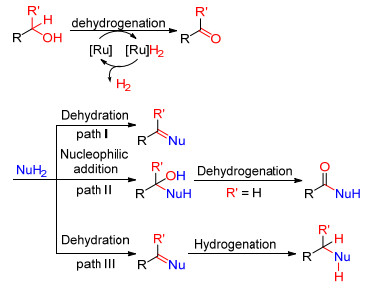
 图式 1
基于钌催化醇类化合物脱氢的偶联反应
Scheme1.
Ruthenium-catalyzed dehydrogenation coupling reactions with alcohol
图式 1
基于钌催化醇类化合物脱氢的偶联反应
Scheme1.
Ruthenium-catalyzed dehydrogenation coupling reactions with alcohol
引用本文:
曾明, 宋婵, 崔冬梅. 基于钌催化醇类化合物脱氢的C—N/C—C偶联反应的研究进展[J]. 有机化学,
2017, 37(6): 1352-1367.
doi:
10.6023/cjoc201701027

Citation: Zeng Ming, Song Chan, Cui Dongmei. Progress in Ruthenium-Catalyzed Dehydrogenation C—C/C—N Bonds Coupling Reactions from Alcohols[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1352-1367. doi: 10.6023/cjoc201701027
Citation: Zeng Ming, Song Chan, Cui Dongmei. Progress in Ruthenium-Catalyzed Dehydrogenation C—C/C—N Bonds Coupling Reactions from Alcohols[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1352-1367. doi: 10.6023/cjoc201701027
基于钌催化醇类化合物脱氢的C—N/C—C偶联反应的研究进展
English
Progress in Ruthenium-Catalyzed Dehydrogenation C—C/C—N Bonds Coupling Reactions from Alcohols
Abstract:
Ruthenium and its complex possess various catalytic activities such as oxidation and reduction. Ruthenium as a cheap and efficient catalyst was also widely used in such field as C—H activation. Considerable attention has been paid to it for its great applications in organic chemistry. The last decade's ruthenium-catalyzed deydrogenation C—N/C—C coupling reactions from acohols classified by their machanisms are summarized in this paper. Creative C—N/C—C coupling reactions are expected be designed by means of dehydrogenation catalyzed by ruthenium from acohols.
-
Key words:
- ruthenium-catalyzed
- / dehydrogenation
- / C—N coupling
- / C—C coupling
-
钌(Ru)是一种硬而脆呈浅灰色的多价稀有金属, 地壳中的含量仅为十亿分之一, 是最稀有的金属之一, 尽管如此, 钌相对铂族元素铂(Pt)、钯(Pd)等而言, 价格相对便宜, 因而被广泛地作为金属催化剂替代铂、钯等元素参与有机合成反应.由于钌4d75s1核外电子排布特点, 钌元素在不同的化合物中常常会呈现低价的还原态或高价的氧化态, 因而钌催化剂在有机合成主要表现为氧化脱氢和还原加氢两大反应类型, 在C—H活化、不对称合成、烯醇异构化等[1]方面中有着重要的用途.近年来, 基于过渡金属催化醇脱氢的“氢转移”[2]反应受到了广泛关注, 基于钌催化醇类化合物脱氢的C—N偶联、C—C偶联等反应成为了科研工作者研究讨论的热点话题之一, 该类偶联反应一般具有底物稳定、催化活性高、条件温和、绿色环保、原子经济性高等特点.
醛酮化学性质比较活泼, 因其化学性质的不稳定, 在参与有机反应过程中容易产生很多副反应.在有机合成中, 以醇作为醛酮的前体可以减少副反应的发生, 提高反应收率.醇的价格相对低廉、化学性质稳定、易于大量保存.近年来, 大量文献报道了基于钌催化醇脱氢的“氢转移”反应在有机合成中的应用, 其偶联反应机理主要可以概括为以下三类[3](Scheme 1): (1) 首先钌催化剂与醇络合, “携氢”离去, 发生脱氢氧化转化为醛或酮, 然后醛或酮与亲核试剂发生脱水缩合反应, 最后钌催化剂在储氢载体或配体作用下释放出氢气, 完成催化剂的循环(Path Ⅰ); (2) 醇在钌催化剂条件下转化为醛或酮, 然后醛或酮与亲核试剂发生亲核加成反应, 最后亲核加成产物在钌催化剂体系中发生β消除[4]脱氢形成含酮羰基的衍生物(Path Ⅱ); (3) 在钌催化剂作用下, 储氢载体或配体暂时对醇分子中的氢进行“储存”, 同时醇转化为醛或酮, 然后, 储存的氢被释放出来, 在钌催化剂作用下对不饱和键进行加成(Path Ⅲ).本文将从以上反应机理角度对近十年来钌催化醇类化合物脱氢的C—N、C—C偶联反应进行综述.
图式 1
基于钌催化醇类化合物脱氢的偶联反应
Scheme1.
Ruthenium-catalyzed dehydrogenation coupling reactions with alcohol
1 基于钌催化醇的氧化-脱水缩合偶联反应
C—N、C—C键的构建在有机合成中非常重要, 通过卤代烃和胺类化合物或芳香硼酸在过渡金属Cu、Rh、Ni、Pd等[5]催化发生偶联反应是构建C—N、C—C键最重要的方法之一.随着社会的进步和发展, 开发符合绿色化学的理念要求的新型的构建C—N、C—C键的方法面临着新的挑战.科研人员发现基于过渡金属Ni、Pd、Rh、Co、Ir、Mn等[6]催化醇脱氢的“氢转移”反应是形成C—N、C—C键的方法之一.近年来, 以金属钌作为催化剂参与醇脱氢的“氢转移”反应构建C—N、C—C键的方法也受到了广泛的关注.
1.1 C—N偶联反应
醇在钌催化剂条件下转化为醛或酮, 然后与胺类衍生物发生脱水缩合反应是形成C—N双键重要途径之一.亚胺是一类具有高活性的反应中间体, 在加成、缩合、不对称合成等有机反应中应用非常广泛. 2014年Ramesh等[7]以伯醇和伯胺为原料, 以甲苯为溶剂在钌催化剂1条件下得到亚胺衍生物, 收率高达96% (Eq. 1). 2014年Nishibayashi等[8]设计合成了钌催化剂2, 并对该催化剂的催化活性进行了考察.他们发现在2存在条件下, 苄胺和苄醇在甲苯中加热回流也可以得到亚胺, 反应收率79%.
2012年Schomaker等[9]报道了烯醇在钌催化剂3条件下转化为烯醛, 然后烯醛与伯胺直接通过脱水缩合形成α, β-不饱和亚胺的方法(Eq. 2).
2016年Milstein等[10]报道了伯醇和水合肼在钌催化4条件下通过醇的氢转移反应制备对称的烯肼衍生物的方法, 值得一提的是, 当以取代的苄醇为原料时, 苯环上取代基的位置与电性对反应收率有很大的影响, 当苄醇的对位为卤素(Cl)取代或间位为供电子基团(OMe)取代时, 反应收率分别为35%、50%, 当苄醇对位为供电子基(OMe)时, 收率为99%, 此外, 该反应比较耗时, 平均反应时间在55 h左右.
在钌催化条件下通过醇的脱氢与胺类衍生物发生脱水的反应主要应用于芳香氮杂环类化合物的合成. 1991年, Kondo等[11]首次以邻氨基苯酚或邻苯二胺和醇类化合物为原料在RuCl2(PPh3)3催化体系中加热到215 ℃左右得到苯并噁唑衍生物或苯并吡唑衍生物, 反应温度较高, 收率51%~80% (Eq. 4).
2009年Blacker等[12]对Kondo的研究内容进行了优化, 他们以Ru(PPh3(CO)H2)为催化剂, Xantphos为配体, 吡啶醋酸盐为添加剂, 对甲基苯磺酸或丙烯腈作为储氢载体, 合成得到了苯并吡唑衍生物, 收率43%~85%.
2014年, Panah等[13]对上述反应进行了进一步优化, 以附着Ru2Cl4(CO6)的磁性纳米粒子PFMN (Fe3O4@SiO2@PPh2)为催化剂, 反应在氮气保护下, 以甲苯为溶剂, 反应温度控制在110 ℃左右, 得到苯并噁唑化合物收率为51%~88%.该反应对底物醇(芳香醇、脂肪醇)的适用性十分广泛, 收率较高, 但并未对其他2-氨基-苯酚衍生物的适用性进行考察.该PFMN可以通过简单的物理回收后进行重复使用, 且具有同等的催化活性, 能够有效的减少后处理上膦的污染, 是一种新型、高效、绿色的钌催化剂.
当有机分子中同时含有氨基和醇羟基时, 在钌催化作用下能够发生自身的环合反应形成氮杂环衍生物. 1990年Tsuji等[14]以2-氨基苯乙醇为原料, 以甲苯为溶剂在RuCl2(PPh3)3催化下发生自身缩合形成吲哚衍生物(Eq. 5).
1992年, Izumi等[15]在Tsuji的基础上并对其条件进行适当的改进, 他们以RuH2(PPh3)4催化剂, 丙烯基碳酸甲酯为储氢载体, 得到了2-苯基-吲哚衍生物收率76%~92%, 反应条件相对温和.
2011年Shimura等[16]设计开发了新型的Ru/CeO2催化剂, 对Tsuji反应条件进行了进一步优化, 他们以均三甲苯为溶剂, 反应体系加热至140 ℃左右得到吲哚, 收率高达99%, Ru/CeO2催化剂回收重复使用时, 仍然具有较好的催化活性.
喹唑啉类衍生物通常以苯胺类化合物和醛、酮为原料发生缩合反应制备[17]. 2014年, Jiang等[18]报道了在Ru3(CO)12/Xantphos催化条件下2-氨基芳香甲醇和取代的苯甲腈在叔戊醇中加热至130 ℃下反应16 h得到2-芳基喹唑啉衍生物方法(Eq. 6).该反应对脂肪族的腈类化合物具有一定局限性, 当底物为丁腈时, 得到目标产物收率仅为18%, 当芳环上带有F、Cl、Br、CF3等吸电子基团反应收率明显高于Me、OMe、N(Me)2等供电子基团.
Jiang等推测该反应可能按如Scheme 2机理进行的:首先, 邻氨基苄醇的氨基对氰基碳原子进行亲核加成, 形成含脒结构中间体Ⅲ或异构化中间体Ⅲ'; 其次, 醇羟基在Ru3(CO)12/Xantphos/t-BuOK体系中脱去一分子氢气转化为含有醛基的中间产物Ⅳ; 最后, 该中间产物发生分子内的脱水缩合反应得到目标产物Ⅴ.该反应操作简单, 适用性范围相对较广, 没有其他副产物产生, 原子利用率高, 是一种高效的合成2-芳基喹唑啉的方法.
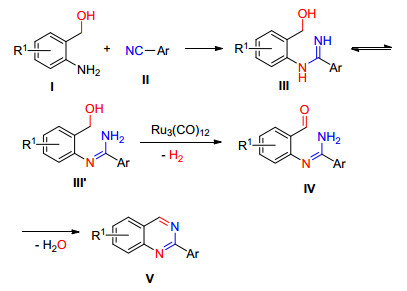 图式 2
钌催化合成2-芳基喹唑啉可能的反应机理
Scheme2.
Proposed mechanism for the synthesis of 2-arylquinazolines catalyzed by Ruthenium
图式 2
钌催化合成2-芳基喹唑啉可能的反应机理
Scheme2.
Proposed mechanism for the synthesis of 2-arylquinazolines catalyzed by Ruthenium
2011年Monrad等[19]报道了以1, 3-二醇类化合物和芳香伯胺衍生物为原料, 以PBu3为配体, 以无水的均三甲苯为溶剂, 在RuCl3·xH2O/MgBr2·OEt2催化体系中得到喹啉衍生物(Eq. 7).
Monrad等提出了如Scheme 3的反应机理, 他们认为在该催化体系中[Ru]能选择性的使1, 3-二醇中的伯醇脱氢转化为为醛, 使仲醇脱水转化为不饱和双键, 形成α, β不饱和醛, 然后再与苯胺发生Michael加成反应, 最后在的作用下发生Doebner-Miller反应得到喹啉衍生物.
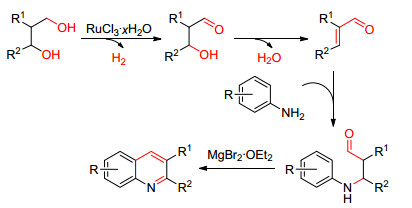 图式 3
钌催化喹啉衍生物的合成
Scheme3.
Ruthenium synthesis of quinoline derivatives
图式 3
钌催化喹啉衍生物的合成
Scheme3.
Ruthenium synthesis of quinoline derivatives
2012年Taddei等[20]报道了将醇和取代的苯肼溶解在2-甲基2-丁醇中在Ru3(CO)12/ZnCl2催化下以丙烯腈为储氢载体, BITHEP为配体, 使用微波加热至130 ℃左右即可得到氮取代的吲哚衍生物(Eq. 8).
Taddei等认为该反应的机理是仲醇首先被氧化成酮Ⅱ, 然后Ⅱ与取代的苯肼发生脱水缩合形成中间体Ⅲ, 最后Ⅲ在ZnCl2作用下发生[3,3] Sigmatropic重排, 脱去一分子氨气得到相应的偶联产物Ⅳ (Scheme 4).
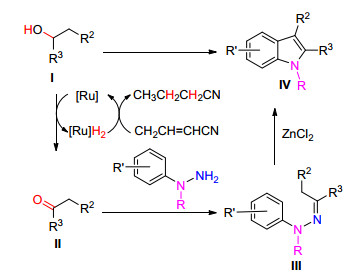 图式 4
钌催化菲舍尔吲哚的合成
Scheme4.
Synthesis Fischer indole derivatives catalyzed by Ruthenium
图式 4
钌催化菲舍尔吲哚的合成
Scheme4.
Synthesis Fischer indole derivatives catalyzed by Ruthenium
2014年Zhang等[21]以苄脒盐酸盐和醇为原料在Ru(p-cymene)Cl2/Cs2CO3催化体系中加热至110 ℃反应16 h制备均三嗪衍生物, 该方法对醇的种类具有一定的局限性, 当使用脂肪醇如正戊醇时, 在该条件下未得到目标产物(Eq. 9)
Zhang等提出了如Scheme 5的反应机理: (1) 芳香醇Ⅰ在Ru(p-cymene)Cl2/Cs2CO3体系中被氧化成芳香醛, 同时苄脒盐酸盐在Cs2CO3作用下被游离物对芳香醛进行缩合形成中间体Ⅲ; (2) 苄基脒对Ⅲ进行亲核加成反应生成Ⅳ, Ⅳ自身脱去一分子氨气形成Ⅴ; (3) Ⅴ再脱氢形成三嗪衍生物Ⅵ.
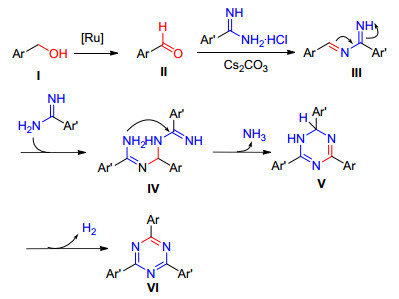 图式 5
钌催化合成2, 4, 6-三芳基-1, 3, 5-三嗪衍生物可能的反应机理
Scheme5.
Proposed mechanism for the synthesis of 2, 4, 6-tri-aryl-1, 3, 5-triazine derivatives catalyzed by Ruthenium
图式 5
钌催化合成2, 4, 6-三芳基-1, 3, 5-三嗪衍生物可能的反应机理
Scheme5.
Proposed mechanism for the synthesis of 2, 4, 6-tri-aryl-1, 3, 5-triazine derivatives catalyzed by Ruthenium
2016年我们课题组[22]以双胍衍生物和醇为原料在Ru(COD)Cl2/t-BuOK的催化体系下, 在二氧六环(除水)中反应得到均三嗪衍生物, 对取代的双胍的适用性比较广泛, 收率较好, 然而在醇的种类方面也存在一定的局限性(Eq. 10).目前, 我们课题组正在进行进一步的相关的底物拓展工作.
1.2 C—C偶联反应
Porcheddu等[23]报道了两分子醇在钌催化剂RuH2-CO(PPh3)3作用下, 以2-丁烯腈作为氢受体, 通过一锅法在微波条件下加热至120 ℃反应制备反式的α, β-不饱和醛的方法.反应过程中氨基硅作为“氢转移”载体增加了底物的化学选择性, 避免了副产物的发生(Eq. 11).
Porcheddu等提出了以下可能的反应机理(Scheme 6), 首先苯甲醇、脂肪醇同时被氧化成相应的醛; 然后氨基硅选择性地与苯甲醛反应得到亚胺中间体, 最后, 脂肪醛的α位亚甲基与硅取代的亚胺发生亲核加成反应, 脱去一分子氨基硅得到α, β-不饱和醛.
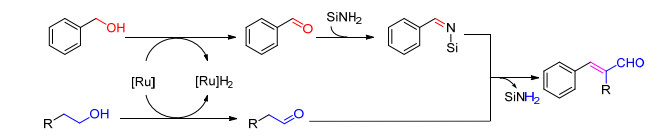 图式 6
钌催化醇的C—C偶联合成α, β-不饱和醛可能的反应机理
Scheme6.
Proposed mechanism for ruthenium catalyzed synthesis of α, β-unsaturated aldehydes based on primary alcohols
图式 6
钌催化醇的C—C偶联合成α, β-不饱和醛可能的反应机理
Scheme6.
Proposed mechanism for ruthenium catalyzed synthesis of α, β-unsaturated aldehydes based on primary alcohols
2013年Saito等[24]以β-氨基醇和取代的乙酮为原料, 在钌催化剂5/t-BuOK作用下无溶剂加热至165 ℃, 发生aldol缩合反应得到多取代的吡咯衍生物.反应过程中, 在钌催化作用下, 取代的乙酮作为储氢载体促进β-氨基醇转化为β-氨基醛, 反应没有无机盐产生, 后处理简单.该方法经济环保、原子利用率高(Eq. 12).
他们认为该反应按Scheme 7所示的机理进行:首先β-氨基醇在钌催化剂条件下脱去一分子氢气转化为β-氨基醛Ⅱ, 然后醛和取代的乙酮在碱性条件下发生aldol缩合形成中间体Ⅴ, 中间体Ⅴ发生分子内的脱水缩合反应得到Ⅵ, 最后Ⅵ脱去一分子水得到2, 3, 5-三取代的吡咯衍生物Ⅶ.
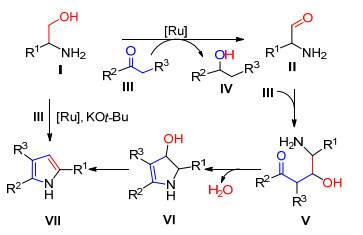 图式 7
钌催化合成吡咯衍生物可能的反应机理
Scheme7.
Proposed mechanism for ruthenium catalyzed synthesis of pyrroles
图式 7
钌催化合成吡咯衍生物可能的反应机理
Scheme7.
Proposed mechanism for ruthenium catalyzed synthesis of pyrroles
2013年Milstein等[25]报道了以β-氨基醇和仲醇为原料, 在钌催化剂6/t-BuOK甲苯中回流反应得到2, 5-二取代的吡咯衍生物, 收率50%~83%, 当以γ-氨基醇为原料时, 反应得到吡啶衍生物, 收率45%~80% (Eq. 13).
Milstein等认为该反应机理与Saito的报道有所不同, 他们认为6优先与仲醇作用将其氧化为甲基酮衍生物Ⅱ, 然后β氨基醇与酮进行脱水缩合形成Ⅲ, 然后在6作用下Ⅲ的羟基被氧化成醛Ⅳ, 在t-BuOK作用下Ⅳ活泼的甲基H被夺取形成Ⅴ的碳负离子活性中间体, 然后Ⅴ发生自身的亲核进攻, 最后脱水得到吡咯衍生物如Scheme 8.
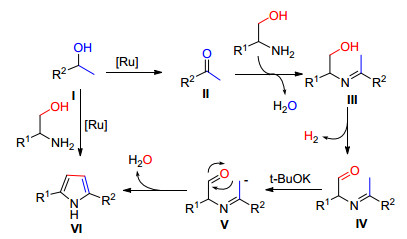 图式 8
钌催化吡咯衍生物的合成
Scheme8.
Ruthenium catalyzed synthesis of pyrroles derivatives
图式 8
钌催化吡咯衍生物的合成
Scheme8.
Ruthenium catalyzed synthesis of pyrroles derivatives
2013年, Beller等[26]报道了在t-BuOK/[Ru2Cl2(p-cymene)]2/Xantphos或K2CO3/Ru3(CO)12/Xanphos催化体系中, 伯胺、α-取代苯乙酮、邻二醇在正丁醇中加热至130 ℃左右发生三分子偶联反应, 高选择性地合成氮取代的吡咯衍生物的方法(Eq. 14).
2016年Sun等[27]报道了在催化剂7作用下γ氨基醇与仲醇在V(toluene)/V(THF)=4/1的混合溶剂中发生C—C偶联形成吡啶衍生物, 收率高达92% (Eq. 15).
Krische等[28]报道了以RuH2(CO)(PPh3)3为催化剂, dppf为配体, 在手性酸8或9的条件下芳香伯醇与1, 3-丁二烯通过脱氢、羟烃烷基化等过程形成反式的芳香仲醇, 收率高达97%.他们发现当以(S)-SEGPHOS (5, 5'-二-(二苯基膦)-4, 4'-二-1, 3-苯并二氧杂环戊)为配体, 在手性酸的条件下可以得到顺式的脂肪族仲醇, 当以dCypp为配体时, 在TsCl共同作用下, 氨基丁醇与1, 3-丁二烯反应得到反式的环合产物如Scheme 9.
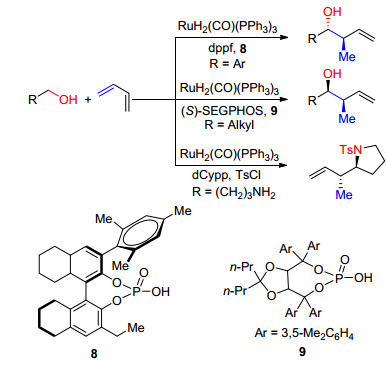 图式 9
钌催化丁二烯的羟烃烷基化反应
Scheme9.
Ruthenium catalyzed hydrohydroxyalkylation of butadiene
图式 9
钌催化丁二烯的羟烃烷基化反应
Scheme9.
Ruthenium catalyzed hydrohydroxyalkylation of butadiene
2 基于钌催化醇的氧化-β脱氢偶联反应
2009年, Williams等[29]报道了钌催化条件下醇作为氮酰基化试剂的反应.他们发现伯醇和伯胺在[Ru(p-cymene)Cl2]2/t-BuOK催化下, 以dppb或dpePhos为配体, 丙酮或3-甲基-2-丁酮储氢载体, 在正丁醇中回流反应24h可以得到酰胺衍生物(Eq. 16).
他们提出了如Scheme 10的反应机理:首先醇在钌催化剂作用下脱去一分子氢气形成醛, 然后伯胺与醛发生亲核加成反应形成Ⅲ, Ⅲ再次在钌催化剂条件下发生β消除脱氢得到酰胺衍生物Ⅳ.
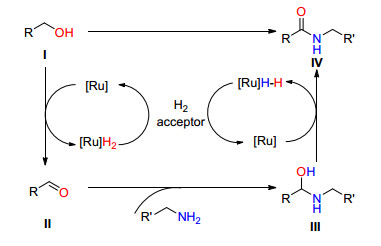 图式 10
钌催化“氢转移”醇与胺氧化偶联反应
Scheme10.
Oxidative coupling of alcohols with amines using hydrogen transfer catalyzed by ruthenium
图式 10
钌催化“氢转移”醇与胺氧化偶联反应
Scheme10.
Oxidative coupling of alcohols with amines using hydrogen transfer catalyzed by ruthenium
2010年Williams等[30]对上述反应进行了改进, 他们将碱改为Cs2CO3, 在油浴加热或微波加热条件下得到酰胺类衍生物, 收率提高到93%, 进一步扩大了伯醇和胺的适用性.
2013年Glorius等[31]也对上述条件进行了优化, 他们以Ru(cod)(2-methylallyl)2为催化剂, ICy·HCl为配体, 苯乙烯为储氢载体, 将甲醇和伯胺置于的甲苯溶液中加热至75~120 ℃反应得到甲酰胺衍生物, 收率44%~95%.
2014年, Guan等[32]对Williams的反应也进行优化, 他们发现在钳形催化剂Ru-Macho/KOH催化体系中伯醇与伯胺、仲胺反应也可以得到酰胺衍生物, 收率高达95%, 同时, 当以仲醇和伯胺为原料时, 产物为亚胺衍生物, 收率40%~85%.该反应条件简单, 收率较高, 反应过程不需要储氢载体的参与.
2010年Schley等[33]通过密度泛函数(DFT)对钌催化条件下氨基醇发生分子内环合转化为内酰胺的影响因素进行了研究, 并对该机理进行了阐述.他们以N-取代氨基戊醇为原料考察了钌催化剂10对该环合反应的催化活性, 结果表明在9条件下, 以KOH为碱, N-取代氨基戊醇在甲苯中加热4 h即可转化为对应的内酰胺, 收率高达95% (Eq. 17).
2014年Hong等[34]报道了1, 4-丁二醇与腈的衍生物在RuH2(PPh3)4作用下以1, 3-二异丙基-(1H)-咪唑3-溴化铵为配体通过“氢转移”反应的环合制备环内酰胺的反应, 收率43%~86% (Eq. 18).
他们推测该反应可能按Scheme 11机理进行的, 在[Ru]作用下, 1, 4-丁二醇脱氢、分子内的环合生成内酯, 同时苯甲腈作为氢的接受体与[Ru]H2结合被还原成苄胺, 然后苄胺与内酯发生亲核取代反应生成环内酰胺衍生物.
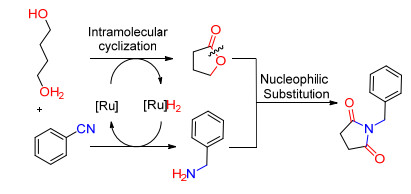 图式 11
钌催化合成环内酰胺可能的反应机理
Scheme11.
Proposed mechanism for the synthesis of cyclic imides catalyzed by ruthenium
图式 11
钌催化合成环内酰胺可能的反应机理
Scheme11.
Proposed mechanism for the synthesis of cyclic imides catalyzed by ruthenium
3 基于钌催化醇的氧化-还原氢化偶联反应
3.1 C—N偶联反应
2007年Williams等[35]报道了伯醇和伯胺在Ru(p-cymene)Cl2/dppf条件下通过醇的氢转移反应可以得到伯胺的N烷基化产物, 该反应底物适用性广泛, 反应收率高达100%. 2009年Williams等[36]对底物进行了进一步的拓展工作, 他们发现仲胺在该催化体系中与伯醇发生氢转移反应可以得到叔胺衍生物, 收率62%~100%, 该反应体系催化剂、配体用量降低到原来的50%.科研者们发现在钌催化下无机氨也可以通过醇的“氢转移”C—N偶联反应转化为有机胺类化合物. 2010年, Yamaguchi等[37]报道了以无机氨NH4HCO3、(NH4)2CO3、NH4HPO4、NH3 (28%. aq.)等为氮源, 在Ru(OH)x/TiO2混合催化剂作用下, 醇类化合物在均三甲苯中加热至140 ℃左右发生“氢转移”C—N偶联反应合成对称叔胺的方法, 收率62%~97%.他们发现, 当以具有空间位阻的异丙醇等为原料时, 生成物为对称的仲胺(Eq. 19). 2010年Beller等[38]发现当以吲哚衍生物为氮源时, 伯醇在钌催化剂条件下与吲哚衍生物反应得到N-取代的吲哚衍生物, 收率68%~93%.
2011年Beller等[39]报道了钌催化条件下醇类化合物在氨气氛围中发生C—N偶联反应制备伯胺的方法.他们以Ru(CO)ClH(PPh3)3为催化剂, Xantphos为配体, 在氛围下氨气将伯醇溶于叔戊醇中加热至130~170 ℃制得伯胺的衍生物收率48%~97%.当以1, 1-二醇(R'=OH)为底物时, 在该条件下的偶联产物为1, 1-二胺, 转化率100% (Eq. 20).
2014年Milstein等[40]对Beller的工作进行了改进, 他们设计合成了新型钳形钌催化剂11, 他们发现在11的催化条件下脂肪醇与氨气在甲苯中加热至135 ℃反应选择性得到对称的仲胺, 收率48%~88%.他们认为可能的原因是脂肪醇与氨气反应生成脂肪族伯胺, 然后伯胺与氨气竞争性的对中间体脂肪醛进行亲核进攻, 脱水缩合形成亚胺, 最后亚胺再在11的作用下通过“氢转移”加氢还原形成对称的仲胺, 当以芳香醇为底物时, 生成物为的芳香亚胺(Eq. 21).
2011年Beller等[41]报道了以α羟基酰胺衍生物和伯胺为原料在Ru3(CO)12/DCPE条件下醇的氢转移反应制备α氨基酸酰胺衍生物的方法, 收率42%~91% (Eq. 22).
2011年Waston等[42]对Beller的工作进行了改进, 首次报道微波加热无溶剂条件下, 伯醇与伯胺、仲级胺等在上述催化体系下反应3 min即可得到C—N偶联的叔胺衍生物, 该方法得到叔胺收率为54%~96%, 大大的缩短了反应时间.他们发现当底物分子中伯醇和叔醇同时存在时, 仲胺选择性地与伯醇发生偶联反应得到相应的叔胺衍生物, 收率61%~90%.
芳香硼酸酯在有机合成中是非常重要的中间体之一. 2013年Williams等[43]以硼酸酯取代的苄醇和胺原料, 以二甲苯为溶剂, dpePhos为配体, 在RuCl2(p-cymene)2催化下加热至150 ℃左右得到相应的具有硼酸取代的仲胺或叔胺衍生物, 收率52%~80%, 反应时间约在24 h左右(Eq. 23).
2014年Jiang等[44]发现吡啶甲醇和2-氨基苯腈在Ru3(CO)12/Binap体系中反应得到对应的仲胺衍生物, 收率高达80%. 2014年Moasser等[45]对上述方法进行了优化和改进, 在RuCl2(p-cymene)2/12/t-BuOK的催化体系中醇与伯胺反应得到仲胺类衍生物的方法, 收率60%~97%, 该方法反应条件温和, 配体结构简单, 适用范围较广.
2015年Shafir等[46]对Moasser等的反应条件进行了优化, Shafir等发现当以N, N'-二(二苯基膦)-N, N'-二甲基-丙二胺为配体, 醇类化合物与芳香伯胺在甲苯中加热至120 ℃可以得到仲胺衍生物, 收率35%~95%, 该反应RuCl2(p-cymene)2的用量降低至1.25 mol%, 底物的适用性广.
2015年Seayad等[47]对Mosser的反应条件进行了进一步的改进, Seayad等发现在[RuCp*Cl2]2/dpePhos/ t-BuOLi甲醇溶液中, 加热至40~100 ℃甲醇与芳香伯胺发生C—N偶联反应, 生成N-甲基化的胺类衍生物, 收率73%~98%, 当以脂肪伯胺为底物时, 生成物为N'N-二甲基取代的胺类衍生物(Eq. 24).
Seayad等推测该反应是按如Scheme 12的机理进行的, 首先甲醇在t-BuOLi条件下进行醇盐交换形成MeOLi, MeOLi与钌催化剂的配合物络合, 然后失去氢原子得到中间体甲醛; 然后甲醛与氨发生缩合形成亚胺, 亚胺再次与[Ru]H络合得到氢原子; 最后, 裸露的氮负离子从甲醇中夺取氢原子形成N-甲基化的产物, 该反应不需要储氢载体.
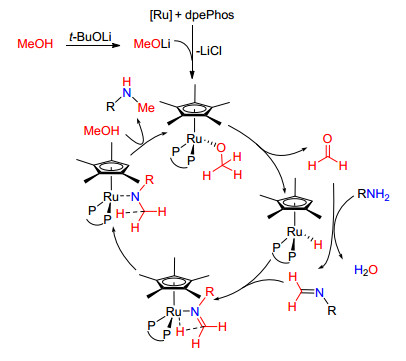 图式 12
钌催化胺的N-甲基化反应
Scheme12.
N-Methylation of amines catalyzed by ruthenium
图式 12
钌催化胺的N-甲基化反应
Scheme12.
N-Methylation of amines catalyzed by ruthenium
2015年Seayad等[48]设计合成了如Scheme 13的含有苯并咪唑结构钌催化剂13, 他们发现13的对醇与胺的偶联反应具有高效的催化活性.在无溶剂条件下, 醇与苯胺在13催化下反应得到N-烷基化的仲胺衍生物, 收率为45%~95%, 该催化剂能广泛地应用于芬太尼、阿尔维林等药物的合成. 2015年Bruneau等[49]设计合成了含有磺酸盐螯合物的钌催化剂14, 他们发现在钌催化剂14条件下, 苄醇作为烷基化试剂与哌啶发生C—N偶联选择性的形成N-取代的哌啶, 收率为91%. 2015年Sahin等[50]对RuCl2(p-cymene)2的结构进行了改造与修饰, 设计合成了钌催化剂15, 并将其应用于上述反应中, 研究表明吗啉与芳香醇在15和樟脑磺酸(CSA)共同作用下对C—N偶联反应的选择性最佳, 催化剂15用量仅为1 mol%, 选择性地生成C—N偶联的产物, 收率高达96%. 2016年Takacs等[51]设计合成了具有手性结构的钌催化剂16, 在钌催化剂16条件下, 仲醇和伯胺在甲苯中加热至110 ℃得到仲胺衍生物, 反应收率高达94%.钌催化剂13~16性质稳定, 易于保存, 均具有较好的催化活性.
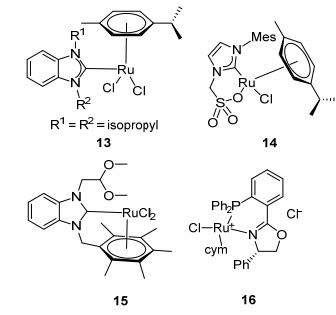 图式 13
钌催化剂13~16的结构
Scheme13.
Structure of ruthenium catalyst 13~16
图式 13
钌催化剂13~16的结构
Scheme13.
Structure of ruthenium catalyst 13~16
N-烷基取代的丙三醇衍生物在有机合成中具有广泛的应用前景[52]. Kann等[53]报道了丙三醇衍生物和仲胺为原料在RuCl2(cymene)2/dpePhos或dppf催化体系中, 在甲苯或叔丁醇中加热至130 ℃左右, 得到C—N偶联的产物, 收率64%~97%.在该催化体系, 他们对镇咳药羟苯哌嗪的中间体进行了合成, 收率86%.
2009年Beller等[54]报道了在负载钌催化剂的磁性Fe3O4纳米颗粒催化条件下, 醇与磺酰胺衍生物在弱碱K3CO3环境下加热至150~180 ℃制备N-取代磺酰胺衍生物的方法, 高达97%.反应催化剂TON(按Fe3O4算)=3~5, 钌催化剂易回收进行重复使用(Eq. 25).
钳形铱[55]、钌催化剂在醇的手性催化反应中也有一定的应用前景. 2014年Guan等[56]报道了伯醇和叔丁基磺胺在钌催化剂作用下不对称合成α取代叔丁基磺胺类衍生物的方法, 收率31%~89%, ee值高达95%, 他们对该反应结果的解释是Ru-Macho对中间体叔丁基磺亚胺的空间结构较为敏感, 受其空间结构的影响, 在“氢转移”加氢还原过程中, 氢能够选择性的加成到C—N双键的同侧(Eq. 26).
Zhao等[57]报道了以外消旋的1, 2-二醇和胺为原料, 在RuCl2(p-cymene)2/17催化条件下对映选择性地合成1, 2-氨基醇的研究, 反应在二氧六环和苯甲酸体系中进行, 他们从动力学不对称合成的角度对该反应的机理进行了研究, 认为添加剂苯甲酸在该不对称合成中起到了协同作用(Eq. 27).
2012年Williams等[58]以邻氨基磺酰胺或邻氨基苯甲酰胺为原料, 与醇在Ru(PPh3)3(CO)(H)2/Xanphos作用下得到杂环衍生物Ⅰ.他们发现当在反应体系中加入添加剂NH4Cl时, 主要产物则为Ⅱ, 实验结果表明NH4Cl的加入并没有影响到醇向醛的转化(Eq. 28).
2016年Zhang等[59]报道了在Ru3(CO)12/Xanphos为催化体系下通过2-氨基3-吡啶甲醇和醇制备1, 2, 3, 4-四氢萘吡啶衍生物的方法, 收率高达90% (Eq. 29).值得一提的是, 当2-氨基3-吡啶甲醇的4、6位均被苯环取代时, 产物主要为Ⅱ.
2016年Zhang等[60]报道了二醇和2, 3-二氨基吡啶在Ru3(CO)12/Xanphos催化体系中在叔戊醇中加热至130 ℃反应, 选择性制备四氢吡啶并吡嗪衍生物的方法, 收率41%~86% (Eq. 30).
Zhang等推测该反应是按如Scheme 14的机理进行:首先二醇在钌催化条件下转化为二酮, 再与2, 3-二氨基吡啶发生脱水缩合反应形成中间体吡啶并吡嗪衍生物, 最后该中间体在[Ru]H2作用下被还原成四氢吡啶并吡嗪衍生物.
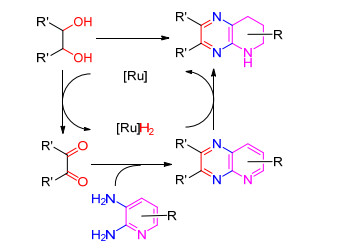 图式 14
钌催化“氢转移”四氢吡嗪衍生物的合成
Scheme14.
Ruthenium-catalyzed synthesis of tetrahydro fused-pyrazine derivatives by hydrogen borrowing method
图式 14
钌催化“氢转移”四氢吡嗪衍生物的合成
Scheme14.
Ruthenium-catalyzed synthesis of tetrahydro fused-pyrazine derivatives by hydrogen borrowing method
2016年, Beller等[61]报道了以1, 2-二醇和酰胺衍生物为原料, 在Ru3(CO)12催化下得到2-噁唑烷酮衍生物, 收率45%~76%, 他们发现, 当二醇R1、R2均为苯基时, 得到的偶联产物为不饱和的2-噁唑烷酮, 收率43% (Eq. 31).
Beller等提出了如Scheme 15所示可能的反应机理:首先醇羟基对酰胺的羰基进行进攻, 发生亲核取代反应形成含酯的结构的中间体Ⅰ; 然后在Ru3(CO)12/dppf共同作用下, Ⅰ的醇羟基下脱去一分子氢气形成酮Ⅱ, Ⅱ自身缩合脱去一分子水, 形成不饱和的2-噁唑烷酮Ⅲ; 最后, Ⅲ的C—N双键在Ru3(CO)12/dppf作用下被还原成2-噁唑烷酮衍生物.
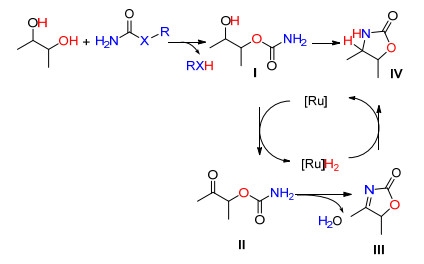 图式 15
钌催化“氢转移”2-噁唑烷酮衍生物的合成
Scheme15.
Synthesis of oxazolidin-2-ones by hydrogen borrowing method catalyzed by ruthenium
图式 15
钌催化“氢转移”2-噁唑烷酮衍生物的合成
Scheme15.
Synthesis of oxazolidin-2-ones by hydrogen borrowing method catalyzed by ruthenium
钌催化“氢转移”反应能够将醇分子中的“转移”一些不饱和基团(CN, NO2)进行加氢还原[62], 如2010年Li等[63]报道, 在Ru(PPh3)3(CO)(H)2/18作用下通过活化醇的氢转移反应对硝基苯进行氢化还原, 继而发生C—N偶联制备叔胺衍生物的方法(Eq. 32).
在反应过程中同时监测到了亚胺、仲胺等中间体, 故他们提出了如Scheme 16可能的反应机理: (1) 醇在[Ru]作用下被氧化成醛, [Ru]与氢络合离去, 对硝基化合物进行加氢还原成胺类化合物; (2) 胺类化合物对醛进行亲核进攻缩合生成中间体亚胺, 继而被[Ru]与氢络合加氢还原成仲胺, (3) 仲胺与醛进一步反应生成叔胺衍生物.
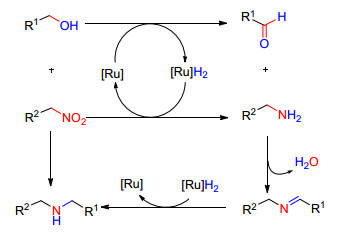 图式 16
钌催化剂醇与硝基类化物合成叔胺衍生物
Scheme16.
Synthesis of tertiary amine catalyzed by ruthenium from alcohols and nitroarenes
图式 16
钌催化剂醇与硝基类化物合成叔胺衍生物
Scheme16.
Synthesis of tertiary amine catalyzed by ruthenium from alcohols and nitroarenes
2015年Wu等[64]以2-硝基苯甲醛和醇为原料, 在Ru(PPh3)Cl2/K2CO3氢转移作用, 经过官能团转化C—N偶联制备喹啉衍生物(Eq. 33).
Wu等认为该反应的机理与上述反应相似, 即首先醇在Ru(PPh3)3Cl2/K2CO3体系中氧化脱氢转化为醛, 同时2-硝基苯甲醛(酮)作为储氢载体在[Ru]H2催化剂配合物作用下被还原成活性中间体2-氨基苯甲醛(酮); 最后, 通过Friedländer缩合得到喹啉衍生物.
2015年Jiang等[65]对Wu的工作进行了改进, 他们发现将2-硝基苯甲醛改为2-硝基苯甲醇时, 以Ru3(CO)12/dppf为催化体系也可以得到喹啉衍生物, 催化剂用量降低为1 mol%, 收率可达82% (Eq. 34).
2015年Jian等[66]报道了以2-硝基苯胺和二醇类化合物为原料, 在Ru3(CO)12/DPPP的催化条件下制备苯并吡嗪衍生物的方法, 收率高达87%.
3.2 C—C偶联反应
在过渡金属钴[67]、钌等催化下伯醇能与羰基α位发生氢转移C—C偶联反应. 2002年Chul Shim等[68]以甲基酮与伯醇为原料, RuCl2(PPh3)3为催化剂, 当在反应体系中加入储氢载体时, 反应选择性的得到α烷基取代的酮, 收率48%~83% (Eq. 35). 2014年, Zhang等[69]发现将催化剂体系改为RuCl2(p-cymene)/Xanphos时, 生成α烷基酮的收率高达92%.
2012年Ryu等[70]以甲苯为溶剂, RuHCl(CO)(PPh3)3为催化剂加热至140 ℃反应得到α烷基或芳基取代的酮的衍生物, 该反应对伯醇和甲基酮的适用性很广, 当原料为芳香乙酮时, 芳环上取代基的位置与电性对反应收率影响不大, 收率70%~92%, 然而当正己醇为底物时, 收率仅为22%, 值得注意的是, 当在反应体系内添加1, 10-菲啰啉时, 反应收率可以提高到88%.
2013年Ryu等[71]以乙酰胺衍生物为原料对上述反应的适用性进行了考察, 当反应体系内添加配体19 (Scheme 17)时, 伯醇与乙酰胺在钌催化条件下反应得到烷基取代的乙酰胺, 收率45%~89%. 2016年Glorius等[72]进一步对上述反应进行了优化, 他们发现伯醇与酮在Ru/20/t-BuOLi催化条件下, 在V(t-AmOH)/V(n-hexane)=1.25:1的混合溶剂中反应也可以得到C—C偶联的产物, 收率52%~99%, 该反应进一步拓展了酮的适用性, 同时, 该催化体系能应用于治疗阿尔兹海默症药物多奈哌齐的合成.
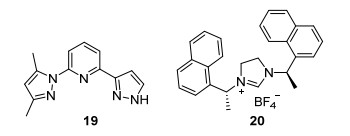 图式 17
配体19~20的结构
Scheme17.
Structures of 19~20
图式 17
配体19~20的结构
Scheme17.
Structures of 19~20
2016年Gnanaprakasam等[73]以21为催化剂对上述反应进行了优化研究.反应在无溶剂条件下加热至140 ℃反应可以得到α烷基(芳基)取代的乙酰胺衍生物, 收率可达70%, 另外, 二氢吲哚酮与伯醇在该条件下得到的产物为3-羟基吲哚酮衍生物, 收率48%~77% (Eq. 36).该反应条件相对温和, 催化剂的活性高, TON=460.
2016年Taddei等[74]研究了Ru(CO)3(PPh3)3HCl催化α, γ-二羰基化合物与伯醇进行C—C偶联的反应, 反应以甲苯为溶剂, 加热至160 ℃进行的, 当加入储氢载体22时反应生成物为β烷基取代的α, γ-二羰基化合物, 他们发现邻氨基苄醇与α, γ-二羰基化合物得到苯并吡啶衍生物, 该过程不需要22的参与(Scheme 18).
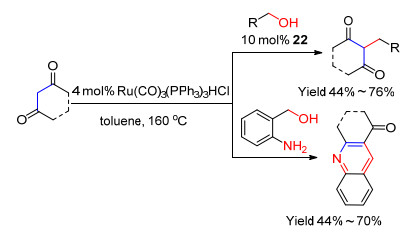 图式 18
钌催化醇与α, γ-二羰基化合物的C—C偶联反应
Scheme18.
Ruthenium catalyzed C—C coupling reactions between alcohols and α, γ-dicarboxylic compounds
图式 18
钌催化醇与α, γ-二羰基化合物的C—C偶联反应
Scheme18.
Ruthenium catalyzed C—C coupling reactions between alcohols and α, γ-dicarboxylic compounds
他们认为该反应按以下机理Scheme 19进行: (1) 伯醇Ⅰ在Ru(CO)3(PPh3)3HCl条件下被氧化成醛, [Ru]与氢形成络合物, α, γ-二羰基化合物的β位对醛进行亲核进攻, 发生aldol缩合反应, 形成中间体Ⅳ; (2) 22'作为氢供体对Ⅳ的双键进行还原加氢, 得到目标化合物Ⅴ, 22脱氢形成22', 22'与[RuH2]加成完成[Ru]和22的循环.
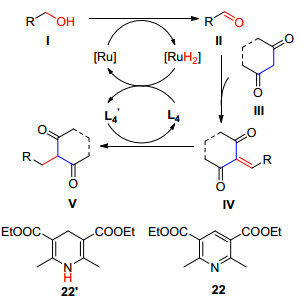 图式 19
钌催化醇与α, γ-二羰基化合物的C—C偶联反应可能的反应机理
Scheme19.
Proposed mechanism of C—C coupling reactions between alcohols and α, γ-dicarboxylic compounds catalyzed by ruthenium
图式 19
钌催化醇与α, γ-二羰基化合物的C—C偶联反应可能的反应机理
Scheme19.
Proposed mechanism of C—C coupling reactions between alcohols and α, γ-dicarboxylic compounds catalyzed by ruthenium
2001年Sang等[75]报道了苯乙酮与脂肪族伯醇在RuCl2(PPh3)3条件下通过醇的“氢转移”反应形成C—C键制备烷基醇的方法, 收率77%~85% (Eq. 37).
2014年Beller等[76]对上述方法进行了改进, 使用甲醇为甲基化试剂, 在钌催化剂Ru-Macho和23作用下选择性的在芳基乙醇的α位进行甲基化取代, 收率59%~87% (Eq. 38).
他们认为该反应按Scheme 20的机理进行:在混合催化剂条件下, 2-芳基乙醇和甲醇分别同时转化为相应的醛, 然后甲醛与苯乙烯醇发生亲核加成、脱水、加氢等反应, 最后得到2-芳基丙醇.
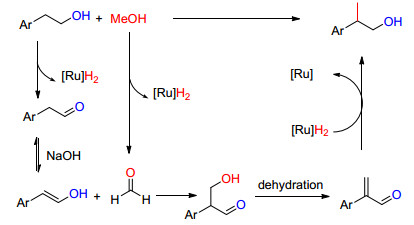 图式 20
钌催化醇的C—C偶联反应
Scheme20.
Synthesis of alcohols via C—C bond coupling catalyzed by ruthenium from alcohols
图式 20
钌催化醇的C—C偶联反应
Scheme20.
Synthesis of alcohols via C—C bond coupling catalyzed by ruthenium from alcohols
2016年Yu等[77]研究了以伯醇作为烷基化试剂与仲醇在钌催化剂24作用下发生C—C偶联制备β烷基取代的仲级醇, 收率52%~91%, 该反应催化剂TOF=15 h-1, 原子利用率高, 条件温和, 适用范围广(Eq. 39). 2016年Kundu等[78]对Yu的工作进行了优化, 他们发现在钌的复合物25催化下, 以NaOH为碱, 同样得到了β烷基取代的仲醇, 收率可达96%, 反应时间缩短为1.5 h, TOF=650 h-1.
2013年Ilhyong等[79]报道了以RuHCl(CO)(PPh3)3为催化剂, 将伯醇溶于乙腈中加热至110 ℃反应得到乙腈的烷基化产物(Eq. 40).
Beller等[80]报道了在钳形钌催化剂26条件下, 以1, 2-二醇衍生物和丙二酸二乙酯为原料可以转化为γ-戊内酯衍生物的方法.他们发现当R3被氢取代时, 生成物为饱和的γ-戊内酯衍生物; 当R3为烷基或芳基取代时, 生成物为不饱和的γ-戊内酯衍生物, 并没有对产生该结果的原因进行详尽的解释(Eq. 41).
Beller等提出了如Scheme 21的反应机理, 首先1, 2二醇在[Ru]作用下选择性氧化为羰基醇Ⅱ, 然后在碱性条件下丙二酸酯的活泼亚甲基对羰基进行亲核进攻, 脱去一分子水形成Ⅲ, 同时在[Ru]H2作用下Ⅲ的C—C双键被还原得到Ⅳ, 最后在碱性条件下Ⅳ发生自身的酯交换、脱羧等反应得到γ-戊内酯衍生物Scheme 21.
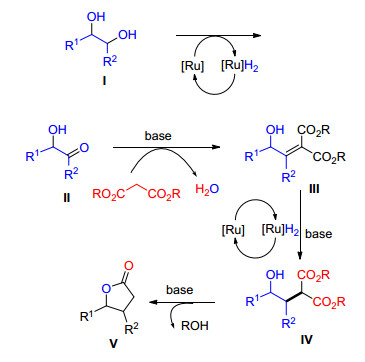 图式 21
钌催化二醇与丙二酸酯偶联反应合成γ-戊内酯衍生物可能的反应机理
Scheme21.
Proposed mechanism for ruthenium catalyzed synthesis of γ-butyrolactones from alcohols and malonates
图式 21
钌催化二醇与丙二酸酯偶联反应合成γ-戊内酯衍生物可能的反应机理
Scheme21.
Proposed mechanism for ruthenium catalyzed synthesis of γ-butyrolactones from alcohols and malonates
4 展望
综上所述, 钌作为一种经济高效的催化剂, 对于有机合成中的C—N、C—C键的构建形成有着广泛的运用, 具有较好的的应用前景.近年来, 研究人员通过对配体的设计和反应条件的改变, 不断地拓展了钌催化剂在醇的“氢转移”反应领域的底物适用性.然而, 参与钌催化醇脱氢的“氢转移”反应的配体往往结构复杂、价格昂贵、不易获得.因此, 开发催化活性高、易于回收利用、价格相对低廉的钌催化剂面临的巨大的挑战, 也是科研者不断探索的方向之一.通过对钌催化醇脱氢的C—N、C—C偶联反应详尽的综述以及对其反应机理的深刻探讨, 我们相信钌催化剂在杂环构建、不对称合成等领域一定会有着更广阔的用途.
-
-
[1]
(a) Li, R. -Q. ; Fu, Y. ; Liu, L. ; Guo, Q. -L. Chin. J. Org. Chem. 2004, 24, 1004 (in Chinese).
(李蕊琼, 傅尧, 刘磊, 郭庆祥, 有机化学, 2004, 24, 1004. )
(b) Zhong, Y. -X. ; Ren. K. ; Xie, X. -M. ; Zhang, Z. -G. Chin. J. Org. Chem. 2016, 36, 258 (in Chinese).
(钟业辛, 任凯, 谢小敏, 张兆国, 有机化学, 2016, 36, 258. )
(c) Gao, A. -L. ; Ye, Q. -S. ; Yu, J. Chin. J. Org. Chem. 2017, 37, 47 (in Chinese).
(高安丽, 叶青松, 余娟, 有机化学, 2017, 37, 47. )
(d) Wang, Y. -J. ; Zhang, Z. -F. ; Zhang, W. -B. Chin. J. Org. Chem. 2015, 35, 528 (in Chinese).
(王英杰, 张振锋, 张万斌, 有机化学, 2015, 35, 528. )
(e) Du, R. -F. ; Tan, X. -H. ; Fan, Y. -Q. Acta Chim. Sinica 2016, 74, 503 (in Chinese).
(窦镕飞, 谭晓荷, 范义秋, 化学学报, 2016, 74, 503. )
(f) Zuo, X. ; Wu, W. -L. ; Su, W. -P. Acta Chim. Sinica 2015, 73, 298 (in Chinese).
(左璇, 吴文亮, 苏伟平, 化学学报, 2015, 73, 298. )
(g) Xu, Y. ; Zhang, H. -Z. ; Wang, X. -Y. ; Liu, G. -Y. Chin. J. Chem. 2015, 33, 1393.
(h) Wang, R. -J. ; Jia, P. -F. ; Yang, Y. -Y. ; An, N. ; Zhang, Y. -D. ; Wu, H. -Y. ; Hu. Z. -A. Chin. J. Chem. 2016, 34, 114.
(i) Liu, Y. -X. ; Yang, N. ; Chu, C. -H. ; Liu, R. -H. Chin. J. Chem. 2015, 33, 1101. -
[2]
(a) Delgadorebollo, M.; Cansecogonzalez, D.; Hollering, M.; Muellerbunz, H.; Albrecht, M. Dalton Trans. 2014, 43, 4462.
(b) Mastalir, M.; Tomsu, G.; Pittenauer, E.; Allmaier, G.; Kirchner, K. Org. Lett. 2016, 18, 3462.
(c) Shiraishi, Y.; Fujiwara, K.; Sugano, Y.; Ichikawa, S.; Hirai, T. ACS Catal. 2013, 3, 312.
(d) Dang, T. T.; Ramalingam, B.; Shan, S. P.; Seayad, A. M. ACS Catal. 2013, 3, 2536.
(e) Zhang, Y.; Qi, X.-J.; Cui, X.-J.; Shi, F.; Deng, Y.-Q. Tetrahedron Lett. 2011, 52, 1334.
(f) Shimizu, K. I.; Shimura, K.; Nishimura, M.; Satsuma, A. RSC Adv. 2011, 1, 1310.
(g) Shimizu, K. I.; Imaiida, N.; Kon, K.; Siddiki, S. M. A. H.; Satsuma, A. ACS Catal. 2013, 3, 998.
(h) Rawlings, A. J.; Diorazio, L. J.; Wills, M. Org. Lett. 2015, 17, 1086.
(i) Bhat, S.; Sridharan, V. Chem. Commun. 2012, 48, 4701.
(j) Yang, Q.; Wang, Q.-F.; Yu, Z.-K. Chem. Soc. Rev. 2015, 44, 2305.
(k) Xiong, B.; Zhang, S.-D.; Jiang, H.-F.; Zhang, M. Org. Lett. 2016, 18, 724. -
[3]
(a) Nandakumar, A.; Midya, S. P.; Landge, V. G.; Balaraman, E. Angew. Chem., Int. Ed. 2015, 54, 11022.
(b) Huang, F.; Liu, Z.; Yu, Z. Angew. Chem., Int. Ed. 2016, 55, 862. -
[4]
Tseng, K.-N. T.; Kampf, J. W.; Szymczak, N. K. ACS Catal. 2015, 5, 5468. doi: 10.1021/acscatal.5b00952
-
[5]
(a) Patil, N. M.; Kelkar, A. A.; Nabi, Z.; Chaudhari, R. V. Chem. Commun. 2004, 35, 2368.
(b) Hirai, Y.; Uozumi, Y. Chem. Commun. 2010, 46, 1103.
(c) Moghaddam, F. M.; Tavakoli, G.; Moafi, A.; Saberi, V.; Rezvani, H. R. ChemCatChem 2015, 6, 3474.
(d) Li, S.-G.; Deng, G.-J.; Yin, F.-F.; Li, C.-J.; Gong, H. Org. Chem. Front. 2017, 4, 417.
(e) Fairlamb, I. J.; Kapdi, A. R.; Lee, A. F.; Mcglacken, G. P.; Weissburger, F.; de Vries, A. H.; Schmieder-Van, D. V. L. Chem.-Eur. J. 2006, 12, 8750.
(f) Li, J.-X.; Hu, W.-G.; Li, C.-S.; Yang, S.-R.; Wu, W.-Q.; Jiang, H.-F. Org. Chem. Front. 2017, 4, 373.
(g) Barot, N.; Patel, S. B.; Kaur, H. J. Mol. Catal. A: Chem. 2016, 423, 77.
(h) Zhang, C.; Li, T.-L.; Wang, L.-G.; Rao, Y. Org. Chem. Front. 2017, 4, 386.
(i) Dutta, J.; Richmond, M. G.; Bhattacharya, S. Dalton Trans. 2015, 44, 13615.
(j) Lin, W.-H.; Wu, W.-C.; Selvaraju, M.; Sun, C.-M. Org. Chem. Front. 2017, 4, 392. -
[6]
(a) Chan, L. K. M.; Poole, D. L.; Shen, D.; Healy, M. P.; Donohoe, T. J. Nat. Chem. 2011, 3, 287.
(b) Serk, K. M.; Namdu, K.; Hyeok, S. S.; Soo, P. I.; Kumar, C. R.; Jaiwook, P. Angew. Chem., Int. Ed. 2005, 44, 6913.
(c) Anxionnat, B.; Gomez Pardo, D.; Ricci, G.; Rossen, K.; Cossy, J. Org. Lett. 2013, 15, 3876.
(d) Chan, L. K. M.; Poole, D. L.; Shen, D.; Healy, M. P.; Donohoe, T. J. Angew. Chem., Int. Ed. 2014, 53, 761.
(e) Alonso, F.; Riente, P.; Sirvent, J. A.; Yus, M. Appl. Catal. A 2010, 378, 42.
(f) Fujita, K. I.; Yoshida, T.; Imori, Y.; Yamaguchi, R. Org. Lett. 2011, 13, 2278.
(g) Yu, X.; Wang, Q.-Y.; Wu, Q.-J.; Wang, D.-W. Russ. J. Gen. Chem. 2016, 86, 178.
(h) Rösler, S.; Ertl, M.; Irrgang, T.; Kempe, R. Angew. Chem., Int. Ed. 2015, 54, 15046.
(i) Saidi, O.; Blacker, A. J.; Farah, M. M.; Marsden, S. P.; Williams, J. M. J. Angew. Chem., Int. Ed. 2009, 48, 7375.
(j) Qu, P.-P.; Sun, C.-L.; Ma, J.; Li, F. Adv. Synth. Catal. 2014, 356, 447.
(k) Michlik, S.; Kempe, R. Angew. Chem., Int. Ed. 2013, 52, 6326.
(l) Kawahara, R.; Fujita, K.-i.; Yamaguchi, R. Adv. Synth. Catal. 2011, 353, 1161.
(m) Wang, R.-Z.; Ma, J.; Li, F. J. Org. Chem. 2015, 80, 10769.
(n) Wang, D.-W.; Zhao, K.-Y.; Xu, C.-Y.; Miao, H.-Y.; Ding, Y.-Q. ACS Catal. 2014, 4, 3910.
(o) Saidi, O.; Blacker, A. J.; Farah, M. M.; Marsden, S. P.; Williams, J. M. Chem. Commun. 2010, 46, 1541.
(p) Hille, T.; Irrgang, T.; Kempe, R. Angew. Chem., Int. Ed. 2017, 56, 371.
(q) Elangovan, S.; Sortais, J. B.; Beller, M.; Darcel, C. Angew. Chem., Int. Ed. 2016, 54, 14483.
(r) Yu, X.-L.; Zhao, R.-R.; Wan, H.-D.; Yang, Y.-C.; Wang, D.-W. Tetrahedron Lett. 2016, 57, 4588.
(s) Yamaguchi, R.; Zhu, M.-W.; Kawagoe, S.; Asai, C.; Fujita, K. I. Synthesis 2009, 1220.
(t) Wang, D.-W.; Zhao, K.-Y.; Yu, X.; Miao, H.-Y.; Ding, Y.-Q. RSC Adv. 2014, 46, 42924.
(u) Saidi, O.; Blacker, A. J.; Lamb, G. W.; Marsden, S. P.; Taylor, J. E.; Williams, J. M. J. Org. Process Res. Dev. 2010, 14, 1046.
(v) Lu, L.; Ma, J.; Qu, P.-P.; Li, F. Org. Lett. 2015, 17, 2350.
(w) Yamaguchi, R.; Kawagoe, S.; Asai, C.; Fujita, K. Org. Lett. 2008, 10, 181.
(x) Li, F.; Ma, J.; Wang, N. J. Org. Chem. 2014, 46, 10447.
(y) Deibl, N.; Ament, K.; Kempe, R. J. Am. Chem. Soc. 2015, 137, 12804.
(z) Kawahara, R.; Fujita, K.; Yamaguchi, R. J. Am. Chem. Soc. 2010, 132, 15108.
(aa) Li, F.; Shan, H.; Chen, L.; Kang, Q.; Zou, P. Chem. Commun. 2011, 43, 603.
(ab) Peña-López, M.; Piehl, P.; Elangovan, S.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2016, 55, 14967.
(ac) Elangovan, S.; Neumann, J.; Sortais, J. B.; Junge, K.; Darcel, C.; Beller, M. Nat. Commun. 2016, 7, 12641.
(ad) Deibl, N.; Kempe, R. Angew. Chem., Int. Ed. 2017, 56, 1663.
(ae) Wang, N.-N.; Zou, X.-Y.; Ma, J.; Li, F. Chem. Commun. 2014, 50, 8303.
(af) Michlik, S.; Kempe, R. Nat. Chem. 2013, 5, 140. -
[7]
Sindhuja, E.; Ramesh, R. Tetrahedron Lett. 2014, 55, 5504. doi: 10.1016/j.tetlet.2014.08.035
-
[8]
Tanabe, Y.; Kuriyama, S.; Arashiba, K.; Nakajima, K.; Nishibayashi, Y. Organometallics 2014, 33, 5295. doi: 10.1021/om5006116
-
[9]
Rigoli, J. W.; Moyer, S. A.; Pearce, S. D.; Schomaker, J. M. Org. Biomol. Chem. 2012, 10, 1746. doi: 10.1039/c2ob06921k
-
[10]
Bauer, J. O.; Leitus, G.; Ben-David, Y.; Milstein, D. ACS Catal. 2016, 6, 8415. doi: 10.1021/acscatal.6b02946
-
[11]
Kondo, T.; Yang, S.; Huh, K. T.; Kobayashi, M.; Kotachi, S.; Watanabe, Y. Chem. Lett. 1991, 20, 1275. doi: 10.1246/cl.1991.1275
-
[12]
Blacker, A. J.; Farah, M. M.; Hall, M. I.; Marsden, S. P.; Saidi, O.; Williams, J. M. J. Org. Lett. 2009, 11, 2039. doi: 10.1021/ol900557u
-
[13]
Khalafi-Nezhad, A.; Panahi, F. ACS Catal. 2014, 4, 1686. doi: 10.1021/cs5000872
-
[14]
Tsuji, Y.; Kotachi, S.; Huh, K. T.; Watanabe, Y. J. Org. Chem. 1990, 55, 580. doi: 10.1021/jo00289a036
-
[15]
Izumi, T.; Yokota, T. J. Heterocycl. Chem. 1992, 29, 1085. doi: 10.1002/jhet.v29:5
-
[16]
Shimura, S.; Miura, H.; Wada, K.; Hosokawa, S.; Yamazoe, S.; Inoue, M. Catal. Sci. Technol. 2011, 1, 1340. doi: 10.1039/c1cy00235j
-
[17]
(a) Marco-Contelles, J.; Pérez-Mayoral, E.; Samadi, A.; Carreiras, M. C.; Soriano, E. Chem. Rev. 2009, 109, 2652.
(b) Martínez, R.; Ramón, D. J.; Yus, M. J. Org. Chem. 2008, 73, 9778.
(c) Venkatesan, H.; Hocutt, F. M.; Jones, T. K.; Rabinowitz, M. H. J. Org. Chem. 2010, 75, 3488. -
[18]
Chen, M.-M.; Zhang, M.; Xiong, B.; Tan, Z.-D.; Lv, W.; Jiang, H.-F. Org. Lett. 2014, 16, 6028. doi: 10.1021/ol503052s
-
[19]
Monrad, R. N.; Madsen, R. Org. Biomol. Chem. 2011, 9, 610. doi: 10.1039/C0OB00676A
-
[20]
Porcheddu, A.; Mura, M. G.; De Luca, L.; Pizzetti, M.; Taddei, M. Org. Lett. 2012, 14, 6112. doi: 10.1021/ol3030956
-
[21]
Xie, F.; Chen, M.-M.; Wang, X.-T.; Jiang, H.-F.; Zhang, M. Org. Biomol. Chem. 2014, 12, 2761. doi: 10.1039/C3OB42589D
-
[22]
Zeng, M.; Wang, T.; Cui, D.-M.; Zhang, C. New J. Chem. 2016, 40, 8225. doi: 10.1039/C6NJ01620K
-
[23]
Mura, M. G.; De Luca, L.; Taddei, M.; Williams, J. M. J.; Porcheddu, A. Org. Lett. 2014, 16, 2586. doi: 10.1021/ol500916g
-
[24]
Iida, K.; Miura, T.; Ando, J.; Saito, S. Org. Lett. 2013, 15, 1436. doi: 10.1021/ol4001262
-
[25]
(a) Srimani, D.; Ben-David, Y.; Milstein, D. Angew. Chem., Int. Ed. 2013, 52, 4012.
(b) Srimani, D.; Ben-David, Y.; Milstein, D. Chem. Commun. 2013, 49, 6632. -
[26]
(a) Zhang, M.; Fang, X. J.; Neumann, H.; Beller, M. J. Am. Chem. Soc. 2013, 135, 11384.
(b) Zhang, M.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 597. -
[27]
Pan, B.; Liu, B.; Yue, E.-L.; Liu, Q.-B.; Yang, X.-Z.; Wang, Z.; Sun, W.-H. ACS Catal. 2016, 6, 1247. doi: 10.1021/acscatal.5b02638
-
[28]
(a) Chen, T.-Y.; Tsutsumi, R.; Montgomery, T. P.; Volchkov, I.; Krische, M. J. J. Am. Chem. Soc. 2015, 137, 1798.
(b) Mcinturff, E. L.; Yamaguchi, E.; Krische, M. J. J. Am. Chem. Soc. 2012, 134, 20628.
(c) Zbieg, J. R.; Yamaguchi, E.; Mcinturff, E. L.; Krische, M. J. Science 2012, 336, 324. -
[29]
Watson, A. J. A.; Maxwell, A. C.; Williams, J. M. J. Org. Lett. 2009, 11, 2667. doi: 10.1021/ol900723v
-
[30]
Watson, A. J. A.; Wakeham, R. J.; Maxwell, A. C.; Williams, J. M. J. Tetrahedron 2014, 70, 3683. doi: 10.1016/j.tet.2014.04.017
-
[31]
Ortega, N.; Richter, C.; Glorius, F. Org. Lett. 2013, 15, 1776. doi: 10.1021/ol400639m
-
[32]
Oldenhuis, N. J.; Dong, V. M.; Guan, Z. Tetrahedron 2014, 70, 4213. doi: 10.1016/j.tet.2014.03.085
-
[33]
Nova, A.; Balcells, D.; Schley, N. D.; Dobereiner, G. E.; Crabtree, R. H.; Eisenstein, O. Organometallics 2010, 29, 6548. doi: 10.1021/om101015u
-
[34]
Kim, J.; Hong, S. H. Org. Lett. 2014, 16, 4404. doi: 10.1021/ol501835t
-
[35]
Hamid, M. H. S. A.; Williams, J. M. J. Chem. Commun. 2007, 38, 725.
-
[36]
Hamid, M. H.; Allen, C. L.; Lamb, G. W.; Maxwell, A. C.; Maytum, H. C.; Watson, A. J.; Williams, J. M. J. Am. Chem. Soc. 2009, 131, 1766. doi: 10.1021/ja807323a
-
[37]
Yamaguchi, K.; He, J.; Oishi, T.; Mizuno, N. Chem. Eur. J. 2010, 16, 7199. doi: 10.1002/chem.201000149
-
[38]
Bähn, S.; Imm, S.; Mevius, K.; Neubert, L.; Tillack, A.; Williams, J. M. J.; Beller, M. Chem. Eur. J. 2010, 16, 3590. doi: 10.1002/chem.v16:12
-
[39]
Imm, S.; Baehn, S.; Zhang, M.; Neubert, L.; Neumann, H.; Klasovsky, F.; Pfeffer, J.; Haas, T.; Beller, M. Angew. Chem., Int. Ed. 2011, 50, 7599. doi: 10.1002/anie.201103199
-
[40]
Balaraman, E.; Srimani, D.; Diskin-Posner, Y.; Milstein, D. Catal. Lett. 2014, 145, 139.
-
[41]
Zhang, M.; Imm, S.; Bähn, S.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2011, 50, 11197. doi: 10.1002/anie.v50.47
-
[42]
Watson, A. J. A.; Maxwell, A. C.; Williams, J. M. J. J. Org. Chem. 2011, 76, 2328. doi: 10.1021/jo102521a
-
[43]
Ma, W.-M.; James, T. D.; Williams, J. M. Org. Lett. 2013, 15, 4850. doi: 10.1021/ol402271a
-
[44]
Chen, M.-M.; Zhang, M.; Xie, F.; Wang, X.-T.; Jiang, H.-F. ChemCatChem 2014, 6, 2993. doi: 10.1002/cctc.v6.10
-
[45]
Enyong, A. B.; Moasser, B. J. Org. Chem. 2014, 79, 7553. doi: 10.1021/jo501273t
-
[46]
Broomfield, L. M.; Wu, Y. C.; Martin, E.; Shafir, A. Adv. Synth. Catal. 2015, 357, 3538. doi: 10.1002/adsc.201500562
-
[47]
Dang, T.-T.; Ramalingam, B.; Seayad, A. M. ACS Catal. 2015, 5, 4082. doi: 10.1021/acscatal.5b00606
-
[48]
Shan, S. P.; Xie, X.-K.; Gnanaprakasam, B.; Dang, T. T.; Ramalingam, B.; Huynh, H. V.; Seayad, A. M. RSC Adv. 2015, 5, 4434. doi: 10.1039/C4RA15398G
-
[49]
Rajaraman, A.; Sahoo, A. R.; Hild, F.; Fischmeister, C.; Achard, M.; Bruneau, C. Dalton Trans. 2015, 44, 17467. doi: 10.1039/C5DT02867A
-
[50]
Şahin, Z.; Gürbüz, N.; Özdemir, İ.; Şahin, O.; Büyükgüngör, O.; Achard, M.; Bruneau, C. Organometallics 2015, 34, 2296.
-
[51]
Marichev, K. O.; Takacs, J. M. ACS Catal. 2016, 6, 2205. doi: 10.1021/acscatal.6b00175
-
[52]
Pagliaro, M.; Rossi, M. The Future of Glycerol, Royal Society of Chemistry, 2010.
-
[53]
Said Stålsmeden, A.; Belmonte Vázquez, J. L.; van Weerdenburg, K.; Rae, R.; Norrby, P.-O.; Kann, N. ACS Sustainable Chem. Eng. 2016, 4, 5730. doi: 10.1021/acssuschemeng.6b01659
-
[54]
Shi, F.; Tse, M. K.; Zhou, S.; Pohl, M. M.; Radnik, J.; Hübner, S.; Jähnisch, K.; Brückner, A.; Beller, M. J. Am. Chem. Soc. 2009, 131, 1775. doi: 10.1021/ja807681v
-
[55]
Mako, T. L.; Byers, J. A. Inorg. Chem. Front. 2016, 3, 766. doi: 10.1039/C5QI00295H
-
[56]
Oldenhuis, N. J.; Dong, V. M.; Guan, Z. J. Am. Chem. Soc. 2014, 136, 12548. doi: 10.1021/ja5058482
-
[57]
Yang, L.-C.; Wang, Y.-N.; Zhang, Y.; Zhao, Y. ACS Catal. 2017, 7, 93. doi: 10.1021/acscatal.6b02959
-
[58]
Watson, A. J. A.; Maxwell, A. C.; Williams, J. M. J. Org. Biomol. Chem. 2012, 10, 240. doi: 10.1039/C1OB06516E
-
[59]
Xiong, B.; Li, Y.; Lv, W.; Tan, Z.-D.; Jiang, H.-F.; Zhang, M. Org. Lett. 2016, 47, 4054.
-
[60]
Xiong, B.; Zhang, S.-D.; Chen, L.; Li, B.; Jiang, H.-F.; Zhang, M. Chem. Commun. 2016, 52, 10636. doi: 10.1039/C6CC05329G
-
[61]
Peña-López, M.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2016, 55, 7826. doi: 10.1002/anie.201600698
-
[62]
(a) Yang, H.-Q.; Shen, R.; Deng, G.-J. Chin. J. Org. Chem. 2012, 32, 1725 (in Chinese).
(杨辉琼, 谌儒, 邓国军, 有机化学, 2012, 32, 1725.)
(b) Neumann, J.; Bornschein, C.; Jiao, H.; Junge, K.; Beller, M. Eur. J. Org. Chem. 2015, 2015, 5944. -
[63]
Feng, C.; Liu, Y.; Peng, S.-M.; Shuai, Q.; Deng, G.-J.; Li, C.-J. Org. Lett. 2010, 12, 4888. doi: 10.1021/ol1020527
-
[64]
Li, H.-J.; Wang, C.-C.; Zhu, S.; Dai, C.-Y.; Wu, Y.-C. Adv. Synth. Catal. 2015, 357, 583. doi: 10.1002/adsc.201400898
-
[65]
Xie, F.; Zhang, M.; Chen, M.-M.; Lv, W.; Jiang, H.-F. ChemCatChem 2015, 7, 349. doi: 10.1002/cctc.201402832
-
[66]
Xie, F.; Zhang, M.; Jiang, H.-F.; Chen, M.-M.; Lv, W.; Zheng, A.-B.; Jian, X.-J. Green Chem. 2015, 17, 279. doi: 10.1039/C4GC01316F
-
[67]
Deibl, N.; Kempe, R. J. Am. Chem. Soc. 2016, 138, 10786. doi: 10.1021/jacs.6b06448
-
[68]
Cho, C. S.; Kim, B. T.; Kim, T.-J.; Chul Shim, S. Tetrahedron Lett. 2002, 43, 7987. doi: 10.1016/S0040-4039(02)01625-8
-
[69]
Yan, F.-X.; Zhang, M.; Wang, X.-T.; Xie, F.; Chen, M.-M.; Jiang, H.-F. Tetrahedron 2014, 70, 1193. doi: 10.1016/j.tet.2013.12.065
-
[70]
Kuwahara, T.; Fukuyama, T.; Ryu, I. Org. Lett. 2012, 14, 4703. doi: 10.1021/ol302145a
-
[71]
Kuwahara, T.; Fukuyama, T.; Ryu, I. RSC Adv. 2013, 3, 13702. doi: 10.1039/c3ra42834f
-
[72]
Schlepphorst, C.; Maji, B.; Glorius, F. ACS Catal. 2016, 6, 4184. doi: 10.1021/acscatal.6b01351
-
[73]
Chaudhari, M. B.; Bisht, G. S.; Kumari, P.; Gnanaprakasam, B. Org. Biomol. Chem. 2016, 14, 9215. doi: 10.1039/C6OB01786J
-
[74]
Cini, E.; Petricci, E.; Truglio, G. I.; Vecchio, M.; Taddei, M. RSC Adv. 2016, 6, 31386. doi: 10.1039/C6RA03585J
-
[75]
Sik Cho, C.; Kim, B. T.; Taejeong Kim, A.; Sang, C. S. J. Org. Chem. 2001, 66, 9020. doi: 10.1021/jo0108459
-
[76]
Li, Y.; Li, H. Q.; Junge, H.; Beller, M. Chem. Commun. 2014, 50, 14991. doi: 10.1039/C4CC06933A
-
[77]
Wang, Q.-F.; Wu, K.-K.; Yu, Z.-K. Organometallics 2016, 35, 1251. doi: 10.1021/acs.organomet.6b00130
-
[78]
Chakrabarti, K.; Paul, B.; Maji, M.; Roy, B. C.; Shee, S.; Kundu, S. Org. Biomol. Chem. 2016, 14, 10988. doi: 10.1039/C6OB02010K
-
[79]
Takashi, A. M. J.; Takahide, F.; Ilhyong, R. Chem. Lett. 2013, 42, 1163. doi: 10.1246/cl.130465
-
[80]
Pena-Lopez, M.; Neumann, H.; Beller, M. Chem. Commun. 2015, 51, 13082. doi: 10.1039/C5CC01708D
-
[1]
-

图式 1 基于钌催化醇类化合物脱氢的偶联反应
Scheme 1 Ruthenium-catalyzed dehydrogenation coupling reactions with alcohol

图式 2 钌催化合成2-芳基喹唑啉可能的反应机理
Scheme 2 Proposed mechanism for the synthesis of 2-arylquinazolines catalyzed by Ruthenium

图式 5 钌催化合成2, 4, 6-三芳基-1, 3, 5-三嗪衍生物可能的反应机理
Scheme 5 Proposed mechanism for the synthesis of 2, 4, 6-tri-aryl-1, 3, 5-triazine derivatives catalyzed by Ruthenium

图式 6 钌催化醇的C—C偶联合成α, β-不饱和醛可能的反应机理
Scheme 6 Proposed mechanism for ruthenium catalyzed synthesis of α, β-unsaturated aldehydes based on primary alcohols

图式 7 钌催化合成吡咯衍生物可能的反应机理
Scheme 7 Proposed mechanism for ruthenium catalyzed synthesis of pyrroles

图式 10 钌催化“氢转移”醇与胺氧化偶联反应
Scheme 10 Oxidative coupling of alcohols with amines using hydrogen transfer catalyzed by ruthenium

图式 11 钌催化合成环内酰胺可能的反应机理
Scheme 11 Proposed mechanism for the synthesis of cyclic imides catalyzed by ruthenium

图式 14 钌催化“氢转移”四氢吡嗪衍生物的合成
Scheme 14 Ruthenium-catalyzed synthesis of tetrahydro fused-pyrazine derivatives by hydrogen borrowing method

图式 15 钌催化“氢转移”2-噁唑烷酮衍生物的合成
Scheme 15 Synthesis of oxazolidin-2-ones by hydrogen borrowing method catalyzed by ruthenium

图式 16 钌催化剂醇与硝基类化物合成叔胺衍生物
Scheme 16 Synthesis of tertiary amine catalyzed by ruthenium from alcohols and nitroarenes

图式 18 钌催化醇与α, γ-二羰基化合物的C—C偶联反应
Scheme 18 Ruthenium catalyzed C—C coupling reactions between alcohols and α, γ-dicarboxylic compounds

图式 19 钌催化醇与α, γ-二羰基化合物的C—C偶联反应可能的反应机理
Scheme 19 Proposed mechanism of C—C coupling reactions between alcohols and α, γ-dicarboxylic compounds catalyzed by ruthenium

图式 20 钌催化醇的C—C偶联反应
Scheme 20 Synthesis of alcohols via C—C bond coupling catalyzed by ruthenium from alcohols
-


 下载:
下载:




















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 12
- 文章访问数: 2623
- HTML全文浏览量: 634
 下载:
下载:
