表 1
化合物的结构及其生物活性
Table 1.
Structure and their activity data of compound

蛋白质翻译后修饰是蛋白质功能的调节机制之一,且在蛋白质的各种方面起着重要作用[1, 2]。类泛素化(Neddylation)是蛋白质翻译后修饰的其中一种,是将NEDD8与底物蛋白共价结合的多步级联酶促反应,该反应涉及四种酶:NEDD8激活酶(NAE)、NEDD8结合酶(E2)、NEDD8连接酶(E3)和NEDD8异肽酶[3]。类泛素化对调控细胞功能如DNA损失、细胞增殖等起重要作用,其异常会引起神经退行性疾病和多种肿瘤等的发生[4, 5]。多项研究表明类泛素化中的三种酶在多种癌症细胞内水平过高及过度表达,因此类泛素化是有吸引力的抗肿瘤靶标[6]。目前,类泛素化最有代表性的底物蛋白是Cullin家族成员[7],当Cullin经过类泛素化后可将CRL激活,而CRL是E3泛素连接酶的最大家族,负责许多关键调节因子的泛素化和降解[8]。其在许多生物学过程中发挥作用,如细胞周期、信号传导和肿瘤发生等[9]。由于CRL的过度激活会导致肿瘤的生长和发展,因此针对以Cullin为底物的类泛素化是一种抗癌策略[10]。NAE抑制剂MLN4924的发现,更进一步证实这种策略是可行的。然而MLN4924阻断了类泛素化的整个过程,会导致一定的毒性,且有报道指出癌细胞可通过靶向NAEβ的稀有克隆产生对MLN4924的抗性[11, 12]。为了克服MLN4924的不足,研究人员开始探索类泛素化的其他环节来开发出新的抑制剂。研究表明,DCN1与UBE2M之间的相互作用可作为抑制类泛素化提供新的思路[13]。
近年来,已报道了多种不同骨架结构的靶向DCN1-UBE2M相互作用的抑制剂。Hammill等[14, 15]通过高通量筛选得到一系列哌啶基脲类化合物,能够抑制此相互作用且具有较好的生物活性。然而有关这类化合物的构效关系分析尚未报道,本研究联合运用3D-QSAR、分子对接和分子动力学这三种方法来研究此类化合物的结构与生物活性之间的关系,以此为进一步对这类化合物的结构优化提供理论基础。
本研究以90个哌啶基脲类DCN1-UBE2M相互作用抑制剂作为研究对象,其结构和生物活性来自文献[14, 15]。所有化合物的生物活性数据是通过相同实验所得到。根据化合物的活性范围和结构差异,以近似3∶1的比例随机划分成69个训练集和21个测试集。化合物的结构和生物活性数据见表 1。
所有化合物的分子结构均由ChemBioDraw11.0构建[16],并利用SYBYL-X 2.0的“Minimize”模块进行结构优化,得到稳定构象。具体过程如下:首先选择Tripos力场,在此力场下赋予小分子Gasteiger-Hückel电荷,再将最大迭代次数和能量收敛能级差分别设为1000和0.005kcal·(mol·Å)-1,其余参数均为缺省值。
分子叠合是构建3D-QSAR模型的重要环节,影响模型的优劣。本研究选用基于公共骨架的方法进行叠合,以活性最好的化合物74为模型分子,通过Sybyl-X 2.0中的Align Database完成叠合。模板分子和叠合效果如图 1所示,在模板分子中用红色标明公共骨架。

本文构建的3D-QSAR模型在SYBYL-X 2.0软件中完成。在在CoMFA模型中,利用Lennard-Jones计算立体场,而静电场的计算则是采用Coulomb函数。在计算过程中,将训练集的57个化合物叠合后置于间距为2Å的晶格中,再用sp3杂化的碳原子(静电荷为+1,1.52Å的范德华半径)作探针原子来计算化合物的立体场和静电场。其中这两个场的能量截断值均为30kJ/mol。对于CoMSIA模型,除了对立体场和静电场进行计算,还增加对疏水场、氢键供体及受体场这三个分子场的计算。计算时所使用的探针是一个半径为1.52Å,疏水性、氢键供体和受体强度以及电荷均为+1的sp3杂化碳原子。同时与CoMFA模型所使用的计算方法不同,CoMSIA模型采用高斯函数计算分子场的能量,该函数与距离相关,因此无需设定能量截断值。另外衰减因子α为0.3。
利用偏最小二乘法(PLS)建立了分子场和生物活性之间的数学模型;利用留一法执行交叉验证获得交叉验证相关系数(q2)和最佳组分数(N);再根据N执行非交叉验证得到相关系数r2、标准估计误差(SEE)和F检验值。q2可通过下面的计算方程得到[17]:
|
|
其中,Ypred,Yactual和Ymean均为训练集化合物的生物活性,分别代表预测值、实验值和平均值。
为了评价由训练集所构建的3D-QSAR模型是否具有良好的预测能力,本研究引入外部验证法,该法可得外部验证系数
|
|
式中,SD为测试集化合物的生物活性预测值与训练集化合物生物活性实验值的平均值之差平方和,而PRESS则指测试集化合物的生物活性预测值与实验值之差平方和。
分子对接研究所使用的软件为Molegro Virtual Docker 5.5(MVD)。DCN1靶蛋白的晶体结构(PDB ID:6BG3)来自PDB数据库。进行对接之前,先将靶蛋白进行预处理,修补部分氨基酸残基和保留部分水分子。先提取原配体DOJ_1301进行自身对接验证所用对接方法的可靠性,再将所有化合物与靶蛋白对接。对接时所设置的参数如下:以Moldock函数作为评分,最大迭代次数设为500,最终输出构象30个,其余的参数不变。
为了更清楚地了解化合物与靶蛋白之间的作用机制,运用GROMACS软件包对其复合物完成50ns的分子动力学模拟研究[18]。模拟前选用GROMOS96力场作为DCN1靶蛋白的力场,由于该力场无法识别小分子配体,因此通过PRODRG在线服务器处理配体[19],可获取到对应的拓扑文件。然后将复合物体系放置在正十二面体的盒子中,其边界大小为1nm。采用周期性边界条件,用SPC水模型填充于蛋白质-配体以外的区域。为了使体系保持电中性,根据verlet切断法随机将Na+或者Cl-加入到复合物体系中[20]。接着先将体系再进行2500步的最陡下降法和2500步的共轭梯度法对体系进行优化。完成优化后进入100ps的NVT和NPT平衡阶段,具体参数设置如下:利用V-rescale恒温耦合法使体系升温至300K,进行NVP平衡时体系存在1000kJ·mol-1·nm-2的限制力,为了平衡溶剂分子;在NPT平衡中,温度一直为300K,压强为1个标准大气压。最终在恒温恒压下进行50ns的分子动力学模拟。
在表 2中列出了CoMFA和CoMSIA模型的统计结果。当3D-QSAR模型的统计参数满足以下条件:q2>0.5,r2>0.9,rpred2> 0.5,则表明模型具有可靠性且预测能力较好[21, 22]。从表 2可知,所构建的CoMFA模型的统计参数如下:交叉验证系数q2=0.686,最佳组分数N=10,非交叉验证系数r2=0.966,SEE=0.168,F检验值=164.086,
 下载:
导出CSV
下载:
导出CSV
| Model | N | q2 | r2 | SEE | rpred2 | F | S | E | H | D | A |
| CoMFA-SE | 10 | 0.686 | 0.966 | 0.168 | 0.879 | 164.086 | 0.569 | 0.431 | - | - | - |
| CoMSIA-SE | 6 | 0.514 | 0.852 | 0.338 | 59.615 | 0.404 | 0.596 | - | - | - | |
| CoMSIA-SH | 7 | 0.605 | 0.905 | 0.273 | 83.109 | 0.354 | - | 0.646 | - | - | |
| CoMSIA-SD | 9 | 0.585 | 0.869 | 0.326 | 43.542 | 0.705 | - | - | 0.295 | - | |
| CoMSIA-SA | 10 | 0.619 | 0.892 | 0.299 | 47.723 | 0.666 | - | - | - | 0.334 | |
| CoMSIA-EH | 6 | 0.514 | 0.861 | 0.328 | 63.750 | - | 0.433 | 0.567 | - | - | |
| CoMSIA-ED | 3 | 0.428 | 0.551 | 0.575 | 26.615 | - | 0.614 | - | 0.386 | - | |
| CoMSIA-EA | 3 | 0.485 | 0.649 | 0.508 | 40.149 | - | 0.499 | - | - | 0.501 | |
| CoMSIA-HD | 7 | 0.610 | 0.895 | 0.289 | 74.247 | - | - | 0.767 | 0.233 | - | |
| CoMSIA-HA | 10 | 0.634 | 0.937 | 0.227 | 86.789 | - | - | 0.735 | - | 0.265 | |
| CoMSIA-DA | 10 | 0.503 | 0.705 | 0.494 | 13.854 | - | - | - | 0.551 | 0.449 | |
| CoMSIA-SEH | 6 | 0.552 | 0.880 | 0.304 | 76.033 | 0.219 | 0.348 | 0.433 | - | - | |
| CoMSIA-SED | 6 | 0.555 | 0.844 | 0.347 | 55.837 | 0.281 | 0.466 | - | 0.254 | - | |
| CoMSIA-SEA | 3 | 0.516 | 0.693 | 0.476 | 48.807 | 0.171 | 0.411 | - | - | 0.417 | |
| CoMSIA-SHD | 8 | 0.625 | 0.912 | 0.266 | 77.457 | 0.282 | - | 0.538 | 0.180 | - | |
| CoMSIA-SHA | 9 | 0.664 | 0.929 | 0.240 | 85.914 | 0.244 | - | 0.501 | - | 0.255 | |
| CoMSIA-SDA | 10 | 0.630 | 0.888 | 0.304 | 46102 | 0.514 | - | - | 0.276 | 0.210 | |
| CoMSIA-EHD | 6 | 0.543 | 0.857 | 0.333 | 61.706 | - | 0.335 | 0.429 | 0.236 | - | |
| CoMSIA-EHA | 3 | 0.538 | 0.721 | 0.453 | 56.010 | - | 0.331 | 0.312 | - | 0.357 | |
| CoMSIA-HAD | 9 | 0.665 | 0.929 | 0.240 | 86.164 | - | - | 0.622 | 0.181 | 0.197 | |
| CoMSIA-EDA | 4 | 0.519 | 0.709 | 0.467 | 38.902 | - | 0.366 | - | 0.254 | 0.380 | |
| CoMSIA-SEHD | 7 | 0.572 | 0.900 | 0.280 | 78.394 | 0.176 | 0.292 | 0.368 | 0.164 | - | |
| CoMSIA-SEHA | 5 | 0.540 | 0.826 | 0.364 | 59.741 | 0142 | 0.260 | 0.302 | - | 0.295 | |
| CoMSIA-SHDA | 9 | 0.682 | 0.931 | 0.237 | 0.875 | 87.986 | 0.201 | - | 0.457 | 0.153 | 0.189 |
| CoMSIA-SEDA | 4 | 0.539 | 0.736 | 0.444 | 44.660 | 0.139 | 0.314 | - | 0.218 | 0.329 | |
| CoMSIA-EHDA | 5 | 0.569 | 0.821 | 0.369 | 57.817 | - | 0.236 | 0.273 | 0.216 | 0.276 | |
| CoMSIA-SEHDA | 7 | 0.599 | 0.900 | 0.280 | 78.857 | 0.219 | 0.214 | 0.300 | 0.156 | 0.200 |
将CoMSIA模型的五种分子场进行排列组合并选出最佳模型。通过对比发现CoMSIA(SHDA)模型是最佳模型,其交叉验证系数q2为0.682,最佳组分数N为9,r2为0.931,SEE为0.237,F为87.986,rpred2为0.875。模型中立体场、疏水场、氢键供体场和氢键受体场的贡献值分别为0.201、0.457、0.153和0.189。
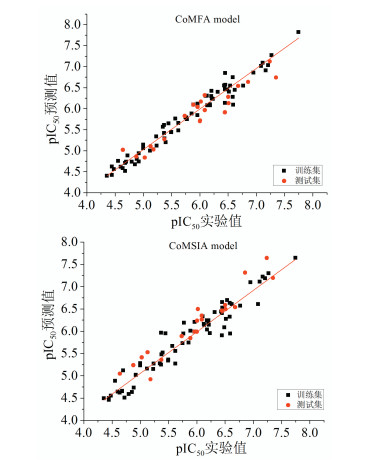
通过所建立的两个3D-QSAR模型,对训练集和测试集中化合物的生物活性进行预测。为了能够更清楚训练集和测试集这两个数据集化合物生物活性的实验值和预测值之间的相关性,以实验值作为横坐标,预测值则为纵坐标,做出相关模型的散点图来描述相关性。从图 2可知,CoMFA和CoMSIA模型的散点图具有一定的线性关系,两数据集的回归曲线比较接近,并且散点均位于回归曲线的两侧,进一步表明这两个3D-QSAR模型具有可靠性以及较好的预测能力。
等势图可以直观地解释建立的3D-QSAR模型中分子场对化合物生物活性的影响。以化合物74为模板,CoMFA模型的立体场和静电场等势图结果如图 3所示。图 3(a)为CoMFA模型立体场,绿色区域代表在此处引入体积较大的基团有利于提高化合物活性,而黄色区域相反。从图中可知,绿色区域覆盖在R2取代基萘环右侧的两个氢原子附近,说明这里引入较大体积的基团有利于化合物生物活性的提高,如化合物74的活性(pIC50=7.745)高于化合物72(pIC50=7.114),这是由于在化合物74中R2取代基为萘环,而化合物72的R2取代基是苯并环戊烷。其黄色区域主要出现在哌啶基两侧以及萘环附近,表示在这些位置引入较小体积基团时有可能将化合物活性提高,可以尝试在此处引入一些较小体积的基团。由于萘环附近既有黄色区域又有绿色区域,因此应结合考虑引入的基团大小。

图 3(b)表示CoMFA模型的静电场等势图,图中的红色区域说明在这区域引入负电性基团有利于化合物活性提高,而蓝色区域则说明正电性基团能够提高化合物活性。从图中观察到有一小块蓝色在2-戊基取代基中的2位甲基附近,表示这里引入正电性基团可能使化合物活性增强,化合物39(pIC50=4.444)的活性明显低于化合物28(pIC50=6.585),两者化合物活性差异大是因为化合物39在此位置引入了一个氧原子。另一块蓝色是在萘环上的一个氢原子上方,说明可尝试在该处引入正电性基团。红色区域分布在萘环和哌啶基附近,提示这些区域适当的引入负电性基团有利于提高活性。
在CoMSIA模型中,立体场等势图与CoMFA模型的相似,在这里不做过多阐述。疏水场等势图如图 4(b)所示,黄色区域代表增加疏水性基团能够使化合物活性增加,而灰色区域恰好相反。可以看到,在2-戊基的2位甲基有一块黄色区域,表明在甲基处引入疏水性基团可提高活性,化合物39(pIC50=4.444)将2-戊基的2位甲基替换成2位羰基,而化合物28(pIC50=6.585)在这个位置并没有引入任何取代基,这一区别是导致两化合物活性相差很大的原因。此外还有一块黄色区域包裹在萘环最右侧的一个碳原子,提示可以在此处进行结构优化来增加活性。在羰基附近、R3取代基苄基中的亚甲基附近以及萘环中间的碳原子有灰色区域,意味着引入亲水基团有利于活性提高。进行结构优化时可尝试在这些位置增加亲水性基团来提高活性。

图 4(c)是氢键供体场等势图,青蓝色区域说明氢键供体在这里有利于化合物活性,当它位于紫色区域时起到相反的作用。有一块青蓝色区域出现在哌啶基附近,表明在该处含有氢键供体基团对化合物活性起增强作用。一大块青蓝色区域不仅覆盖了2-戊基中的烷烃链,也覆盖了亚氨基以及萘环中与其相连的苯环,说明有氢键供体基团有利于增强活性。其中亚氨基可能是氢键供体,能与DCN1靶蛋白的氨基酸产生氢键相互作用。在对接分析中,亚氨基与Gln1114相互作用形成一个氢键,验证了前面的推测。
图 4(d)为氢键受体场等势图,在该图中红色区域表示当该处存在氢键受体会降低活性,紫色则表明有氢键受体会使活性提高。在R1取代基的2位甲基和R3取代基的亚甲基周围有一块红色区域,提示在这里有氢键受体会导致活性的降低,如化合物35(pIC50=4.444)引入羰基后明显低于化合物26(pIC50=6.585)。紫色区域分布于哌啶基、羰基、萘环的右侧以及2-戊基中的烷烃链附近,表示这些位置有氢键受体的存在对化合物活性有利。
90个化合物都能与DCN1靶蛋白的活性口袋相结合,且形成了3~5个氢键,其中与Gln1114所形成的氢键是共有的,故Gln1114是关键氨基酸残基。为了进一步阐明化合物与DCN1靶蛋白的结合模式,选取化合物74来进行分析,其对接图如图 5所示。从图中可知,靶蛋白的活性口袋由Ile1083、Ile1086、Phe1089、Pro1097、Ala1098、Ile1105、Ala1106、Phe1109、Gln1114、Glu1116、Phe1164、Met1177、Tyr1181和Leu1184等氨基酸残基所组成。化合物74与DCN1形成了5个氢键,分别是:化合物74的亚氨基与Gln1114形成氢键相互作用,其羰基上的氧原子和水分子1558、1631以及氨基酸Tyr1181产生3个氢键相互作用,还有一个由哌啶基上的氮原子与水分子1482所形成。其中与Tyr1181产生的氢键相互作用较弱,故而不考虑其对活性的影响。
为了进一步了解在真实环境中化合物与靶蛋白的结合模式以及验证对接结果的可靠性,选择化合物12、74与DCN1靶蛋白的复合物为初始构象,分别进行了50ns的分子动力学模拟。均方根偏差(RMSD)能够用于评判模拟过程中复合物体系是否达到平衡,两复合物体系的RMSD在模拟过程中的变化如图 6所示。从图中可观察到在25000ps以前74-6BG3复合物体系的RMSD值在波动,表明此时的体系是不稳定的。随后达到较稳定的状态,其平均RMSD值为0.542nm。而12-6BG3复合物体系一直在波动,这可说明化合物12与化合物74生物活性差异的这一现象。

均方根涨落(RMSF)描述了在模拟过程中受体蛋白中每一个氨基酸的波动情况,可用来评价各个氨基酸的稳定性。计算出两个复合物体系中每个氨基酸的RMSF值,其结果如图 7所示。为了使图形美观,在图中将6BG3中的1062~1250号氨基酸修改为165~353。在两个复合物体系中,结合口袋的氨基酸如Ile1083、Phe1109、Gln1114、Tyr1181、Met1177等都较稳定,表明化合物12、74均与此口袋结合。进一步对比发现,74-6BG3复合物体系中的氨基酸Gln1114和Tyr1181的RMSF低于12-6BG3体系。推测是因为,相比于化合物12与氨基酸Gln1114产生的氢键相互作用,化合物74与氨基酸Gln1114形成了较强的氢键相互作用。氨基酸Tyr1181与化合物74产生氢键相互作用,而与化合物12则没有,这与前面的对接结果相吻合。此外,疏水口袋中的Ile1086、Phe1089、Phe1109、Thr1113、Phe1164这些氨基酸的RMSF值在74-6BG3体系中要低于12-6BG3体系,可能是化合物74的疏水作用强于化合物12的,再一次验证了分子对接的结果,同时与两个化合物的生物活性差异相吻合。

为了更加清楚地阐明复合物体系的相互作用和稳定性,选择模拟过程较稳定的25~50 ns的轨迹,每隔100ps截取瞬时构象,共250个构象,利用MM-PBSA的方法计算出12-6BG3和74-6BG3两个复合物体系的结合自由能,结果列于表 3。通常结合自由能越低则表明复合物体系越稳定,从表可知,74-6BG3体系的结合自由能低于12-6BG3体系的,说明74-6BG3体系更加稳定,这与实验数据相符合。另外还发现范德华作用能明显大于其他能量,提示在结合过程中疏水作用起重要作用。
下载:
导出CSV
| Compd. | Van der Waals energy/(kJ·mol-1) | Electrostattic energy/(kJ·mol-1) | Polar solvation energy/(kJ·mol-1) | Non-Polar solvation energy/(kJ·mol-1) | Binding Energy/(kJ·mol-1) |
| 12 | -46.123±23.860 | -0.669±0.419 | -6.476±8.419 | -3.796±1.919 | -57.552±19.937 |
| 74 | -226.538±2.361 | -18.160±1.335 | 57.030±1.388 | -18.361±0.221 | -205.924±2.370 |
类泛素化是一个有吸引力的抗癌靶标,阻断DCN1-UBE2M相互作用为抑制类泛素化提供新的思路。本研究对哌啶基脲类化合物开展了3D-QSAR、分子对接和分子动力学模拟的研究。基于公共骨架叠合构建了可靠且具有较好预测能力的CoMFA模型和CoMSIA模型。根据其三维等势图可了解对化合物活性有影响的结构特征。分子对接研究表明所有化合物均与Gln1114形成氢键相互作用,故Gln1114是关键氨基酸残基,与之结合能够使化合物与靶蛋白的结合稳定性增强。分子动力学研究结果表明氢键相互作用和疏水作用在结合过程中起重要作用。这些研究结果为这类化合物进一步结构优化提供了理论基础。
Han Z J, Feng Y H, Gu B H, et al. Int. J. Oncol., 2018, 52(4): 1081~1094.
Venne A S, Kollipara L. Proteomics, 2014, 14(4-5): 513~524. doi: 10.1002/pmic.201300344
Zhao Y, Morgan M A, Sun Y. Antioxid. Redox Sign., 2014, 21(17): 2383~2400. doi: 10.1089/ars.2013.5795
Chen Y, Neve R L, Liu H. J. Cell. Mol. Med., 2012, 16(11): 2583~2591. doi: 10.1111/j.1582-4934.2012.01604.x
Zhou L, Zhang W, Sun Y, et al. Cell. Signal., 2018, 44: 92~102. doi: 10.1016/j.cellsig.2018.01.009
Li L, Kang J, Zhang W, et al. EBioMedicine, 2019, 45: 81~91. doi: 10.1016/j.ebiom.2019.06.005
Zheng N, Schulman B A, Song L Z, et al. Nature, 2002, 416(6882): 703~709. doi: 10.1038/416703a
Petroski M D, Deshaies R J. Nat. Rev. Mol. Cell. Biol., 2005, 6(1): 9~20.
Zhao Y, Sun Y. Curr. Pharm. Des., 2013, 19(18): 3215~3225. doi: 10.2174/13816128113199990300
Wu S, Yu L. Cytotechnology, 2016, 68(1): 1~8. doi: 10.1007/s10616-015-9870-0
Milhollen M A, Thomas M P, Narayanan U, et al. Cancer Cell, 2012, 21(3): 388~401. doi: 10.1016/j.ccr.2012.02.009
Toth J I, Yang L, Dahl R, et al. Cell Rep., 2012, 1(4): 309~316. doi: 10.1016/j.celrep.2012.02.006
Yu Q, Jiang Y, Sun Y. Acta Pharm. Sin. B, 2020, 10(5): 746~765. doi: 10.1016/j.apsb.2019.09.005
Hammill J T, Scott D C, Min J, et al. J. Med. Chem., 2018, 61(7): 2680~2693. doi: 10.1021/acs.jmedchem.7b01277
Hammill J T, Bhasin D, Scott D C, et al. J. Med. Chem., 2018, 61(7): 2694~2706. doi: 10.1021/acs.jmedchem.7b01282
Milne G W A. J. Chem. Inf. Model., 2010, 50: 2053~2053. doi: 10.1021/ci100385n
张玲, 刘鹰翔, 赵钟祥. 等. 化学通报, 2018, 81(2): 148~157. https://www.cnki.com.cn/Article/CJFDTOTAL-ZGXH201902002.htm
Abraham M J, Murtola T, Schulz R, et al. SoftwareX, 2015, (1-2): 19~25.
Schüttelkopf A W, Van Aalten D M F. Acta Crystallogr. D, 2004, 60(8): 1355~1363. doi: 10.1107/S0907444904011679
刘冬琳, 刘鹰翔, 李耿, 等. 化学通报, 2019, 82(7): 642~648. http://www.ccspublishing.org.cn/article/id/85c9a7b1-db18-41d8-80a2-f981df3e1181
宋昱, 任聪, 杨彭真, 等. 化学通报, 2019, 82(5): 446~451. http://www.ccspublishing.org.cn/article/id/45f88d30-c68c-4f69-8209-2345594d8399
Oluić J, Nikolic K, Vucicevic J, et al. Comb. Chem. High. T. Screen., 2017, 20(4): 292~303.

图 1 模板分子74(a)和基于公共骨架叠加图(b)
Figure 1 Docking template compound 74 (a) and common substructure-based alignment of the dataset (b)

图 3 CoMFA模型的立体场等势图(a)和静电场等势图(b)
Figure 3 Steric contour maps (a) and electrostetic contour maps (b) for CoMFA model

图 4 CoMSIA模型的立体场(a)、疏水场(b)、氢键供体场(c) 和氢键受体场等势图(d)
Figure 4 Steric(a), hydrophobic(b), H-bond donor(c) and H-bond acceptor(d) contour maps for CoMSIA

图 6 12-6BG3(黑色)和74-6BG3(红色)的RMSD变化图
Figure 6 RMSD of the 12-6BG3 (black) and 74-6BG3 (red)

图 7 12-6BG3(黑色)和74-6BG3(红色)的RMSF变化图
Figure 7 RMSF of the 12-6BG3 (black) and 74-6BG3 (red)
表 2 CoMFA和CoMSIA模型的统计参数
Table 2. Statistical parameters of CoMFA and CoMSIA models
| Model | N | q2 | r2 | SEE | rpred2 | F | S | E | H | D | A |
| CoMFA-SE | 10 | 0.686 | 0.966 | 0.168 | 0.879 | 164.086 | 0.569 | 0.431 | - | - | - |
| CoMSIA-SE | 6 | 0.514 | 0.852 | 0.338 | 59.615 | 0.404 | 0.596 | - | - | - | |
| CoMSIA-SH | 7 | 0.605 | 0.905 | 0.273 | 83.109 | 0.354 | - | 0.646 | - | - | |
| CoMSIA-SD | 9 | 0.585 | 0.869 | 0.326 | 43.542 | 0.705 | - | - | 0.295 | - | |
| CoMSIA-SA | 10 | 0.619 | 0.892 | 0.299 | 47.723 | 0.666 | - | - | - | 0.334 | |
| CoMSIA-EH | 6 | 0.514 | 0.861 | 0.328 | 63.750 | - | 0.433 | 0.567 | - | - | |
| CoMSIA-ED | 3 | 0.428 | 0.551 | 0.575 | 26.615 | - | 0.614 | - | 0.386 | - | |
| CoMSIA-EA | 3 | 0.485 | 0.649 | 0.508 | 40.149 | - | 0.499 | - | - | 0.501 | |
| CoMSIA-HD | 7 | 0.610 | 0.895 | 0.289 | 74.247 | - | - | 0.767 | 0.233 | - | |
| CoMSIA-HA | 10 | 0.634 | 0.937 | 0.227 | 86.789 | - | - | 0.735 | - | 0.265 | |
| CoMSIA-DA | 10 | 0.503 | 0.705 | 0.494 | 13.854 | - | - | - | 0.551 | 0.449 | |
| CoMSIA-SEH | 6 | 0.552 | 0.880 | 0.304 | 76.033 | 0.219 | 0.348 | 0.433 | - | - | |
| CoMSIA-SED | 6 | 0.555 | 0.844 | 0.347 | 55.837 | 0.281 | 0.466 | - | 0.254 | - | |
| CoMSIA-SEA | 3 | 0.516 | 0.693 | 0.476 | 48.807 | 0.171 | 0.411 | - | - | 0.417 | |
| CoMSIA-SHD | 8 | 0.625 | 0.912 | 0.266 | 77.457 | 0.282 | - | 0.538 | 0.180 | - | |
| CoMSIA-SHA | 9 | 0.664 | 0.929 | 0.240 | 85.914 | 0.244 | - | 0.501 | - | 0.255 | |
| CoMSIA-SDA | 10 | 0.630 | 0.888 | 0.304 | 46102 | 0.514 | - | - | 0.276 | 0.210 | |
| CoMSIA-EHD | 6 | 0.543 | 0.857 | 0.333 | 61.706 | - | 0.335 | 0.429 | 0.236 | - | |
| CoMSIA-EHA | 3 | 0.538 | 0.721 | 0.453 | 56.010 | - | 0.331 | 0.312 | - | 0.357 | |
| CoMSIA-HAD | 9 | 0.665 | 0.929 | 0.240 | 86.164 | - | - | 0.622 | 0.181 | 0.197 | |
| CoMSIA-EDA | 4 | 0.519 | 0.709 | 0.467 | 38.902 | - | 0.366 | - | 0.254 | 0.380 | |
| CoMSIA-SEHD | 7 | 0.572 | 0.900 | 0.280 | 78.394 | 0.176 | 0.292 | 0.368 | 0.164 | - | |
| CoMSIA-SEHA | 5 | 0.540 | 0.826 | 0.364 | 59.741 | 0142 | 0.260 | 0.302 | - | 0.295 | |
| CoMSIA-SHDA | 9 | 0.682 | 0.931 | 0.237 | 0.875 | 87.986 | 0.201 | - | 0.457 | 0.153 | 0.189 |
| CoMSIA-SEDA | 4 | 0.539 | 0.736 | 0.444 | 44.660 | 0.139 | 0.314 | - | 0.218 | 0.329 | |
| CoMSIA-EHDA | 5 | 0.569 | 0.821 | 0.369 | 57.817 | - | 0.236 | 0.273 | 0.216 | 0.276 | |
| CoMSIA-SEHDA | 7 | 0.599 | 0.900 | 0.280 | 78.857 | 0.219 | 0.214 | 0.300 | 0.156 | 0.200 |
 下载: 导出CSV
下载: 导出CSV
表 3 12-6BG3和74-6BG3在模拟过程中的结合自由能
Table 3. Binding free energy of 12-6BG3 and 74-6BG3 during simulation process
| Compd. | Van der Waals energy/(kJ·mol-1) | Electrostattic energy/(kJ·mol-1) | Polar solvation energy/(kJ·mol-1) | Non-Polar solvation energy/(kJ·mol-1) | Binding Energy/(kJ·mol-1) |
| 12 | -46.123±23.860 | -0.669±0.419 | -6.476±8.419 | -3.796±1.919 | -57.552±19.937 |
| 74 | -226.538±2.361 | -18.160±1.335 | 57.030±1.388 | -18.361±0.221 | -205.924±2.370 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: