400MHz,DMSO-d 6 ) δ :7.38~7.27 (m,10H),4.78 (d,J =6.8Hz,2H),4.64 (d,J =6.8Hz,2H),3.51 (s,4H);ESI-MS,m /z : 279.17 [M+H]+ 。
2.
结果与讨论
2.1
化合物1的合成
以异吲哚啉(2a )、氧杂环丁-3-酮(3)和TMSCN反应合成3-(异吲哚啉-2-基)氧杂环丁烷-3-腈(1a )的反应为模型,研究反应条件对产物收率的影响,筛选优化的反应工艺条件。
2.1.1
物料比对1a 收率的影响
异吲哚啉(2a )、氧杂环丁-3-酮(3 )和TMSCN为原料,三组分“一锅法”合成1a ,反应中三者的物料摩尔比对产物的收率具有重要的影响。考虑原料成本因素,选择异吲哚啉(2a )和TMSCN过量。研究发现(表 1 n (2a ):n (3):n (TMSCN)=1.0:1:1.0时,产物1a 收率仅为20.1%,提高异吲哚啉(2a )和TMSCN的用量,产物1a 收率明显提高,当三者的物料比为2.0:1:2.0时,1a 收率达到66.5%,继续增加TMSCN的用量为2.5倍时,产物1a 收率达到78.3%,但继续增加TMSCN的用量为3.0倍,产物收率没有明显变化。增加异吲哚啉(2a )的用量,三者比例为2.5:1:2.5,收率略有降低,所以确定优化物料摩尔比为n (2a ):n (3 ):n (TMSCN)=2.0:1:2.5。
表 1
2.1.2
反应溶剂对1a 收率的影响
随后考察了反应溶剂对产物收率的影响(表 2 [20 ,选择乙酸作为反应溶剂,然而并没有得到预期产物1a 。继续筛选三种溶剂甲苯、甲醇、乙醇,发现在甲苯中反应,产物收率较低,而在甲醇和乙醇中反应收率显著提高。其中,在甲醇中反应,产物1a 收率达到78.3%。因此确定适宜反应溶剂为无水甲醇。
表 2
2.1.3
反应温度对1a 收率的影响
接着考察反应温度对产物1a 收率的影响(表 3 1a 收率为49.5%,继续升高温度达到65℃(甲醇的回流温度)时,收率升高到78.3%。所以,确定适宜反应温度为65℃。
表 3
2.1.4
反应时间对1a 收率的影响
在确定了优化的物料比、反应溶剂、反应温度后,考察反应时间对产物1a 收率的影响(表 4 1a 收率为67.9%,延长反应时间至6h时,收率明显提高至78.3%,但继续延长反应时间至7h和8h,收率没有明显变化。所以确定反应时间为6h。
表 4
2.2
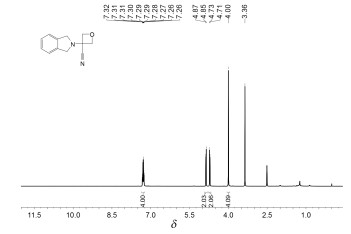
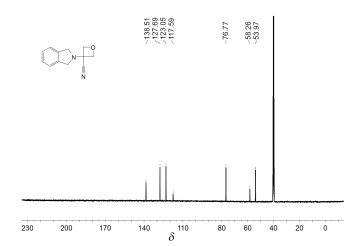
化合物1a的核磁谱分析
化合物1a 的1 H NMR谱(图 1 δ 7.36~7.23的多重峰(积分为4H)是异吲哚啉芳环上4个Ar-H;δ 4.86及δ 4.72的两个双峰(积分均为2H)耦合常数都是6.8Hz,为氧杂环丁烷结构上与氧原子相连接的两个亚甲基;δ 4.00的单峰(积分为4H)是吲哚啉结构上与氮原子相连接的两个亚甲基,由于化学环境相同而发生重合。化合物1a 的13 C NMR谱(图 2 δ 138.51、127.69、123.05为吲哚啉结构上苯环碳,由于结构对称因素,所以显示三个峰;δ 117.59为氰基碳原子峰;δ 76.77为氧杂环丁烷结构上与氧原子相连接的两个亚甲基碳发生重合;δ 58.26是与氰基相连接的季碳原子的峰;δ 53.97是吲哚啉结构上与氮原子相连接的两个亚甲基的重合峰。通过对1 H NMR、13 C NMR分析,结合质谱分析数据,可以确证目标化合物的结构。
图 1
图 2
2.3
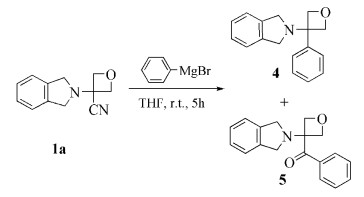
目标产物的应用
为了进一步研究目标化合物在有机合成方面的应用,将化合物1a 与苯基溴化镁发生反应,可以得到2-(3-苯基氧杂环丁烷-3-基)异吲哚啉(4 )和[3-(异吲哚啉-2-基)氧杂环丁烷-3-基]苯甲酮(5 ),其结构均经1 HNMR和ESI-MS确证,合成路线如图式 2
图式 2
格氏试剂与氰基加成然后水解制备酮类化合物是一种常见的有机合成单元反应[21 。格氏试剂与三级腈发生取代反应已有文献报道[22 。在本实验中,苯基溴化镁与3-(异吲哚啉-2-基)氧杂环丁烷-3-腈(1a )结构中的氰基发生加成,生成一个带负电的亚胺离子,然后加水淬灭,发生水解得到酮化合物5 。而对于化合物4的生成,分析原因,可能是由于化合物1a 结构中氰基作为吸电子基团离去,形成较为稳定的三级碳正离子,然后格氏试剂作为亲核试剂进攻该碳正离子,经过SN 1亲核取代反应过程,得到化合物4 。目前,关于3-仲氨基氧杂环丁烷-3-腈衍生物与格氏试剂反应的研究,包括反应条件对两种产物收率的调控研究,正在本实验室进行中。
合成方法:无水、氮气保护条件下,在50mL的单口瓶中加入600mg(3mmol)化合物1a 和5mL四氢呋喃,搅拌均匀后,在0℃条件下缓慢滴加21mL(21mmol)苯基溴化镁溶液(1.0mol/L的四氢呋喃溶液),反应体系在室温下搅拌5h,TLC监测反应结束后,反应体系中加入1mL水淬灭,再加入10mL饱和氯化钠水溶液稀释,乙酸乙酯萃取(3×10mL),有机相经无水硫酸钠干燥后减压旋干,粗品经硅胶柱层析分离纯化(石油醚/乙酸乙酯,体积比10:1),得到151mg化合物4 和264mg化合物5 。
2-(3-苯基氧杂环丁烷-3-基)异吲哚啉(4 ):白色固体,收率40.1%。1 H NMR(400MHz,CDCl3 ) δ: 7.62~7.53 (m,5H),7.41~7.29 (m,4H),5.05 (d,J =6.8Hz,2H),4.96 (d,J =6.8Hz,2H),4.09 (s,4H)。ESI-MS,m /z :252.18 [M+H]+ 。
[3-(异吲哚啉-2-基)氧杂环丁烷-3-基]苯甲酮(5 ):白色固体,收率31.5%。1 H NMR (400MHz,CDCl3 ) δ :8.02 (d,J =7.2Hz,2H),7.87 (d,J =8.4Hz,2H),7.62~7.57 (m,1H),7.39~7.30 (m,4H),4.86 (d,J =7.2Hz,2H),4.72 (d,J =7.2Hz,2H),4.11 (s,4H)。ESI-MS,m /z :280.16 [M+H]+ 。
3.
结论
以氧杂环丁-3-酮和三甲基氰硅烷为原料,分别和4种仲胺反应,一锅法合成得到目标化合物3-仲氨基氧杂环丁烷-3-腈衍生物(1a~1d ),产物结构经1 H NMR、13 C NMR和ESI-MS表征。并以异吲哚啉、氧杂环丁-3-酮和三甲基氰硅烷的反应为模型反应,考察影响产物1a 收率的主要因素,确定优化反应条件为:物料摩尔比为n (异吲哚啉):n (氧杂环丁-3-酮):n (三甲基氰硅烷)=2.0:1:2.5;反应溶剂为无水甲醇,在65℃反应6h。在优化反应条件下,化合物1a 收率78.3%。对于目标化合物的应用研究发现,化合物1a 与苯基溴化镁在四氢呋喃溶剂中室温反应5h,得到2-(3-苯基氧杂环丁烷-3-基)异吲哚啉(4 )和[3-(异吲哚啉-2-基)氧杂环丁烷-3-基](苯基)甲酮(5 ),收率分别为40.1%和31.5%。该反应工艺具有原料廉价易得、反应时间短、收率较高等优点。为3-仲氨基氧杂环丁烷-3-腈化合物的合成提供了一种简便可行的工艺方法。

 下载:
下载:

 下载:
下载:





 下载:
下载:
