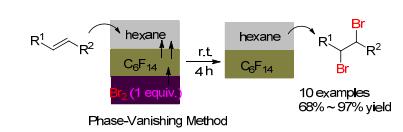
 图 图式1
相位消失法研究烯烃的双溴化加成
Figure 图式1.
Phase-Vanishing dibromination of alkenes
图 图式1
相位消失法研究烯烃的双溴化加成
Figure 图式1.
Phase-Vanishing dibromination of alkenes
引用本文:
何天雄, 曾祥华. 烯烃双卤化加成反应的研究进展[J]. 有机化学,
2017, 37(4): 798-809.
doi:
10.6023/cjoc201611041

Citation: He Tianxiong, Zeng Xianghua. Recent Advances in Dihalogenation of Alkenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 798-809. doi: 10.6023/cjoc201611041
Citation: He Tianxiong, Zeng Xianghua. Recent Advances in Dihalogenation of Alkenes[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 798-809. doi: 10.6023/cjoc201611041
烯烃双卤化加成反应的研究进展
English
Recent Advances in Dihalogenation of Alkenes
Abstract:
The vicinal dihalogens structure motif can be found in a variety of natural products and pharmaceuticals. The dihalogenation of alkenes is a commonly employed strategy for the rapid construction of carbon-halogen bonds in organic synthesis. In recent years, main progress has been achieved in the dihalogenation of alkenes. Based on our work and research interests, the aim of this review is to give an overview of the progress on the diverse synthetic methodologies of the dihalogenation of alkenes since 2000. Additionally, research trends of this area are also discussed.
-
Key words:
- alkene
- / dihalogenation
- / synthetic method
-
含卤素的有机化合物大量存在于天然产物和药物中, 并且是化工原料或药物中间体合成的重要合成子[1].正因为如此, 科学家开发了多种多样构建碳卤键的方法, 有通过碳氧键、碳氮键及烯烃的卤化加成反应等.其中, 由于烯烃的卤化加成反应类型多样 (包括卤胺化、卤羟化、卤醚化、卤酯化和双卤化等), 反应模式多变, 得到了有机化学家的广泛关注[2].
早在1938年, 烯烃的双卤化加成反应已有报道[3], 其卤源包括氟气、氯气和液溴, 然而, 氟气、氯气和液溴的毒性大, 给实验操作带来困难.近年来, 随着绿色化学和有机合成的快速发展, 化学工作者们在烯烃双卤化加成反应方面的研究也取得了迅猛的进展[4], 其研究集中在两个方面:一是寻找安全、易合成操作的卤源; 二是研究各种功能化烯烃的催化体系.
通过原位氧化金属卤化物生成卤素或者直接用卤素为卤源的烯烃双卤化综述已有相关报道[4], 故本文主要对2000年以来发展的新合成方法、卤化试剂、烯烃双卤化类型及烯烃底物类型进行了梳理, 介绍烯烃在双溴化、双氯化、溴氯化及双氟化的研究现状, 以及包含烯烃的不对称双卤化加成反应.对以上反应的机理进行了阐述, 并对今后烯烃的双卤化加成反应的发展方向进行了展望.
1 烯烃的双溴化加成反应
烯烃双溴化加成反应的研究较多, 根据溴化试剂的不同大致可分为无机溴盐、有机溴盐和含溴有机物.首先介绍液溴为溴化试剂的烯烃双溴化加成反应.
2002年, Ryu课题组[5]报道了相消失法研究烯烃的双溴化加成 (Scheme 1).该体系采用全氟正己烷 (FC-72) 为分界相, 将正己烷溶剂和烯烃与液溴层分开, 然后液溴慢慢向上渗透与烯烃反应, 经过2 d后液溴层消失.此反应体系探讨了脂肪烯烃、苯乙烯和丙烯酸乙酯的双溴化加成反应, 可以获得68%~97%产率. 2005年, Patel等[6]报道了采用吡啶乙烷溴酸盐负载液溴并用无溶剂法研究烯烃的双溴化加成 (Eq. 1).值得说明的是, 该体系液溴也可以用溴化钾和Oxone反应生成, 然后与吡啶乙烷溴酸盐生成DPTBE溴化试剂, 而且吡啶乙烷溴酸盐可以回收循环使用.在双溴化加成反应中, 底物的普适性广和反应时间均在30 min以内, 可以获得87%~96%产率.同年, Toda等[7]也报道了吡啶溴酸盐和季铵溴酸盐负载液溴作为溴化试剂, 无溶剂法研究了查尔酮和1, 2-二苯乙烯的双溴化加成.之后, 其他课题组对含氮载体负载液溴做了进一步研究, 如聚 (N-乙烯基吡咯烷酮)[8]、咪唑盐离子液体[9]和十六烷剂三甲基铵盐[10].
图 图式1
相位消失法研究烯烃的双溴化加成
Figure 图式1.
Phase-Vanishing dibromination of alkenes
2006年, Shi课题组[11]报道了将有机溴化试剂 (NBS) 和无机溴化试剂LiBr联用, 可以在2~30 min内快速双溴化苯乙烯和环己烯类烯烃, 并获得56%~94%产率 (Eq. 2). 2011年, Yan等[12]对该体系进行了拓展, 然而, 当把LiBr换成NaBr时, 需要加入10%的FeBr3作催化剂和加热.
2009年, Iskra课题组[13]报道了将廉价易得的氢溴酸水溶液作为溴化试剂, 研究了苯乙烯类底物的双溴化加成反应.该体系用NaNO2为催化剂及空气中的氧气为氧化剂, 室温下原位生成Br2, 然后进行烯烃的双溴化加成.在不同苯乙烯底物中, 可以获得35%~95%的产率和良好的非对映选择性 (d.r.), 但是对不饱和烯烃肉桂酸甲酯的非对映选择性不好, 并且作者对反应机理进行了研究 (Scheme 2).之后, Magolan[14]和Jiao[15]等几乎同时发现, 用廉价的DMSO/HBr体系也可实现脂肪烯烃和苯乙烯的双溴化加成 (Scheme 3).此反应中, DMSO既是溶剂又是氧化剂, 而且对含各种官能化烯烃都有良好的容忍性.作者推测其反应机理可能是: DMSO与氢溴酸发生亲电加成, 然后失去水分子生成硫盐BDMS, 接着溴正离子与烯烃生成溴鎓离子, 最后溴负离子进攻溴鎓离子开环生成二溴化合物. 2012年, Nama等[16]报道了NH4Br/Oxone在乙腈溶剂中研究苯乙烯和脂肪烯烃的双溴化加成, 但是该体系对一些不饱和羰基烯烃底物不合适, 如香豆素.同年, Wang课题组[17]报道了NaBr/ Oxone无溶剂球磨法研究不饱和羰基化合物 (包括查尔酮和肉桂酸甲酯) 的双溴化加成反应, 该体系可以在40 min内快速获得78%~95%产率的二溴化产物且有很好的非对映选择性 (Eq. 3).之后, Et4NBr/HBrO3[18]和KBr/I2O5[19]原位氧化生成液溴的体系也相继报道, 研究了肉桂酸酯、脂肪烯烃、查尔酮和苯乙烯类底物.
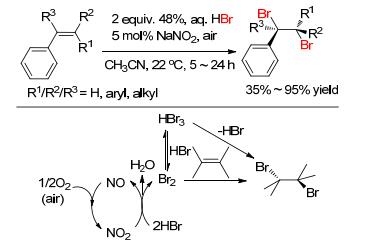 图 图式2
亚硝酸钠催化烯烃的双溴化加成及可能的反应机理
Figure 图式2.
Sodium nitrite-catalyzed dibromination of alkenes and proposed mechanism
图 图式2
亚硝酸钠催化烯烃的双溴化加成及可能的反应机理
Figure 图式2.
Sodium nitrite-catalyzed dibromination of alkenes and proposed mechanism
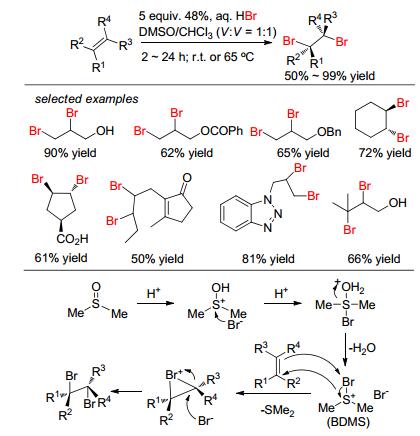 图 图式3
DMSO/HBr体系促进的烯烃双溴化加成及可能反应机理
Figure 图式3.
Dibromination of olefins with HBr/DMSO system and proposed mechanism
图 图式3
DMSO/HBr体系促进的烯烃双溴化加成及可能反应机理
Figure 图式3.
Dibromination of olefins with HBr/DMSO system and proposed mechanism
2010年, Zhao等[20]首先报道了一例有机胺催化α, β-不饱和醛的双溴化加成 (Eq. 4).此体系通过吡咯烷和2, 5-吡咯烷酮为共同催化剂、NBS为溴化试剂、氯仿为溶剂, 在60 ℃下反应2 h, 可获得中等产率的双溴化产物, 并且d.r.都大于25:1.有趣的是, 研究者发现该体系对苯乙烯和脂肪烯烃的催化效果一般.之后, Liang小组[21]发现, 用苯甲酸为催化剂, 在室温条件下也可实现α, β-不饱和羰基化合物查尔酮的双溴化加成, 并研究了其反应机理 (Scheme 4).更重要的是, 将NBS换成NCS氯化试剂, 同样可实现双氯化加成.在此基础之上, Gulder等[22]重点研究催化剂对溴化试剂的影响, 合成了一系列邻碘苯甲酰胺催化剂.研究者发现通过氯化铵促进邻碘苯甲酰胺催化剂与NBS原位反应生成高价碘溴 (Iodine (Ⅲ)) 催化剂, 从而催化烯烃的双溴化加成, 而且烯烃底物的普适性广和获得优秀的非对映选择性 (Scheme 5).但是, 该体系把NBS换成NCS氯化试剂后, 只有脂肪烯烃可实现双氯化加成, 而不饱和烯烃只是生成了烯烃的单氯取代产物.
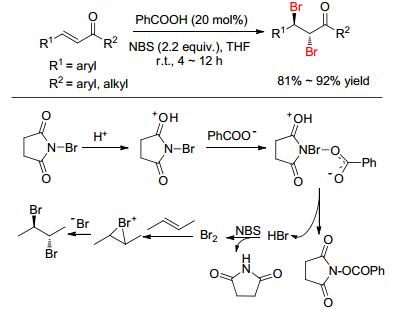 图 图式4
苯甲酸催化α, β-不饱和羰基化合物的双溴化加成
Figure 图式4.
Benzoic acid-catalyzed dibromination of α, β-unsaturated carbonyl compounds
图 图式4
苯甲酸催化α, β-不饱和羰基化合物的双溴化加成
Figure 图式4.
Benzoic acid-catalyzed dibromination of α, β-unsaturated carbonyl compounds
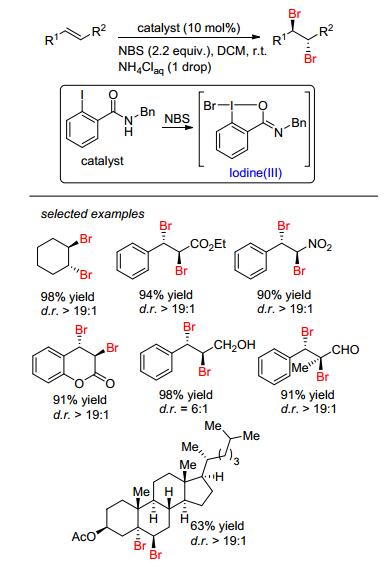 图 图式5
三价碘催化烯烃的双溴化加成
Figure 图式5.
Iodine (Ⅲ)-catalyzed dibromination of alkenes
图 图式5
三价碘催化烯烃的双溴化加成
Figure 图式5.
Iodine (Ⅲ)-catalyzed dibromination of alkenes
由于有机小分子催化剂具有绿色环保的优点, 有机小分子催化烯烃双溴化加成方面的研究不断有新的报道. 2012年, Barbas课题组[23]报道了硫脲催化烯烃的双溴化加成 (Eq. 5), 而且底物的普适性较广, 包括缺电子/富电子烯烃、三取代烯烃和2-吲哚酮烯烃.此体系用的溴化试剂是反应活性比N-溴代丁二酰亚胺 (NBS) 更高的1, 3-二溴-5, 5-二甲基海因 (DBDMH), 在烯烃双溴化加成产物的产率和非对映选择性方面都有良好结果.
随着不对称催化的快速发展, 烯烃不对称双溴化加成方面也有了突破. 2013年, Burns等[24]报道了首例丙烯醇的不对称双溴化加成 (Eq. 6).此体系用二溴丙二酸二乙酯 (DEDBM) 和三异丙氧基溴化钛为溴化试剂、酒石酸衍生的手性二醇为催化剂或诱导剂可以获得中等产率和80%~91% ee的手性双溴化加成产物.非常有趣的是, 作者发现, 当用化学剂量的手性诱导剂时明显比催化剂量的效果更好, 可以提高5%~10% ee值.因此, 作者推测其反应机理是 (Scheme 6):首先烯丙醇与两种溴化试剂生成六配位的钛化合物 (其中三异丙氧基溴化钛的溴负离子与钛配位), 然后2, 2-二溴丙二酸二乙酯发生烯醇化失去一个溴正离子与双键生成溴鎓离子, 接着与钛配位的溴负离子进攻溴鎓离子开环, 最后生成不对称双溴化加成.虽然丙烯醇的不对称双溴化加成的产率不高, 但是该研究给烯烃的对对称双溴化加成提供了新的思路, 尤其是选择溴化试剂和手性催化剂. 2016年, 该课题组[25]进一步研究了此反应 (Eq. 7), 选择了手性希夫碱为催化剂、N-溴代丁二酰亚胺 (NBS) 和三异丙醇溴化钛[BrTi (Oi-Pr)3]为溴化试剂、正己烷为溶剂, -20 ℃反应4~12 h, 可以获得80%~87% ee值和75%~86%产率, 而且脂肪类丙烯醇也适合该体系.
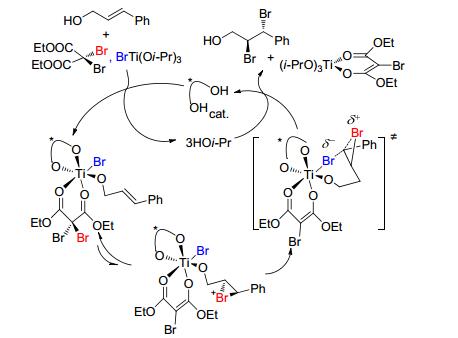 图 图式6
丙烯醇不对称双溴化的可能反应机理
Figure 图式6.
Proposed mechanism of asymmetric dibromination of allylic alcohols
图 图式6
丙烯醇不对称双溴化的可能反应机理
Figure 图式6.
Proposed mechanism of asymmetric dibromination of allylic alcohols
通常, 三苯基氧膦 (Ph3PO) 是Wittig反应的一个副产物, 然而, 2014年, Xu等[26]发现Ph3PO能高效催化不饱和羰基化合物的双溴化加成, 这是该研究的一个亮点.此体系用草酰溴为溴化试剂和分子筛为添加剂, 对α, β-不饱和酯可获得83%~96%的产率和大于19:1的d.r.值双溴化产物; 但β, γ-不饱和α-酮酸酯类底物, 只能获得中等的d.r.值 (Scheme 7), 而且作者进一步研究了α, β, γ, δ-不饱和酯溴化加成, 发现延长反应时间后, 可以获得69%产率四溴化加成产物和10:1的d.r.值 (Scheme 7).此外, 该体系对苯乙烯类底物也获得中等产率.通过控制实验, 作者推测其反应机理是 (Scheme 7):首先三苯基氧膦与草酰溴反应生成中间体溴鏻盐, 然后溴鏻盐与烯烃反应生成双溴化加成产物同时释放三苯基膦, 最后三苯基膦与草酰溴反应生成溴鏻盐, 从而完成催化循环过程.受此机理的启发, 该研究小组进一步研究了苯甲醛与Wittig试剂反应生成烯烃[27], 然后加入草酰溴, 实现了一锅法完成双溴化或四溴化加成 (Scheme 8), 省去了分离烯烃和将副产物三苯基氧膦直接作为催化剂.
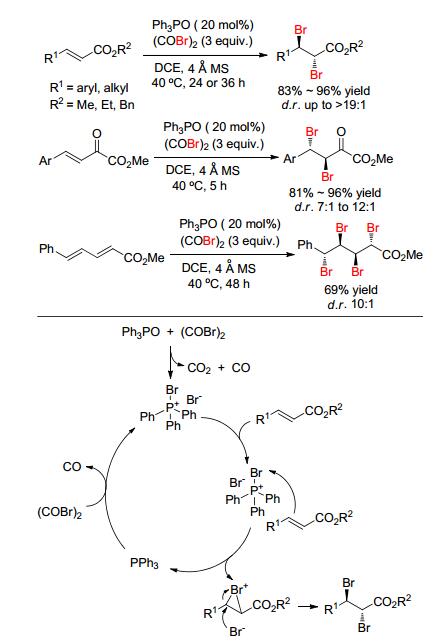 图 图式7
三苯基氧膦催化不饱和羰基化合物双溴化加成及反应机理
Figure 图式7.
Triphenylphosphine oxide-catalyzed dibromination of unsaturated compounds and proposed mechanism
图 图式7
三苯基氧膦催化不饱和羰基化合物双溴化加成及反应机理
Figure 图式7.
Triphenylphosphine oxide-catalyzed dibromination of unsaturated compounds and proposed mechanism
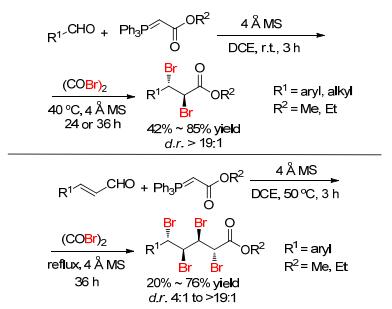 图 图式8
一锅法合成双溴化和四溴化化合物
Figure 图式8.
One-pot method for the synthesis of dibromination and tetrabromination compounds
图 图式8
一锅法合成双溴化和四溴化化合物
Figure 图式8.
One-pot method for the synthesis of dibromination and tetrabromination compounds
2 烯烃的双氯化加成反应
对于烯烃的双氯化加成反应, 氯气是最理想的氯化试剂, 因为百分之百的原子经济性, 所以一直有文献报道.如2003年, Iskra等[28]报道了将氯气通入全氟正辛烷 (FC-77) 后逐渐渗透到反应相溶剂与烯烃反应, 此体系对苯乙烯、脂肪烯烃可以获得88%~97%产率, 但肉桂酸乙酯只可获得77%产率和1:1 d.r.值. 2009年, Snyder等[29]报道全合成Napyradiomycin A1时, 通过烯烃的双氯化反应可以快速构建碳氯键.尽管氯气作为氯化试剂具有以上优点, 但是由于其反应活性太强, 需要在-78 ℃反应才能获得良好的非对映选择性, 还有氯气的毒性问题, 这些给实际合成操作带来不便[30].基于以上考虑, 近年来, 化学工作者们在研究其它氯化试剂和催化体系, 并拓展烯烃的范围.
2011年, Zou等[31]报道了醋酸锰氧化盐酸原位生成氯气研究查尔酮类烯烃的双氯化加成, 可以获得73%~ 85%的产率, 但是没有报道其产物的d.r.值.之后, Kitamura课题组[32]报道了碘氧苯氧化盐酸原位生成氯气研究苯乙烯类烯烃的双氯化加成, 在40 ℃反应2~5 h后可获得56%~93%的产率. 2013年, Tong课题组[33]报道了Oxone氧化氯化钠原位生成氯气研究丙烯醇类烯烃的双氯化加成, 可以获得53%~99%产率和1:1到大于20:1 d.r.值 (Eq. 8).在此基础上, Narender课题组[34]报道了相同反应条件下, 室温下研究苯乙烯和脂肪类烯烃的双氯化加成, 可以获得69%~85%产率.另外, Yoshimitsu等[35]也报道了三苯基膦与NCS原位反应生成氯气研究此类烯烃的双氯化加成, 可以获得57%~ 96%产率. 2016年, Egami等[36]报道了4-苯基吡啶-N-氧化物氧化高价碘二氯化苯乙烯类底物双氯化加成 (Eq. 9), 该体系尽管是高价的碘氯试剂, 如果不加入4-苯基吡啶-N-氧化物, 此反应不能进行.
相比于用无机盐做氯化试剂时, 需要加入化学剂量的氧化剂, 而有机氯化试剂则只需加入少量的催化剂. 2011年, Nicolaou课题组[37]报道了金鸡纳碱催化丙烯醇类烯烃的不对称双氯化加成, 选用二氯碘苯为氯化试剂 (Eq. 10).该有机催化体系中, 对芳香丙烯醇类底物可获得81% ee值, 但是在苯环上引入不管是富电子基团 (如甲基) 或贫电子基团 (如氯、溴、氟、三氟甲基和萘基) 时, 其产物ee值都有所下降; 尤其是当羟基被保护后, 其产物几乎是消旋体, 这说明其催化剂跟底物的羟基形成了氢键作用, 从而诱导手性的产生 (Scheme 9).之后, Burns课题组[25]在全合成Deschloromytilipin A和Danicalipin A时, 对丙烯醇类烯烃的不对称双氯化加成进行了深入研究 (Eq. 11).此反应体系选用手性醇烯胺为催化剂、叔丁基醇氯 (t-BuOCl) 和三异丙醇氯化钛[ClTi (Oi-Pr)3]为氯化试剂、正己烷为溶剂, -20 ℃反应4~12 h, 可以获得80%~91% ee值, 并且脂肪类丙烯醇也适合该体系, 最高可获得61%产率和90% ee值.
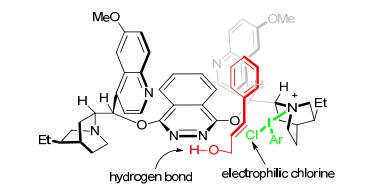 图 图式9
不对称称双氯化诱导模型
Figure 图式9.
Proposed stereoinduction model for enantioselective dichlorination
图 图式9
不对称称双氯化诱导模型
Figure 图式9.
Proposed stereoinduction model for enantioselective dichlorination
2014年, Xu课题组[38]报道了三苯基氧膦催化β, γ-不饱和α-酮酸酯的双氯化加成, 非常有趣的是, 该反应用草酰氯作为氯化试剂时, 得到反式1, 3-双氯化加成产物而不是1, 2-双氯化加成 (Scheme 10).此反应体系中, 在苯环上含有富电子基和贫电子基都能获得优秀的产率, 且最高可得到Z/E值大于20:1, 但是此反应不适合脂肪酮酸酯.作者也推测了其反应机理:首先三苯基氧膦与草酰氯反应生成季膦氯盐并释放二氧化碳和一氧化碳, 然后季膦氯盐与酮酸酯形成络合物并发生双键迁移, 最后得到1, 3-双氯化加成产物并释放三苯基氧膦进入下一次催化循环.
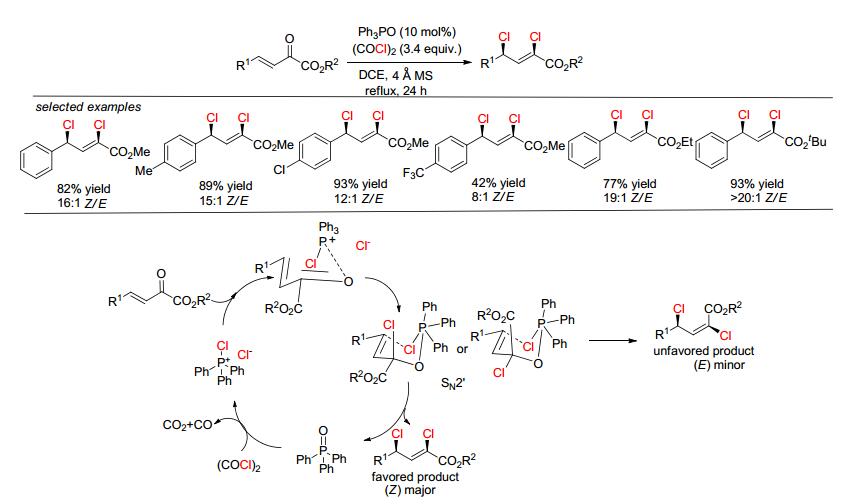 图 图式10
三苯基氧膦催化酮酸酯的1, 3-双氯化加成及可能反应机理
Figure 图式10.
Triphenylphosphine oxide-catalyzed 1, 3-dichlorination of unsaturated ketoesters and proposed mechanism
图 图式10
三苯基氧膦催化酮酸酯的1, 3-双氯化加成及可能反应机理
Figure 图式10.
Triphenylphosphine oxide-catalyzed 1, 3-dichlorination of unsaturated ketoesters and proposed mechanism
一般情况, 烯烃的双卤化加成主要生成反式产物, 顺式产物很少或者最多不超过50%产率.至今, 有关烯烃的双卤化顺式加成研究较少, 而且难度很大. 2015年, Denmark等[39]报道了首例二苯基二硒催化脂肪烯烃的顺式二氯化加成 (Eq. 12).该体系的底物普适性很广, 包括顺式烯烃、反式烯烃、丙烯醇、环烷烯烃、不饱和酯烯烃等, 产物可获得40%~91%产率和最高大于99:1 d.r.值, 最后作者也推测了反应机理 (Scheme 11).
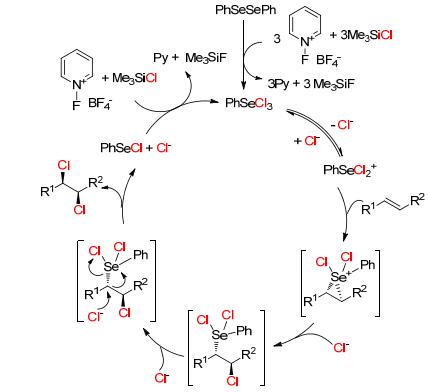 图 图式11
硒催化烯烃的顺式二氯化加成可能反应机理
Figure 图式11.
Proposed catalytic cycle for the selenium-catalysed syn-dichlorination of alkenes
图 图式11
硒催化烯烃的顺式二氯化加成可能反应机理
Figure 图式11.
Proposed catalytic cycle for the selenium-catalysed syn-dichlorination of alkenes
与烯烃的双溴化加成相比, 烯烃双氯化加成的研究集中在脂肪烯烃, 而官能化不饱和烯烃的研究很少, 且未报道d.r.值.直到2016年, 我们课题组[40]报道了α, β-不饱和酯和β, γ-不饱和α-酮酸酯等不饱和羰基烯烃的双氯化加成, 并获得了中等到优秀d.r.值 (Scheme 12).
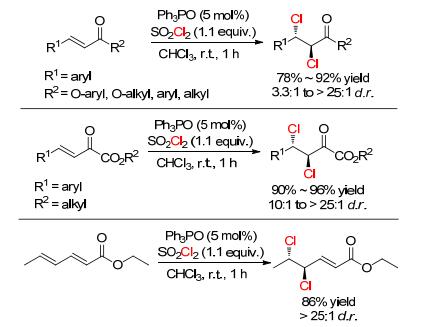 图 图式12
三苯基膦氧催化不饱和羰基烯烃的二氯化加成
Figure 图式12.
Triphenylphosphine oxide-catalyzed 1, 2-dichlorination of unsaturated carbonyl compounds
图 图式12
三苯基膦氧催化不饱和羰基烯烃的二氯化加成
Figure 图式12.
Triphenylphosphine oxide-catalyzed 1, 2-dichlorination of unsaturated carbonyl compounds
该体系用三苯基膦氧为催化剂、磺酰氯为氯化试剂、氯仿为溶剂, 室温下反应1 h可获得优秀产率的二氯化产物.非常有趣的是, 当用磺酰氯替换草酰氯时, β, γ-不饱和α-酮酸酯生成1, 2-二氯化加成产物, 而不是Xu等[38]报道的1, 3-二氯化加成产物; 而且该体系也适合二烯烃, 如山梨酸乙酯, 比文献报道的方法更简便.之后, 作者还研究了α, β-磷酸酯的双氯化加成[41], 此反应选用二苯基二硒为催化剂 (Scheme 13), 可以获得88%~92%产率和最高8:1 d.r.值, 并用单晶结构表征了产物的构型.通过一系列的控制实验, 作者提出的反应机理和Denmark提出的有一点不同, 具有催化活性中间体硒化合物是二苯基二氯化硒并得到了其单晶结构.
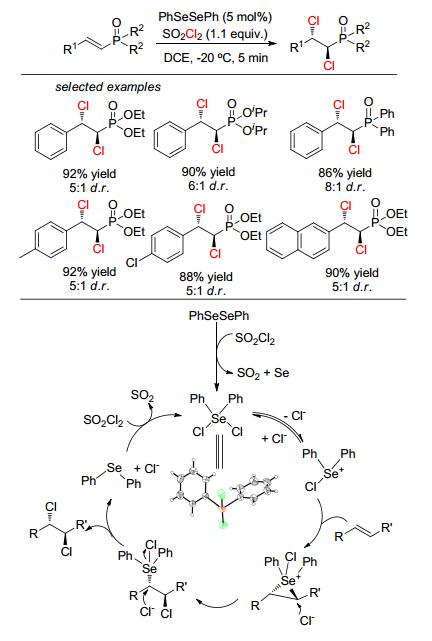 图 图式13
硒催化不饱和磷酸酯二氯化加成及反应机理
Figure 图式13.
Selenium-catalysed dichlorination of unsaturated phosphonates and proposed mechanism
图 图式13
硒催化不饱和磷酸酯二氯化加成及反应机理
Figure 图式13.
Selenium-catalysed dichlorination of unsaturated phosphonates and proposed mechanism
3 烯烃的溴氯化加成反应
虽然烯烃双溴化和双氯化加成的研究相对较多, 机理研究也成熟, 但是如果同时加成两个不同的卤原子, 仍然没有合适的方法[2].直到2015年, Burns等[42]在全合成Halomon、Plocamenone及Isoplocamenone溴氯萜类药物时报道了首例研究丙烯醇类烯烃的溴氯加成.该体系用手性烯胺醇为催化剂、NBS和ClTi (Oi-Pr)3为卤化试剂, 可以获得高达95% ee产物 (Scheme 14), 并且可用于克级规模反应.
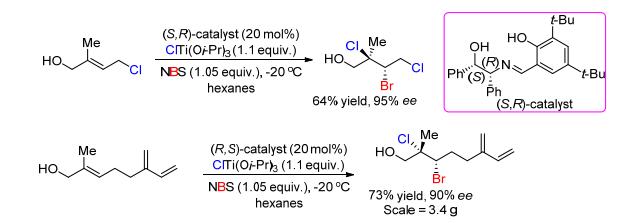 图 图式14
丙烯醇的不对称溴氯化加成
Figure 图式14.
Asymmetric bromochlorination of allylic alcohols
图 图式14
丙烯醇的不对称溴氯化加成
Figure 图式14.
Asymmetric bromochlorination of allylic alcohols
2016年, Xu等[43]对Burns的研究进行了深入探讨, 筛选了一系列手性胺醇催化剂, 将丙烯醇底物的普适性拓展到3-苯基丙烯醇.该研究在筛选催化剂时发现, Burns用的手性烯胺醇仍是此反应最佳催化剂, 而且对3-苯基丙烯醇类底物的化学、区域和立体选择性可以达到中等 (Scheme 15), 并用单晶表征了一产物的绝对构型和推测了其反应机理.
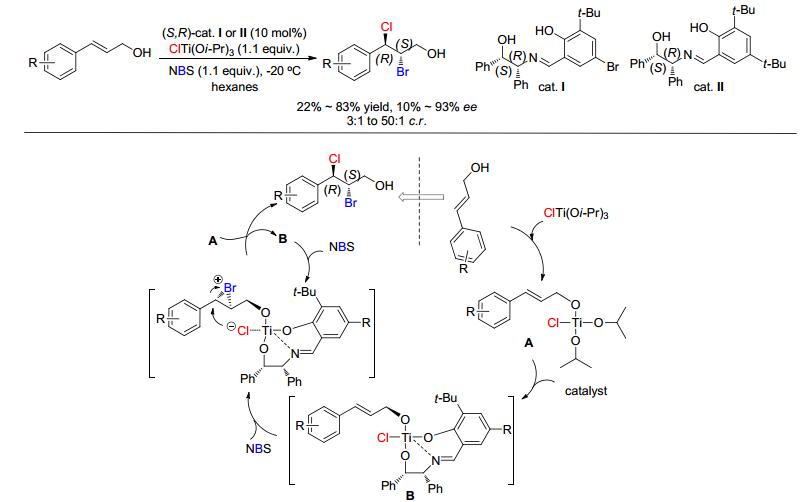 图 图式15
苯丙烯醇的不对称溴氯化加成及可能反应机理
Figure 图式15.
Asymmetric bromochlorination of aromatic allylic alcohols and proposed mechanism
图 图式15
苯丙烯醇的不对称溴氯化加成及可能反应机理
Figure 图式15.
Asymmetric bromochlorination of aromatic allylic alcohols and proposed mechanism
4 烯烃的双氟化加成反应
相比较于烯烃的双溴化和双氯化加成, 烯烃的双氟化加成是非常具有挑战性.通常烯烃的双氟化采用氧化F2或XeF2、Selectfluor、对甲基二氟碘苯/HF等氟化试剂, 然而该方法很难控制其产物的立体选择性[44].鉴于此, 有关烯烃的双氟化反应的研究很少.最近, 烯烃的双氟化加成反应有了新的突破.
2016年, Gilmour等[45]报道了对甲基碘苯催化脂肪末端烯烃的双氟化加成, 该反应选用了HF铵盐和Selectfluor为氟化试剂, 在室温下可以获得中等产率的双氟化产物, 而且烯烃的普适性广 (Scheme 16).作者推测其反应机理是:首先对甲基碘苯与氟化试剂反应生成对甲基二氟碘苯/HF中间体, 然后与烯烃反应生成高碘鎓离子, 最后氟负离子进攻高碘鎓离子开环生成双氟化产物, 同时释放对甲基碘苯进入下一个催化循环过程.
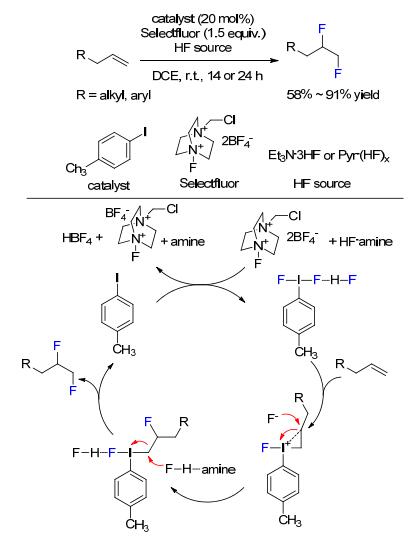 图 图式16
对甲基碘苯催化烯烃的1, 2-双氟化加成及可能反应机理
Figure 图式16.
p-Iodotoluene-catalyzed 1, 2-difluorination of olefins and proposed mechanism
图 图式16
对甲基碘苯催化烯烃的1, 2-双氟化加成及可能反应机理
Figure 图式16.
p-Iodotoluene-catalyzed 1, 2-difluorination of olefins and proposed mechanism
几乎同时, Jacobsen课题组[46]也报道了对甲酸甲酯碘苯衍生物催化脂肪烯烃、三取代烯烃和α, β-不饱和酰胺的双氟化加成, 并获得了优秀d.r.值.该体系用吡啶氢氟酸盐为氟化试剂、间氯过氧苯甲酸为氧化剂和二氯甲烷为溶剂, 室温下反应.更值得说明的是, 作者还合成了手性对甲酸甲酯碘苯衍生物催化α, β-不饱和酰胺的不对称双氟化加成 (Scheme 17), 虽然只得到51%产率, 但ee值可达91%和大于19:1 d.r.值.作者提出的反应机理和Gilmour提出的有点区别, 就是碘苯衍生物首先被间氯过氧苯甲酸氧化生成碘氧苯衍生物, 然后与氟化试剂生成二氟碘苯衍生物和水, 接着二氟碘苯衍生物与烯烃反应.然而, 非常有趣的是, 当作者在优化α, β-不饱和酰胺的不对称双氟化加成反应条件时, 发现在室温下, 得到手性1, 1-二氟-2-苯基酰胺产物 (Scheme 18)[47].之后, 作者进行了详细的研究, 并把底物范围拓展到肉桂酸酯和苯乙烯, 均可获得优秀ee值和可放大至克级规模反应.
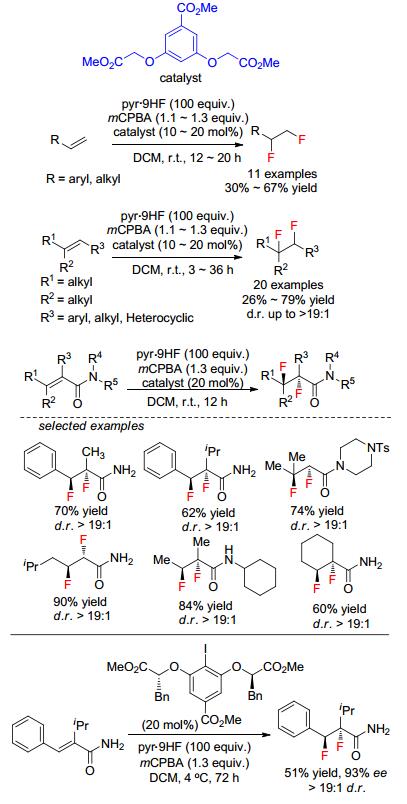 图 图式17
烯烃的1, 2-双氟化加成
Figure 图式17.
1, 2-Difluorination of alkenes
图 图式17
烯烃的1, 2-双氟化加成
Figure 图式17.
1, 2-Difluorination of alkenes
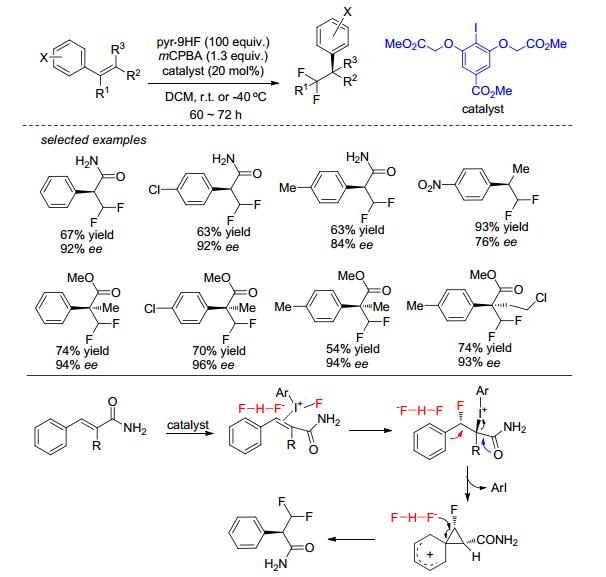 图 图式18
烯烃的不对称1, 1-双氟化反应及可能机理
Figure 图式18.
Asymmetric 1, 1-difluorination of olefins and proposed mechanism
图 图式18
烯烃的不对称1, 1-双氟化反应及可能机理
Figure 图式18.
Asymmetric 1, 1-difluorination of olefins and proposed mechanism
5 总结与展望
烯烃的双卤化反应已经被大量研究和报道, 取得了较大的进展.实现了各种缺电子烯烃及供电子烯烃作为底物的双卤化反应, 与有机小分子催化的结合已经被成功开发, 化学选择性、区域选择性问题、非对映选择性也有所研究.另外, 已出现多种不对称双卤化加成反应的反应模式, 如丙烯醇的双溴化、双氯化和溴氯化等.但是, 烯烃的双卤化反应仍存在诸多不足, 其中较有挑战的是不饱和官能化烯烃的不对称双卤化反应生成双手性中心时, 对映选择性的控制.对于烯烃的不对称双卤化反应模式还较少, 与有机小分子催化和其它金属催化的不对称模式还有待充分发展.烯烃的不对称双卤化反应也刚刚起步, 能实现的不对称双卤化的烯烃很少, 更多的官能团化烯烃, 特别是含贫电子的羰基烯烃还具有很大的挖掘价值.同时, 已报道的反应催化剂用量大, 新手性配体开发少等常见催化反应的问题, 在该类反应中也有体现, 新手性配体的开发毫无疑问将会促进新概念、新反应模式和新反应活性的出现.进一步对该类反应的研究, 将促进烯烃双卤化反应在含卤素手性药物、医药中间体和天然产物全合成等方面的应用.
-
-
[1]
selected representative examples, please see: (a) Butler A., Walker J. V. Chem. Rev., 1993, 93:1937.
(b) Gribble G. W. Acc. Chem. Res., 1998, 31:141.
(c) Gribble G. W. Chemosphere, 2003, 52:289. -
[2]
(a) Denmark S. E., Kuester W. E., Burk M. T. Angew. Chem., Int. Ed., 2012, 51:10938.
(b) Chen J., Zhou L. Synthesis, 2014, 586.
(c) Cheng Y. A., Yu W. Z., Yeung Y. Y. Org. Biomol. Chem., 2014, 12:2333.
(d) Brucks A. P., Treitler D. S., Liu S. A., Snyder S. A. Synthesis, 2013, 1886. -
[3]
Gilman H. Organic Chemistry: An Advanced Treatise, Vol. 1, Wiley, New York, 1938, pp. 36~43.
-
[4]
Cresswell A. J., Eey S. T. C., Denmark S. E. Angew. Chem., Int. Ed., 2015, 54:15642. doi: 10.1002/anie.201507152
-
[5]
Ryu I., Matsubara H., Yasuda S., Nakamura H., Curran D. P. J. Am. Chem. Soc., 2002, 124:12946. doi: 10.1021/ja027965y
-
[6]
Kavala V., Naik S., Patel B. K. J. Org. Chem., 2005, 70:4267. doi: 10.1021/jo050059u
-
[7]
Nakamatsu S., Toyota S., Jones W., Toda F. Chem. Commun., 2005, 41:3808.
-
[8]
Koshy E. P., Zacharias J., Pillai V. N. R. React. Funct. Polym., 2006, 66:845. doi: 10.1016/j.reactfunctpolym.2005.11.012
-
[9]
Primerano P., Cordaro M., Scala A. Tetrahedron Lett., 2013, 54:4061. doi: 10.1016/j.tetlet.2013.05.097
-
[10]
Kumar A., Jamir L., Sinha U. B. Chem. Sci. Trans., 2014, 3:480.
-
[11]
Shao L.-X., Shi M. Synlett, 2006, 1269.
-
[12]
Zheng Y.-F., Yu J., Yan G.-B., Li X., Luo S. Chin. Chem. Lett., 2011, 22:1195.
-
[13]
Podgorsek A., Eissen M., Fleckenstein J., Stavber S., Zupan M., Iskra J. Green Chem., 2009, 11:120. doi: 10.1039/B814989E
-
[14]
Karki K., Magolan J. J. Org. Chem., 2015, 80:3701. doi: 10.1021/acs.joc.5b00211
-
[15]
Song S., Li X., Sun X., Yuan Y., Jiao N. Green Chem., 2015, 17:3285. doi: 10.1039/C5GC00528K
-
[16]
Macharla A. K., Nappunni R. C., Nama N. Tetrahedron Lett., 2012, 53:1401. doi: 10.1016/j.tetlet.2012.01.026
-
[17]
Wang G.-W., Gao J. Green Chem., 2012, 14:1125. doi: 10.1039/c2gc16606b
-
[18]
Das P. J., Sarkar S. Indian J. Chem., 2013, 52B, 802.
-
[19]
Wang Y., Wang J., Xiong Y., Liu Z.-Q. Tetrahedron Lett., 2014, 55:2734. doi: 10.1016/j.tetlet.2014.03.064
-
[20]
Zhu M., Lin S., Zhao G.-L., Sun J., Córdova A. Tetrahedron Lett., 2010, 51:2708. doi: 10.1016/j.tetlet.2010.03.043
-
[21]
Xue H., Tan H., Wei D., Wei Y., Lin S., Liang F., Zhao B. Org. Biomol. Chem., 2013, 11:5382.
-
[22]
Stodulski M., Goetzinger A., Kohlhepp S. V., Gulder T. Chem. Commun., 2014, 50:3435. doi: 10.1039/c3cc49850f
-
[23]
Hernández-Torres G., Tan B., Barbas Ⅲ C. F. Org. Lett., 2012, 14:1858. doi: 10.1021/ol300456x
-
[24]
Hu D. X., Shibuya G. M., Burns N. Z. J. Am. Chem. Soc., 2013, 135:12960. doi: 10.1021/ja4083182
-
[25]
Landry M. L., Hu D. X., Shibuya G. M., Burns N. Z. J. Am. Chem. Soc., 2016, 138:5150. doi: 10.1021/jacs.6b01643
-
[26]
Yu T.-Y., Wang Y., Hu X.-Q., Xu P.-F. Chem. Commun., 2014, 50:7817. doi: 10.1039/c4cc02847c
-
[27]
Yu T.-Y., Wei H., Luo Y.-C., Wang Y., Wang Z.-Y., Xu P.-F. J. Org. Chem., 2016, 81:2730. doi: 10.1021/acs.joc.5b02618
-
[28]
Iskra J., Stavber S., Zupan M. Chem. Commun., 2003, 39:2496.
-
[29]
Snyder S. A., Tang Z.-Y., Gupta R. J. Am. Chem. Soc., 2009, 131:5744. doi: 10.1021/ja9014716
-
[30]
Poutsma M. L. Science, 1967, 157:997. doi: 10.1126/science.157.3792.997
-
[31]
Liu X., Wang L., Zou J. Chin. J. Chem., 2011, 29:2097. doi: 10.1002/cjoc.v29.10
-
[32]
Kitamura K., Tazawa Y., Morshed M. H., Kobayashi S. Synthesis, 2012, 44:1159. doi: 10.1055/s-0031-1290578
-
[33]
Ren J., Tong R. Org. Biomol. Chem., 2013, 11:4312. doi: 10.1039/c3ob40670a
-
[34]
Swamy P., Reddy M. M., Kumar M. A., Naresh M., Narender N. Synthesis, 2014, 46:251.
-
[35]
Kamada Y., Kitamura Y., Tanaka T., Yoshimitsu T. Org. Biomol. Chem., 2013, 11:1598. doi: 10.1039/c3ob27345h
-
[36]
Egami H., Yoneda T., Uku M., Ide T., Kawato Y., Hamashima Y. J. Org. Chem., 2016, 81:4020. doi: 10.1021/acs.joc.6b00295
-
[37]
Nicolaou K. C., Simmons N. L., Ying Y., Heretsch P. M., Chen J. S. J. Am. Chem. Soc., 2011, 133:8134. doi: 10.1021/ja202555m
-
[38]
Yu T.-Y., Wang Y., Xu P.-F. Chem. Eur. J., 2014, 20:98. doi: 10.1002/chem.201303688
-
[39]
Cresswell A. J., Eey S. T.-C.; Denmark S. E. Nat. Chem., 2015, 7:146. doi: 10.1038/nchem.2141
-
[40]
Zeng X., Gong C., Zhang J., Xie J. RSC Adv., 2016, 6:85182. doi: 10.1039/C6RA20101F
-
[41]
Zeng X., Gong C., Zhang J., Xie J. New J. Chem., 2016, 40:7866. doi: 10.1039/C6NJ00658B
-
[42]
Bucher C., Deans R. M., Burns N. Z. J. Am. Chem. Soc., 2015, 137:12784. doi: 10.1021/jacs.5b08398
-
[43]
Huang W.-S., Chen L., Zheng Z.-J., Yang K.-F., Xu Z., Cui Y.-M., Xu L.-W. Org. Biomol. Chem., 2016, 14:7927. doi: 10.1039/C6OB01306F
-
[44]
For an indirect method, see: Olah G. A., Welch J. T., Vankar Y. D., Nojima M., Kerekes I., Olah J. A. J. Org. Chem., 1979, 44:3872. doi: 10.1021/jo01336a027
-
[45]
Molnár I. G., Gilmour R. J. Am. Chem. Soc., 2016, 138:5004. doi: 10.1021/jacs.6b01183
-
[46]
Banik S. M., Medley J. W., Jacobsen E. N. J. Am. Chem. Soc., 2016, 138:5000. doi: 10.1021/jacs.6b02391
-
[47]
Banik S. M., Medley J. W., Jacobsen E. N. Science, 2016, 353:51. doi: 10.1126/science.aaf8078
-
[1]
-

图式2 亚硝酸钠催化烯烃的双溴化加成及可能的反应机理
Scheme 2 Sodium nitrite-catalyzed dibromination of alkenes and proposed mechanism

图式3 DMSO/HBr体系促进的烯烃双溴化加成及可能反应机理
Scheme 3 Dibromination of olefins with HBr/DMSO system and proposed mechanism

图式4 苯甲酸催化α, β-不饱和羰基化合物的双溴化加成
Scheme 4 Benzoic acid-catalyzed dibromination of α, β-unsaturated carbonyl compounds

图式6 丙烯醇不对称双溴化的可能反应机理
Scheme 6 Proposed mechanism of asymmetric dibromination of allylic alcohols

图式7 三苯基氧膦催化不饱和羰基化合物双溴化加成及反应机理
Scheme 7 Triphenylphosphine oxide-catalyzed dibromination of unsaturated compounds and proposed mechanism

图式8 一锅法合成双溴化和四溴化化合物
Scheme 8 One-pot method for the synthesis of dibromination and tetrabromination compounds

图式9 不对称称双氯化诱导模型
Scheme 9 Proposed stereoinduction model for enantioselective dichlorination

图式10 三苯基氧膦催化酮酸酯的1, 3-双氯化加成及可能反应机理
Scheme 10 Triphenylphosphine oxide-catalyzed 1, 3-dichlorination of unsaturated ketoesters and proposed mechanism

图式11 硒催化烯烃的顺式二氯化加成可能反应机理
Scheme 11 Proposed catalytic cycle for the selenium-catalysed syn-dichlorination of alkenes

图式12 三苯基膦氧催化不饱和羰基烯烃的二氯化加成
Scheme 12 Triphenylphosphine oxide-catalyzed 1, 2-dichlorination of unsaturated carbonyl compounds

图式13 硒催化不饱和磷酸酯二氯化加成及反应机理
Scheme 13 Selenium-catalysed dichlorination of unsaturated phosphonates and proposed mechanism

图式15 苯丙烯醇的不对称溴氯化加成及可能反应机理
Scheme 15 Asymmetric bromochlorination of aromatic allylic alcohols and proposed mechanism

图式16 对甲基碘苯催化烯烃的1, 2-双氟化加成及可能反应机理
Scheme 16 p-Iodotoluene-catalyzed 1, 2-difluorination of olefins and proposed mechanism
-


 下载:
下载:

















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 30
- 文章访问数: 3439
- HTML全文浏览量: 857
 下载:
下载:
