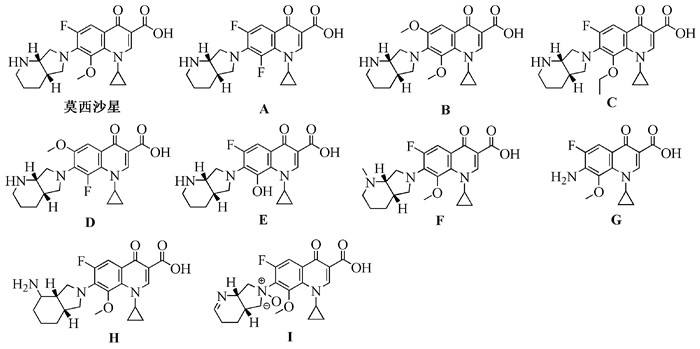
图式 1.
莫西沙星及其杂质的化学结构
Scheme 1.
Chemical structures of moxifloxacin and impurities

莫西沙星(Moxifloxacin)是1999年德国拜耳公司研发上市的第四代氟喹诺酮类广谱抗生素,商品名“拜复乐”,化学名1-环丙基-7-[(S, S)-2, 8-二氮杂双环[4.3.0]壬烷-8-基]-6-氟-8-甲氧基-1, 4-二氢-4-氧亚基-3-喹啉羧酸。莫西沙星与其他喹诺酮类药物的不同之处是在其分子结构的C8位引入了甲氧基,正是这一结构特征使它在保留了第三代喹诺酮类药物抗革兰氏阴性菌的优秀活性外,还对革兰氏阳性菌、非典型病原体如衣原体、支原体、厌氧菌等也有良好的抗菌活性[1, 2]。在临床上,莫西沙星主要用于治疗慢性支气管炎的急性发作、急性窦腺炎、社区获得性肺炎以及皮肤和软组织感染,具有抗菌谱广、抗菌活性强、低毒、不易产生耐药性、无明显光毒性等众多优点,有望成为继环丙沙星后销售最佳的喹诺酮类抗菌药物[3, 4]。
莫西沙星的合成工艺已有不少文献进行了报道[5~11],每条合成路线均有不同的优势,但在大规模工业生成中,由于合成步骤长,部分操作比较复杂,反应时间较长,使得目标产物莫西沙星掺杂了一些杂质。欧洲药典、美国药典、英国药典等多国药典收载的莫西沙星明确要求检测其5个可能存在的杂质A~E(图式 1)。但是除此之外,莫西沙星产品中还存在其他的杂质,例如,Kumar等[12]通过制备液相色谱从莫西沙星原药中分离得到了另一个新的相关物质1-环丙基-6-氟-1, 4-二氢-8-甲氧基-7-([S, S]-2-甲基-2, 8-二氮杂双环[4.3.0]壬烷-8-基)-4-氧亚基-3-喹啉羧酸(化合物F,图式 1),其在莫西沙星产品中的含量大于0.1%;张延峰等[13]通过制备液相色谱也从莫西沙星原药中分离得到了一个新的相关物质1-环丙基-7-(1-氨基-8-氮杂双环[4.3.0]壬烷-8-基)-6-氟-8-甲氧基-1, 4-二氢-4-氧亚基-3-喹啉羧酸(化合物G,图式 1),其在莫西沙星产品中的含量为0.1%。通过紫外光照射莫西沙星也将会产生一种新的杂质[14],即光降解产物1-环丙基-6-氟-7-氨基-8-甲氧基-4-氧亚基-1, 4-二氢喹啉-3-羧酸(化合物H,图式 1)。此外,莫西沙星被氧化剂氧化后还将产生杂质I(图式 1)[15]。尽管莫西沙星相关物质A~E能够在市面上购得,然而其售价非常高,每毫克对照品高达万元以上,而莫西沙星相关物质F~I在市面上还暂无销售,但是这些相关物质都是莫西沙星产品中容易出现的杂质,在进行莫西沙星产品申报生产时以及工业生产过程中的质量控制均需要使用这些莫西沙星相关物质。若直接购买这些相关物质将会增加企业生产莫西沙星的成本,而且相关物质F~I仍难以购得,因此企业通过自己合成这些莫西沙星相关物质不仅能够降低成本,还能制备到市面难以购买的相关物质。本文重点对莫西沙星及其杂质A~I的国内外合成路线进行综述,为今后莫西沙星的工艺优化以及相关物质的合成提供参考。
莫西沙星的合成主要是由其母核1-环丙基-6, 7-二氟-8-甲氧基-4-氧亚基-1, 4-二氢-3-喹啉羧酸(化合物1)C7位氟原子与侧链(S, S)-2, 8-二氮杂双环[4.3.0]壬烷(化合物2)发生取代反应制得(式(1))。但是,在实践反应中莫西沙星母核的C6位氟原子会与C7位氟原子进行竞争性取代,从而产生C6位氟原子被取代的副产物,该副产物难以从产品中进行分离。然而,将母核与螯合剂反应生成螯合物后能够使母核的电子发生转移,增加C7位氟原子活性并钝化C6位氟原子活性,从而提高侧链与母核发生取代反应的选择性。因此,莫西沙星有非螯合和螯合两种制备方法。
|
|
(1) |
非螯合法是将莫西沙星母核C7位氟原子直接与侧链进行反应制得莫西沙星。该法的反应步骤少,操作简单,所需设备少。1992年德国拜耳公司首次公布的莫西沙星合成路线即是采用非螯合法[16]。此外,国内外的其他文献或专利也对该路线进行了优化[17~19],比如使用CuCl2催化剂[20]、改变反应温度[21]、使用不同起始原料[22]等,但是由于C6位和C7位氟原子没有进行活化,缺乏选择性,因此产品的收率均较低并且产品纯化难度较大。
针对传统莫西沙星合成中母核C6位和C7位氟原子缺乏选择性的问题,程青芳等[23]对合成路线进行改进,以1-环丙基-6, 7, 8-三氟-1, 4-二氢-4-氧亚基喹啉-3-羧酸乙酯(化合物3)为母核合成莫西沙星。由于化合物3的C6、C7、C8位均为氟原子,而C6和C8位氟原子的吸电子效应增加了C7位活性,从而使得化合物3的C7位氟原子具有了选择性,其具体的合成路线如式(2)所示。首先,将化合物3用酸进行水解得到1-环丙基-6, 7, 8-三氟-1, 4-二氢-4-氧亚基-3-喹啉羧酸4,接着在K2CO3的催化下将其与拆分后的化合物2反应制得1-环丙基-7-[(S, S)-2, 8-二氮杂双环[4.3.0]壬烷-8-基]-6, 8-二氟-1, 4-二氢-4-氧亚基-3-喹啉羧酸5,最后用甲醇钠将化合物5的C8位氟直接甲氧基化即得到得莫西沙星,该路线总收率为71%,高于传统的合成方法。
|
|
(2) |
采用螯合法制备莫西沙星时能够提高化合物2对母核位置的选择性,能够有效的控制C6位取代杂质的产生,其总反应过程如式(3)所示,首先是将母核的C4位羰基和C3位氧原子用螯合剂进行螯合制得相应的螯合物7,接着与化合物2发生取代反应制得螯合物8,最后进行水解得到莫西沙星。
|
|
(3) |
在螯合法的合成路线中,螯合剂的选择是影响莫西沙星产率的至关重要因素,母核与螯合剂螯合后得到的螯合物吸电子能力越强越有利于C7位氟的反应活性。因此,在螯合法合成莫西沙星的研究中主要是对螯合剂选择的研究。其中HBF4是第一个报道的螯合剂[24],具体的合成路线如式(4)所示。首先将母核1与HBF4反应制得氟硼螯合物9,再与化合物2发生取代反应得到中间体10,最后水解得到莫西沙星,总收率可达70.0%。尽管该合成路线提高了母核C6位氟原子选择性,但是HBF4是一种较贵的试剂并对设备有严重的腐蚀性,不利于工业化大规模生产。随后,Palomo等[25]的专利对该方法进行改进,用廉价并低腐蚀性的BF3·Et2O代替HBF4来制备氟硼螯合物9,制备莫西沙星的总收率达到89%(式(4))。但是,在制备氟硼螯合物9时要先将母核1通过硅化合物转化成中间体11后才能与BF3·Et2O反应,而这步转化过程操作比较复杂。
|
|
(4) |
为了避免HBF4对设备的腐蚀性,Naomi等[26]采用了三乙酸硼代替HBF4来制备硼螯合物中间体,其合成路线如式(5)所示。首先用硼酸和乙酸酐在ZnCl2的催化下制得螯合剂三乙酸硼烷,再与母核1反应得到硼螯合物12,其产率可达到91%。但是,在制备三乙酸硼烷的过程中,ZnCl2催化吸水的能力不足,需要使用过量的乙酸酐。
随后,Chava等[27]对该路线进行优化,不使用ZnCl2催化螯合剂的制备,而是将硼酸与乙酸酐反应后直接与莫西沙星的母核5反应制得硼螯合物12,再与化合物2反应制得含有侧链的螯合物13,其产率可达到90%,最后进行水解得到莫西沙星(式(6))。近些年报道的一些专利以及文献也大多利用硼酸与乙酸酐反应制得的三乙酸硼烷与莫西沙星母核反应形成螯合物后,再在碱性条件下与侧链反应来制备莫西沙星。
|
|
(5) |
|
|
(6) |
除了用乙酸酐制备的三乙酸硼外,目前三丙酸硼烷以及三(三氟乙酸)硼烷也被用作螯合剂[28, 29]。还有一些文献在使用乙酸硼螯合法制备莫西沙星的过程对其反应条件、缚酸剂的选择进行研究[30~35],例如晁阳等[36]采用分批加入硼酸代替一次性加入的方法,同时在缩合后直接酸化,并通过加入食盐水来析出莫西沙星结晶。但是这些优化的方法并没有从根本上改变制备莫西沙星的乙酸硼螯合法,而仅仅是合成路线中的一步或者几步的处理方法有所不同,并未提出具有更强创新性的方法。
Andrea等[37]采用镁盐代替硼螯合剂来制备莫西沙星,具体合成路线如式(7)所示。首先,将莫西沙星母核1与镁盐反应形成螯合物14,接着再与化合物2反应制得含有侧链的螯合物15,最后在酸性条件下进行水解得到莫西沙星,总收率为89%。
徐虹等[38]采用三甲基氯化锡、三丁基氯化锡、三异丙基氯化锡或三苯基氯化锡为螯合剂,先与莫西沙星母核1形成锡螯合物16后,再与化合物2缩合后制得含有侧链的中间体17,最后在酸性条件下进行水解得到莫西沙星(式(8)),总收率可高达92%。但是所用的三烷基氯化锡是剧毒物质,使得生产和操作的难度大;此外,三甲基氯化锡水溶性高,萃取回收较难,而其他烷基锡沸点高,回收纯化也比较困难。因此该合成路线不便于实际的工业化生产。
|
|
(7) |
|
|
(8) |
由于莫西沙星的喹啉环上C6位、C8位以及侧链比较活泼,因此在莫西沙星的合成以及储存过程中容易发生反应产生一些杂质。目前已有9种杂质被报道并能够通过化学合成进行制备。从结构上分析,这些杂质都是莫西沙星的衍生物,是莫西沙星的一个或者几个基团被取代了,因此这些杂质的合成过程与莫西沙星的合成过程基本类似。
从结构上分析,莫西沙星杂质A与莫西沙星的不同之处在于前者C6位是氟原子,后者是甲氧基。因此,杂质A是以2, 3, 4, 5-四氟苯甲酸为原料,经过酰化、酯化、环合等反应制备的[39],其具体合成路线如式(9)所示。首先,将起始原料2, 3, 4, 5-四氟苯甲酸用二氯亚砜进行氯化制得2, 3, 4, 5-四氟苯甲酰氯19;接着采用醇镁法使丙二酸二乙酯19发生亲核取代反应生成2-(2, 3, 4, 5-四氟苯基)马来酸二乙酯20;随后以水作溶剂,将化合物20与对甲苯磺酸进行回流生成3-氧代-3-(2, 3, 4, 5-四氟苯基)丙酸乙酯21,再依次与原甲酸三乙酯、环丙胺反应经过中间体22生成化合物23;在叔丁醇钾的催化下,化合物23进行分子内环化反应从而构建出了喹啉母核即1-环丙基-6, 7, 8-三氟-4-氧亚基-1, 4-二氢-3喹啉羧酸乙酯24,用盐酸进行水解得到1-环丙基-6, 7, 8-三氟-4-氧亚基-1, 4-二氢-3喹啉羧酸25;最后将化合物25与化合物2反应生成1-环丙基-7-(S, S-2, 8-二氮杂环[4.3.0]-壬烷-8-基)-6, 8-二氟-1, 4-二氢-3喹啉羧酸,即莫西沙星杂质A,其总收率为34%。尽管该路线合成步骤较多,但是每步都是经典的化学反应,其操作简单,所合成的产品纯度也较高,能够作为对照品进行研究。
|
|
(9) |
将莫西沙星杂质B与杂质D对比发现两者不同之处是C8位的取代基,杂质B的C8位是甲氧基,而杂质D的C8位是氟原子,因此可以将杂质D的C8位氟原子通过取代反应来制备杂质B[40],具体合成路线如式(10)所示。首先以2, 3, 4-三氟-5-甲氧基苯甲酸为原料通过多步反应制得莫西沙星杂质D(式(13)),然后与甲醇钠反应即可制得杂质B。尽管由杂质D转化成杂质B只有一步反应,但是由于C6位和C1位大的空间位阻使得杂质B的产率较低,为54%。
|
|
(10) |
杂质C与莫西沙星的区别在于前者C8位是乙氧基,而莫西沙星的C8位为甲氧基,因此在合成杂质C时需要用乙基试剂将乙基引入到母核的C位。目前莫西沙星杂质C有两条合成路线,其中闵涛等[41]报道的合成方法是在莫西沙星杂质E的基础上来制备杂质C(式(11))。该合成路线首先将杂质E侧链(S, S)-2, 8-二氮杂双环[4.3.0]壬烷中的仲氨基用Boc进行保护(化合物26),以免乙基试剂在对母核C8位乙基化时也将侧链仲氨基乙基化;接着,Boc保护的化合物26用乙基试剂将C3羧基和C8位羟基同时乙基化(化合物27),从而在母核C8位引入了乙氧基;随后,将化合物27的C3位在碱性条件下进行水解得到喹啉羧酸化合物28;最后,用三氟乙酸将化合物28的保护基团Boc脱去得到1-环丙基-7-(S, S-2, 8-二氮杂环[4.3.0]-壬烷-8-基)-6-氟-8-乙氧基-1, 4-二氢-3喹啉羧酸,即莫西沙星杂质C,其总收率为65%。
上述合成路线是以杂质E为起始原料,通过官能团转化制得杂质C,尽管避免了从苯甲酸衍生物起始一步步构建母核的冗长步骤,但是该路线需要先制备或购买莫西沙星杂质E作为原料,因此该合成方法存在合成路线复杂、成本高等缺点。随后,陈善龙等[42]报道了一条新的合成路线来制备莫西沙星杂质C,该合成路线以莫西沙星母核1为原料按照常见的乙酸硼螯合物法制备莫西沙星的合成路线来制备杂质C,其具体合成步骤如式(12)所示。首先,在脱甲基试剂BBr3的作用下将莫西沙星母核1的甲基脱去得到羟基化合物29;接着,化合物29与烷基化试剂溴乙烷反应得到相应的乙氧基化合物30,再将化合物30的喹诺酮母核与螯合剂硼酸乙酯进行鳌合得到鳌合物31;随后,将化合物31与侧链发生亲核取代得到化合物32;最后将化合物32水解即得到莫西沙星杂质C,其总收率为76%。该合成路线反应条件温和、容易操作,便于莫西沙星杂质C的制备。
|
|
(11) |
|
|
(12) |
|
|
(13) |
莫西沙星杂质D的结构与杂质A类似,仅仅是C6位取代基不同,前者是甲氧基取代,后者是氟原子取代。因此,只需将西沙星杂质A的起始原料2, 3, 4, 5-四氟苯甲酸(化合物18,式(9))用2, 3, 4, -三氟-5-甲氧基苯甲酸(化合物33)代替即可按照杂质A的合成路线来制备杂质D(式(13))[40],总收率在21%左右。
从化学结构上分析,杂质E是莫西沙星的酸性降解产物,源于莫西沙星C8位甲氧基在酸性条件下发生的醚键断裂,暴露出酚羟基随即产生的杂质。因此,杂质E的合成实质上是对莫西沙星C8位进行去甲基化。闵涛等[43]采用三甲基碘硅烷、氢溴酸和三溴化硼作为脱甲基试剂均能够制备莫西沙星杂质E(式(14)),其总收率分别为74%、55%和50%。氢溴酸作为脱甲基试剂是剧烈的反应条件下进行,而且反应时间较长,这会使得莫西沙星原料部分被降解;三溴化硼由于对空气敏感,在滴加过程中容易挥发变成气雾,并且在加水后处理时出现络合物,分离困难。因此氢溴酸和三溴化硼作为脱甲基试剂其反应效率低于反应条件较温和的三甲基碘硅烷。
|
|
(14) |
莫西沙星侧链氮杂环仲氮的氢原子较活泼,在合成莫西沙星的过程中容易形成甲基化产物1-环丙基-6-氟-1, 4-二氢-8-甲氧基-7-([S, S]-N-甲基-2, 8-二氮杂双环[4.3.0]壬烷-8-基)-4-氧亚基-3-喹啉羧酸,即杂质F。因此,在合成杂质F时重点是对其侧链进行甲基化。目前已经有多项专利以及文献报道了其合成方法,按照侧链氮杂环仲氮的甲基化方式可以分为甲基试剂直接甲基化法和甲醛缩合法两种。
侧链氮杂环仲氮的甲基化试剂主要有硫酸二甲酯、碳酸二甲酯和碘甲烷这三种。叶海等[44]采用这三种甲基化试剂均合成了莫西沙星杂质F,具体合成过程如式(15)所示。首先,将化合物2的五元环上仲氨基用Boc进行保护(化合物42),再用甲基化试剂对其另外一个仲氨基进行甲基化生成N-甲基中间体43;接着,用三氟乙酸将Boc保护基团去掉得到N-甲基-(S, S)-2, 8-二氮杂双环[4.3.0]壬烷44;随后,按照莫西沙星的经典制备方法与螯合物12反应制得化合物45;最后,将化合物45水解即得到莫西沙星杂质F。但是,该专利没有报道产品的最终收率以及不同甲基化试剂对产品产率的影响。
上述合成方法步骤较长,操作也较繁琐,在第二步Boc2O保护氨基时还缺乏选择性,给产物的纯化带来困难。此外,由于化合物2没有紫外吸收,其在保护和甲基化反应中不利于反应监测和纯化。为了简化合成步骤,他们又报道了一条新合成路线(见式(16))[45]。该路线以莫西沙星为起始原料,首先直接用甲基化试剂CH3I将侧链仲氮进行甲基化得到化合物46,接着在碱性条件下将C3位甲酯进行水解即可得到莫西沙星杂质F,总收率为51%。
|
|
(15) |
|
|
(16) |
此外,卢建勋等[46]也直接用CH3I甲基化法来制备莫西沙星杂质F。该合成方法与叶海等[44]报道的方法类似,只是首先用异丙醇将莫西沙星的C3位羧基保护制成异丙酯47,然后再用CH3I将侧链氨基进行甲基化制得N-甲基中间体48,最后水解得到莫西沙星杂质F(式(17)),其总收率为34%。尽管上述方法缩短了反应步骤,但是在使用CH3I甲基化时不仅生成单甲基化产物,还生成一些季铵盐副产物,这给产物的分离纯化带来困难。
为了避免因使用甲基化试剂产生季铵盐副产物,郑清四等[47]通过埃施魏勒-克拉克反应在甲酸的存在下用甲醛直接与莫西沙星的侧链氮杂环的仲胺反应生成N-甲基产物莫西沙星杂质F,其产率为87%(式(18))。
|
|
(17) |
|
|
(18) |
针对在合成关键中间体N-甲基-(S, S)-2, 8-二氮杂双环[4.3.0]壬烷44时由于Boc2O对起始原料化合物2的氨基保护(式(15))缺乏选择性造成产率低的缺点,张明光等[48]对该合成路线进行了优化,用(S, S)-8-苄基-2, 8-二氮杂双环[4.3.0]壬烷49为原料来制备莫西沙星杂质F。与侧链原料2相比,化合物49是对原料2五环氮杂环的氨基用苄基进行了保护,该苄基的引入不仅能够提高下一步甲基化反应的选择性,还能使得产物具有紫外吸收,更有利于分离纯化。改进后的具体的合成方法如式(19)所示,首先通过甲醛和硼氢化钠将原料49的仲胺进行甲基化得到(S, S)-8-苄基-2-甲基-2, 8-二氮杂双环[4.3.0]壬烷50;然后,在甲醇溶剂中用Pd/C和氢气脱掉苄基生成甲基化侧链51;最后再与莫西沙星母核1反应得莫西沙星杂质F,总收率为13%。
|
|
(19) |
杂质G是莫西沙星的降解产物,其C7位的(S, S)-2, 8-二氮杂双环[4.3.0]壬烷-8-基被降解脱去变成氨基。目前有报道了三种合成方法,其一是采用叠氮化还原的方法制备[49, 50],如式(20)所示。首先,用叠氮化钠对莫西沙星母核5的C7氟原子进行亲核取代生成叠氮化合物52;接着,用Pd/C和氢气将叠氮基还原成氨基得到化合物53;最后,在碱性条件下水解即得到杂质G总收率在77~84%之间。尽管该路线合成简单,但是叠氮化钠是易燃易爆的剧毒物,操作危险,不利于大规模工业化制备莫西沙星杂质G。
针对使用叠氮化钠危险性,王足兵等[51]使用苄胺取代叠氮钠的新合成方法来制备莫西沙星杂质E,如式(21)所示。首先,用苄胺类试剂对莫西沙星母核5的C7位氟原子进行取代制得莫西沙星苄胺化合物54;接着,用Pd/C和氢气脱去苄基得到氨基化合物53,再用碱将C3位酯基水解基得到莫西沙星杂质G,总收率为20%。
尽管该合成路线避免使用了叠氮化钠带来的危险,但是其在脱去苄基却使用了Pd/C和氢气,这在大规模工业生产中仍然存在危险。随后张明光等[48]对杂质G的合成路线进一步进行了优化,提出了一种更安全、简便、高效的制备方法,如式(22)所示。以叔丁胺作为胺化试剂,与硼螯合物12反应制得55,再依次在酸性和碱性条件性脱去硼螯合酯和叔丁基得到莫西沙星杂质G,总收率为27%。
|
|
(20) |
|
|
(21) |
|
|
(22) |
杂质H可能是由莫西沙星侧基2, 8-二氮杂双环[4.3.0]壬烷-8-基带入1-氨基-8-氮杂双环[4.3.0]壬烷杂质在与母核缩合产生的衍生物。目前已有文献报道了其合成方法[48],如式(23)所示。该合成路线以1-硝基环己烯57为原料,在三氟乙酸的催化下与N-(甲氧甲基)-N-(三甲基硅烷基)苄胺58发生1, 3-双极性环加成反应生成1-硝基-8-苄基-8-氮杂双环[4.3.0]壬烷59,接着通过水合肼还原得到1-氨基-8-苄基-8-氮杂双环[4.3.0]壬烷60;再依次氨基Boc保护、脱苄基反应得到1-叔丁氧羰基胺基-8-氮杂双环[4.3.0]壬烷62;最后与莫西沙星母核硼螯合物12缩合并脱去硼酯制得莫西沙星杂质H,总收率为14%。
莫西沙星的侧基二氮杂双环[4.3.0]壬烷-8-基空间位阻较小,其结构中的两个胺容易被氧化剂氧化,叔胺被氧化成氮氧化合物、仲胺被氧化成羟胺,羟胺在高温的条件下脱去一分子水生成烯烃。因此,莫西沙星杂质I可以直接通过氧化剂氧化莫西沙星制得。谭珍友等[52]使用的氧化剂双氧水或过氧乙酸,其具体合成路线如式(24)所示。首先,按照传统盐酸莫西沙星的制备方法合成盐酸莫西沙星;接着用氧化剂双氧水、过氧乙酸或过氧苯甲酸对盐酸莫西沙星侧链进行氧化得到氧化物63,再将羟胺脱去水分子生成莫西沙星杂质I,从盐酸莫西沙星氧化脱水制得杂质I的收率可达到32%。
|
|
(23) |
|
|
(24) |
莫西沙星作为第四代喹啉酮类抗菌药物,通过抑制DNA Ⅱ和Ⅳ型拓扑异构酶来抑制DNA复制从而产生杀菌作用,其抗菌谱几乎涵盖了呼吸道主要的致病菌,并具有口服生物利用度高、毒性低、疗效好等优点。目前对莫西沙星的合成工艺研究很多,总体上可以分为非螯合法和螯合法两种合成策略,其中螯合法具有更大的优势,部分合成路线具有良好的收率。但是,在螯合法中螯合剂及以侧链与母核缩合中缚酸剂的选择对莫西沙星产品的产率影响甚大,而目前文献报道的螯合剂和缚酸剂在母核螯合步骤与侧链缩合步骤只产生中等产率的产物,因此在今后莫西沙星的工艺研究中螯合剂和缚酸剂的开发仍然是重点。另一方面,莫西沙星在生成、贮藏和运输过程中容易产生副产物、降解产物等特殊杂质。药物在临床上产生不良反应除了与药品本身药理活性有关外,还与其掺杂的杂质有很大关系。为了保证临床用药的安全性,对药品的杂质进行研究是药品质量控制的一项重要内容。莫西沙星杂质市场购买售价高,而且部分杂质难以购得需药品研究人员进行合成。目前9个莫西沙星杂质都已能够通过化学合成制备,但是部分杂质的合成路线仍然比较复杂,产率比较低,不利于大规模的工艺制备。因此,对莫西沙星杂质的合成路线进行线优化仍然是莫西沙星研究中的一项重要和有价值的工作。
刘九雨, 郭慧元. 国外医药抗生素分册, 2002, 23:274~278. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=gwyykssfc200206009
宋丽晓. 中国现代药物应用, 2009, 3:102~103. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=zgxdyyyy200904024
J M Blondeau. J. Antimicrob. Chemother., 1999, 43:1~11. http://www.ncbi.nlm.nih.gov/pubmed/10382869
J A Hoogkamp-Korstanje, J Roelofs-Willemse. J. Antimicrob. Chemother., 2000, 45:31~39. http://www.ncbi.nlm.nih.gov/pubmed/10629010
U Petersen, A Krebs, T Schenke et al. EP 550903,1993-07-14.
M Takemura, Y Kimura, N Matlsuhashi. EP 603887,1994-06-29.
王福东, 李谦和, 彭东明. 药学进展, 2003, 27:217~220. http://med.wanfangdata.com.cn/Paper/Detail?id=PeriodicalPaper_yxjz200304007
刘明亮, 魏永刚, 孙兰英等. 中国医药工业杂志, 2004, 35:129~131. http://d.wanfangdata.com.cn/Periodical/zgyygy200403001
翟红, 常瑜, 相会明等. 化工生产与技术, 2007, 14:15~17. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hgscyjs200706004
江超, 周宜遂. 中国医药工业杂志, 2009, 40:393~396. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=zgyygy200905022
卢定强, 王维胞, 凌岫等. 现代化工, 2014, 34:33~37. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=xdhg201402008
Y R Kumar, V V Prasad Raju, R R Kumar et al. J. Pharm. Biomed. Anal., 2004, 34:1125~1129. doi: 10.1016/j.jpba.2003.11.012
张延峰, 陈磊, 王忠等. 中国医药工业杂志, 2012, 43:890~892. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=zgyygy201211005
U Hubicka, J Krzek, B Z·uromska et al. Photochem. Photobiol. Sci., 2012, 11:351~357. doi: 10.1039/C1PP05259D
谭珍友, 罗统有, 刘道甫等. CN:105566322A, 2016.
P Uwe, K Andress, S Thomas et al. EP:550903, 1993.
J Ludescher, A C Pise, A G Holkar et al. EP:1992626, 2007.
刘运达, 刘世领, 陆晓等. CN:102675306A, 2012.
郭峰. CN:102675313A, 2012.
许士娜, 夏吕玲, 高霞. CN:105524060A, 2016.
杨杨, 朱永强, 王凌燕等. CN:101941969A, 2010.
G Reinhold, M Klaus, H Werner et al. US:6897315, 2005.
程青芳, 王启发, 许兴友等. 中国医药工业杂志, 2010, 41:561~563. http://d.wanfangdata.com.cn/Periodical/zgyygy201008001
D Jianfuederiko, M I Anna. JP:63-198665, 1988.
N Palomo, G Cosme, P Villasante et al. EP:1832587, 2007.
T Naomi, F Hironobu, M Hiroshi. US:5157117, 1996.
H R Chava. WO:2005012285A, 2005.
D R Rao, N R Kankan, P Ravikumar et al. WO:2008059223, 2008.
张柯华, 张柏林, 陈喆等. CN:103159759A, 2013.
江超, 周宜遂. 中国医药工业杂志, 2009, 40:393~395. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=zgyygy200905022
张红雨, 张俊杰, 于振艳等. CN:101973992A, 2011.
王庆娟, 王秀娟, 郭艳玲. 齐鲁制药, 2011, 30:683~684.
彭俊华, 陈婷婷, 车来滨等. CN:103012452A, 2013.
邹美香, 郭建锋, 段桂运等. CN:103172629A, 2013.
刘聪, 蒋狄峰, 田爱荣等. 精细化工中间体, 2013, 43:33~37. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=jxhgzjt201301010
晁阳, 包玉胜, 叶海等. 海峡药学, 2013, 25:16~18. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=haixyx201310006
C Andrea, P Pierluigi, J G Liu et al. US:20110230661, 2011.
徐虹, 王威, 张铮等. CN:103073570, 2013.
朱永强, 王凌燕, 黄国正等. CN:201110163437A, 2011.
闵涛, 车晓明, 陆晨光等. 中国, CN:104211701A, 2014.
闵涛, 陆晨光, 车晓明等. CN:104945399A, 2015.
陈善龙, 谭娟. 中国抗生素杂质, 2016, 41:117~121. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=zgksszz201602006
闵涛, 陆晨光, 车晓明等. CN:104945398A, 2015.
叶海, 晁阳, 曹卫等. CN:102584819A, 2012.
叶海, 晁阳, 闵涛等. CN:103396416A, 2013.
卢建勋, 孙占国, 张锦聪等. CN:103664940A, 2014.
郑清四, 林海芳. CN:103467466A, 2013-12-25.
张明光, 马堰启, 夏正君等. 中国医药工业杂志, 2014, 45:404~408. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=zgyygy201405002
Q R Qi, J Pan, X Q Guo et al. Bioorg. Med. Chem. Lett., 2012, 22:7688~7692. doi: 10.1016/j.bmcl.2012.09.106
魏臣绿, 潘佳, 何菱等. 华西药学杂志, 2010, 25:253~256. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxyxzz201003002
王足兵, 赵超, 郭璇等. CN:104292158A, 2015.
谭珍友, 罗统有, 刘道甫等. CN:105566322A, 2016.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

























 下载:
下载:
 下载:
下载: