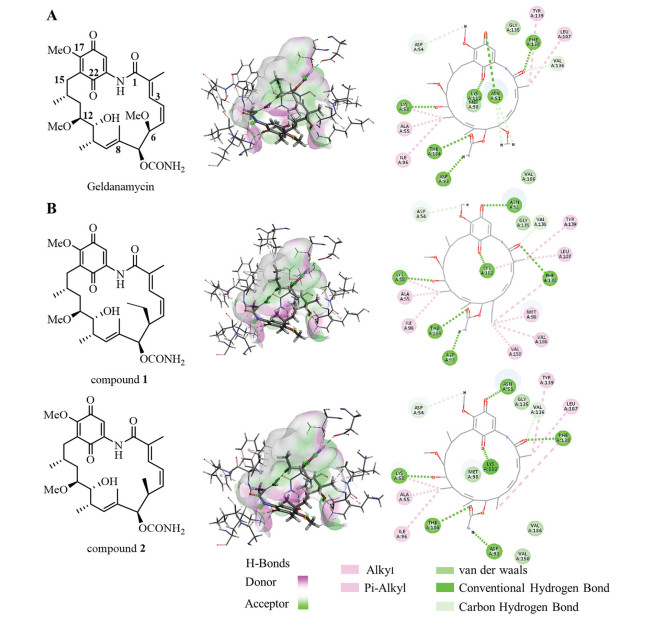
Figure 1.
Cocrystal structures of GA and C6-Alkyl variants. Structures, binding patterns and two-dimensional interaction modes of GA (A), compounds 1 and 2 (B) with Hsp90 (PDB 1YET).
Rational design, synthesis, and biological evaluation of novel C6-modified geldanamycin derivatives as potent Hsp90 inhibitors and anti-tumor agents
Ruxuan Wang , Rentao Zhang , Haoran Yang , Nina Xue , Xiaoguang Chen , Xiaoming Yu

Heat shock protein 90 (Hsp90) is a molecular chaperon first identified in the 1960s [1]. Overexpression of Hsp90 is commonly observed in cancer cells, and many of its client proteins are known to be oncogenic, e.g., Her-2, HIF-1α, VEGFR, p53 [2, 3]. Neckers and coworkers [4-6] revealed in 1995 that benzoquinone ansamycin family natural products inhibit chaperon activity of Hsp90 by binding to its ATP-binding site located within its N-terminal domain, triggering the degradation of several down-stream oncogenic proteins and apoptosis of cancer cells. This finding, and other contemporary studies as well, provoked tremendous efforts in search of clinically useful Hsp90 inhibitors for the treatment of cancer, and early progress in this campaign appeared encouraging [7]. The number of investigational small molecular Hsp90 inhibitors raised quickly in a short period, among which nearly a dozen received clinical trials. However, the most legendary cases are definitely those related to geldanamycin (GA), a representative member of benzoquinone ansamycin, as three semisynthetic analogous of GA, namely 17-AAG, 17-DMAG and IPI-504, had advanced into phase Ⅲ clinical trials [8]. Nevertheless, these agents failed to exhibit clinical benefits, largely due to adversary effects such as hepato- and pulmonary toxicity that were not predicted in their early studies but believed to be associated with the quinone moiety embedded in the molecules [9]. Therefore, we started an attempt aimed at quinone-free analogous with elevated or at least retained Hsp90 affinity and anticancer activity.
On realizing that semisynthetically amenable sites within the molecules of benzoquinone ansamycins are not only very limited themselves but also have been exhausted in early studies [10], we decided to take an unprecedented approach that would combine the techniques of total synthesis and rational drug design. In order to pursue a better planned exploration, we first proved in a preliminary study that quinone-free analogues of benzoquinone ansamycins were capable of exhibiting anticancer effects [11, 12], and in a following study we addressed a challenging synthetic problem concerning the C8-modification of the natural products [13]. Recently, we reported a total synthesis of GA [14] that was expected to be highly compatible to the philosophy of rational drug design. Herein we report the result from our first attempt of rational design directed total synthetic SAR study of GA.
Docking analysis was performed to illustrate the interactions between Hsp90 (PDB 1YET) and GA. As shown in Fig. 1A, when binding to the target, the compound adopted a compressed C-shape conformation. The benzoquinone part served as the top of the "C", while the ansa ring formed the stem and bottom of "C". Although there is extensive surface complementarity between the compact GA structure and the pocket, there remains a buried cavity filled with three water molecules near the C6‑methoxy and the carbamate groups [15]. It is conceivable that replacing the methoxy at the C6 position with 4–6 non-hydrogen bonding atoms may enhance the spatial complementarity between the ligand and the target. Besides, from the electrical distribution of the binding cavity, the pocket becomes increasingly hydrophobic toward the bottom, mainly covering the C5-C10 region of the ansa ring. Therefore, the substitution of C6-OMe to a hydrophobic group may enhance the interaction with the amino acid residues inside the binding cavity, thus enhancing the hydrophobic interaction of GA with Hsp90.
Based on the rational indicated by the cocrystal structure of GA and Hsp90, we designed C6-ethyl and C6-methyl variants of GA in order to confirm the speculations. To verify the binding mode of these compounds, we used Discovery Studio (version 2018) to simulate the virtual docking between compounds 1, 2 and Hsp90. Due to the structural similarity between compounds 1 and 2 and the inhibitor co-crystalized, the 3D structure of Hsp90 was selected and obtained from the Protein Data Bank with the PDB code 1YET for docking study. The protein was prepared by removing water and adding hydrogen atoms. The structures of compounds 1 and 2 were generated and molecular docking was performed with the Discovery Studio 4.5 software package (Accelrys, San Diego, USA). The docking calculation was carried out with the CDOCKER protocol. In the process of CDOCKER docking, the pose cluster radius was set to "0.5". Other parameters were used as default. CDOCKER interaction energy values of the target compounds were shown in Table 1.
The synthesis of compounds 1 and 2 was achieved by the synthetic route slightly modified from our earlier results [13, 14, 16] and was shown in Scheme 1. Detailed experimental procedures and compound characterization data can be found in Supporting information. C5-C12 fragment 4 was prepared in 16 steps for 4a and 15 steps for 4b from crotyl alcohol, and C13-C21 fragment 3 in 15 steps from vanillain [17].
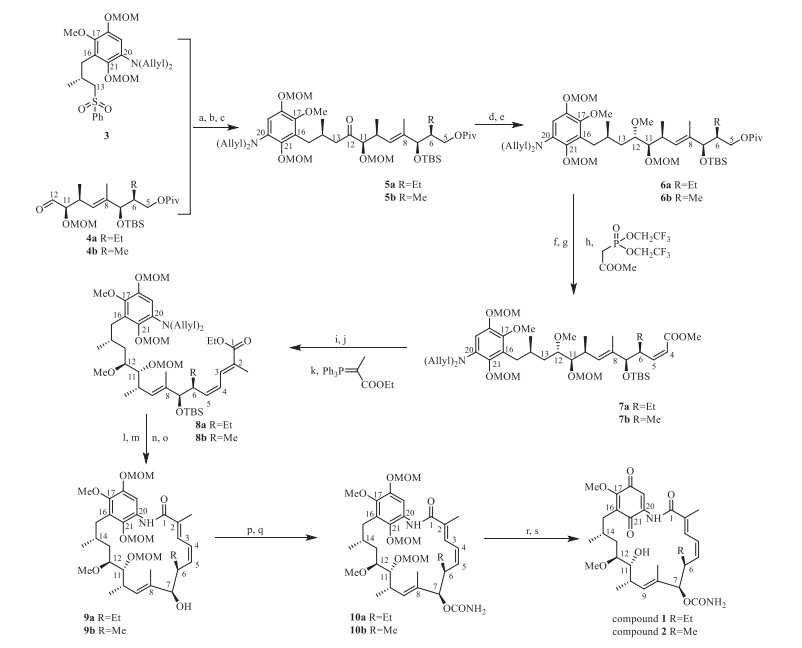
Sulfonyl compound 3, the C13-C21 fragment of the target molecules, was deprotonated at C13 using n‑butyl lithium and reacted with aldehyde 4 to furnish the coupling of the two major building blocks. The resulting β‑hydroxy sulfone was oxidized using Dess-Martin oxidant to give the corresponding α-sulfonyl ketone and then reduced with SmI2 to remove the C13-sulfonyl group to give compound 5 in an overall yield of 80% for 5a and 80% for 5b. Chelation controlled reduction of 5 with zinc borohydride helped secure the anti-stereochemistry between C11 and C12, and newly formed C12-OH was methylated to give 6a and 6b in 66% and 75% yield over 2 steps.
Reductive removal of the pivalyl group in 6 and the following Dess-Martin oxidation of the C5-OH led to an aldehyde that were further subjected to a Z-selective HWE reaction with methyl 2-(bis(2, 2, 2-trifluoroethoxy) phosphoryl) acetate, giving compound 7a in 93% yield and 7b in 86% yield, respectively. The ester group in 7 was reduced to hydroxyl and then oxidized to aldehyde, again using a Dess-Martin oxidant. Wittig reaction of this intermediate gave compound 8 in 75% yield for 8a and 70% for 8b over 3 steps [18].
A 4-step sequence comprising 1) pyridinium hydrofluoride affected TBS removal; 2) Pd catalyzed N-deallylation; 3) basic hydrolysis of the ethyl ester and 4) BOPCl promoted macrocyclization delivered compound 9 with a fully established skeleton. Then C7 carbamoyl group was introduced as previously described to give 10. Total MOM removal was then furnished by using TMSBr, and the resultant hydroquinone was oxidized to the target molecules with air under the catalysis of Pd/C [19, 20].
Compounds 1 and 2 were submitted to a thorough characterization using 1H and 13C NMR, MS and IR. The spectra met well with the designed structures. More importantly, we noticed that compound 2 is a natural product isolated by Ni et al. [21]. A comparison of the spectra of 2 and that of the reported data indicated that the two compounds are identical.
Subsequently, fluorescence polarization assays were performed to determined binding affinity of these compounds towards Hsp90α protein according to our previous described [22], and results were summarized in Fig. 2. GA and NVP-AUY922 were tested in parallel as the control compounds. Likely with GA, compounds 1 and 2 dose-dependently competed with the probes VER00051001 binding toward Hsp90 N-terminal domain. Compound 1 displayed a rather potent Hsp90 binding activity with the IC50 value of 34.26 nmol/L, which is comparable to the reported Hsp90 inhibitor GA (IC50, 10.33 nmol/L). And, the IC50 value of compound 2 is 5-fold more than compound 1. It is assumed that more than one non-hydrogen atoms are essential for Hsp90α binding. When the C6-ethyl was replaced of C6‑methoxy, the Hsp90α binding affinity activity remained at the same level. However, methyl displacement decreased the binding activity.
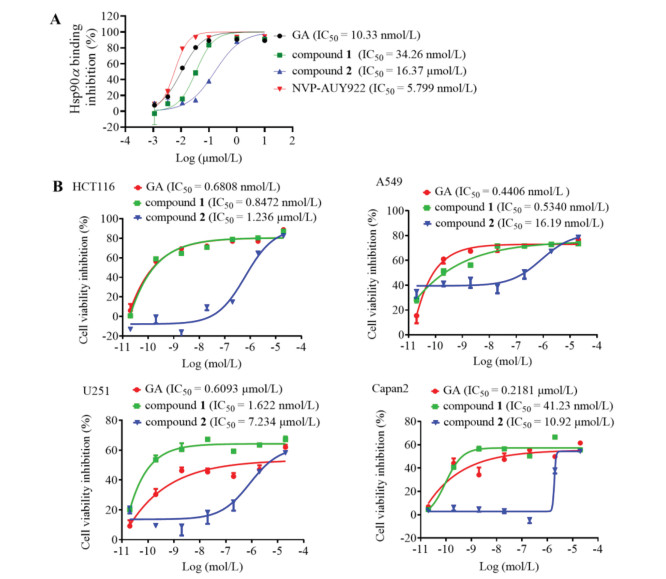
In addition, an MTT assay was performed to detect the anti-proliferation activity of GA derivatives in A549, HCT116, Capan2 and U251 cell lines. As shown in Fig. 2B, like GA, these compounds exhibited potential and widespread anti-tumor effects. Compound 1 showed more potent growth inhibition than positive control GA in human glioblastoma U251 cell and human pancreatic cancer Capan2 cell. SAR investigation suggested that in replacing the ethyl with methoxy, compound 1 with higher lipophilicity and displayed enhanced cytotoxic activity with the IC50 value of 1.622 nmol/L and 41.23 nmol/L in U251 and Capan2 cells. The replacement of methyl with methoxy, showed a decreased activity with an IC50 value of 7.234 µmol/L and 10.92 µmol/L.
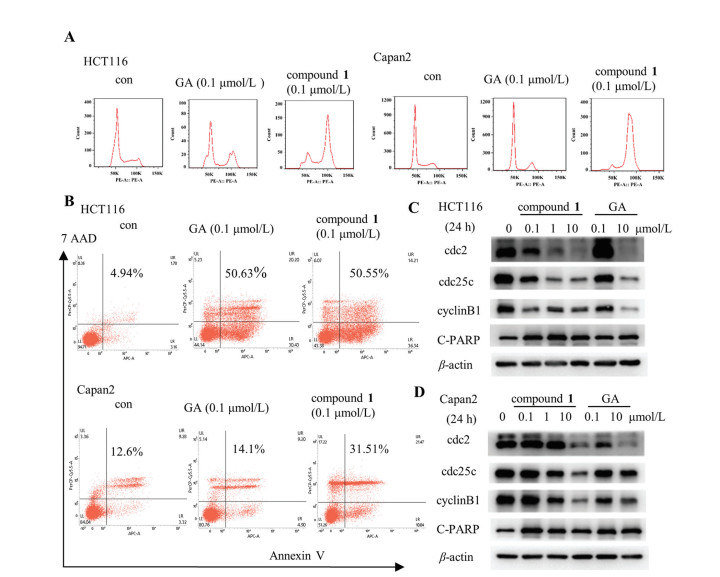
Flow cytometric analysis was performed to determine the cell cycle arrest and apoptosis after the structural modifications. As shown in Fig. 3, 0.1 µmol/L of compound 1 significantly increased the cell distribution of G2/M phase, the percentage of cells in G2/M phase was increased from 19.89% to 87.22% in Capan2 cells and from 20.16% to 57.89% in HCT116 cells. Meanwhile, compound 1 (0.1 µmol/L) dramatically increased the proportion of cell apoptosis, which were increased to 31.51% and 50.55% in Capan2 and HCT116 cells (Figs. 3A and B). The induction of G2/M phase arrest and apoptosis after treatment with compound 1 are more potential than those of equal concentration of GA. Consistently, the expressions of G2/M checkpoints including cdc25c, cdc2 and cyclinB were notably decreased by compound 1 in a dose-dependent manner. Cleaved-PARP, as a key marker of apoptosis, was markedly increased after exposure to compound 1 (Figs. 3C and D, Fig. S2 in Supporting information). These results indicated that compound 1 exhibited a similar mechanism of action to those of control compound GA.
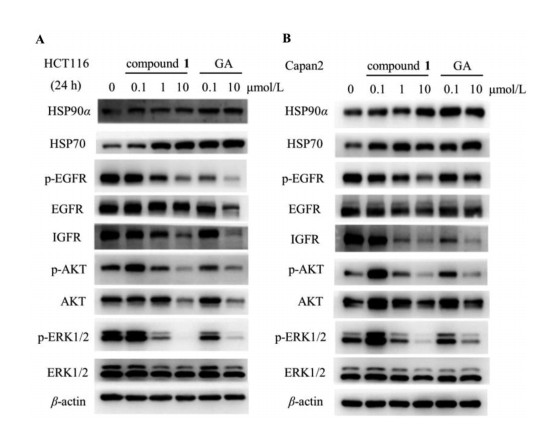
We also investigated the influence of compound 1 on EGFR and IGFR protein expressions, which were commonly clients of Hsp90 chaperon, and their downstream signaling pathways were crucial for proliferation and aggravation of cancer. As shown in Fig. 4 and Fig. S2, compound 1 significantly reduced the levels of EGFR/phosphorylated EGFR and IGF-1Rβ and suppressed downstream AKT and ERK1/2 signaling. Additionally, the reduction of Hsp90 client proteins was concurrent with an increase of Hsp70 and Hsp90, established biomarkers of Hsp90 N-terminal inhibition. Taken together, likely GA, compound 1 targets Hsp90 N-terminal domain and inhibits multiple oncoproteins expressions.
Based on a previously identified Hsp90 inhibitor GA, two C6-alkyl derivatives were rationally designed, synthesized and evaluated. During the synthesis, it was unexpectedly discovered that compound 2 has been reported in the literature as its natural source, which is a branched product of the GA biosynthesis process [21]. The NMR data of the final compound 2 obtained by total synthesis were compared with those reported in the literature, and the structure was verified to be correct, relying on which the structure of compound 1 was also confirmed.
The bioassay results fit well to that predicted by computational analysis, indicating that Discovery Studio is a powerful and reliable tool for molecular design. The obvious decrease of bioactivity of compound 2 implied that a shortened branch at C6 is detrimental, while more lipophilic substituent at this site may help increase the compound's binding affinity.
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests.
This work was supported by the CAMS Innovation Fund for Medical Sciences (Nos. CIFMS 2016-I2M-3–009 and CIFMS 2017- I2M-3–011) and the National Natural Science Foundation of China (No. 21272279).
Supplementary material associated with this article can be found, in the online version, at doi:
F. Ritossa, Cell Stress Chaperones 1 (1996) 97–98. doi: 10.1379/1466-1268(1996)001<0097:DOTHSR>2.3.CO;2
M.Y. Xu, N.N. Xue, D. Liu, et al., Chinese Chem. Lett. 27 (2016) 11–15. doi: 10.1016/j.cclet.2015.09.024
F.H. Schopf, M.M. Biebl, J. Buchner, Nat. Rev. Mol. Cell Biol. 18 (2017) 345–360. doi: 10.1038/nrm.2017.20
D.F. Smith, L. Whitesell, S.C. Nair, et al., Mol. Cell. Biol. 15 (1995) 6804–6812. doi: 10.1128/MCB.15.12.6804
L. Whitesell, E.G. Mimnaugh, B. De Costa, C.E. Myers, L.M. Neckers, Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 8324–8328. doi: 10.1073/pnas.91.18.8324
H.J. Ochel, T.W. Schulte, P. Nguyen, J. Trepel, L. Neckers, Mol. Genet. Metab. 66 (1999) 24–30. doi: 10.1006/mgme.1998.2774
J. Trepel, M. Mollapour, G. Giaccone, L. Neckers, Nat. Rev. Cancer 10 (2010) 537–549. doi: 10.1038/nrc2887
Y. Fukuyo, C.R. Hunt, N. Horikoshi, Cancer Lett. 290 (2010) 24–35. doi: 10.1016/j.canlet.2009.07.010
G. Tudor, P. Gutierrez, A. Aguilera-Gutierrez, E.A. Sausville, Biochem. Pharmacol. 65 (2003) 1061–1075. doi: 10.1016/S0006-2952(03)00013-3
J. Franke, S. Eichner, C. Zeilinger, A. Kirschning, Nat. Prod. Rep. 30 (2013) 1299–1323. doi: 10.1039/c3np70012g
C. Bian, R. Yan, X. Yu, Tetrahedron 70 (2014) 2982–2991. doi: 10.1016/j.tet.2014.03.020
Z. Zhang, N. Xue, C. Bian, et al., Bioorganic Med. Chem. Lett. 26 (2016) 4287–4291. doi: 10.1016/j.bmcl.2016.07.040
L. Jin, R. Zhang, Z. Zhang, C. Bian, X. Yu, Tetrahedron Lett. 60 (2019) 547–551. doi: 10.1016/j.tetlet.2019.01.024
Z. Zhang, Y. Li, R. Zhang, X. Yu, J. Org. Chem. 86 (2021) 15063–15075. doi: 10.1021/acs.joc.1c01582
C.E. Stebbins, A.A. Russo, C. Schneider, et al., Cell 89 (1997) 239–250. doi: 10.1016/S0092-8674(00)80203-2
R. Yan, C. Bian, X. Yu, Org. Lett. 16 (2014) 3280–3283. doi: 10.1021/ol5012899
M.B. Andrus, E.J. Hicken, E.L. Meredith, B.L. Simmons, J.F. Cannon, Org. Lett. 5 (2003) 3859–3862. doi: 10.1021/ol035400g
D.A. Evans, S.J. Miller, M.D. Ennis, P.L. Ornstein, J. Org. Chem. 57 (1992) 1067–1069. doi: 10.1021/jo00030a006
T. Hosoya, E. Takashiro, T. Matsumoto, K. Suzuki, J. Am. Chem. Soc. 116 (1994) 1004–1015. doi: 10.1021/ja00082a023
R. Baker, J.L. Castro, J. Chem. Soc. Perkin Trans. 1 (1990) 47–65.
S. Ni, B. Jiang, L. Wu, et al., J. Antibiot. (Tokyo) 67 (2014) 183–185. doi: 10.1038/ja.2013.94
N. Xue, J. Jin, D. Liu, et al., Curr. Cancer Drug Targets 14 (2014) 671–683. doi: 10.2174/1568009614666140908101523

Figure 1 Cocrystal structures of GA and C6-Alkyl variants. Structures, binding patterns and two-dimensional interaction modes of GA (A), compounds 1 and 2 (B) with Hsp90 (PDB 1YET).

Scheme 1 Synthesis of C6-alkyl variants of GA. Reagents and conditions: (a) n-BuLi, THF, −78 ℃, 30 min; (b) Dess-Martin, NaHCO3, DCM, 0 ℃ to r.t., 1 h; (c) SmI2, MeOH/THF (3:2, v/v), −78 ℃, 15 min; (d) Zn(BH4)2, cyclohexene, 2-methoxyproene, Et2O, 0 ℃ to r.t., 3 h; (e) Me3OBF4, proton sponge, DCM, 0 ℃ to r.t., 1 h; (f) DIBAL-H, DCM, −78 ℃, 30 min; (g) Dess-Martin, NaHCO3, DCM, 0 ℃ to r.t., 2 h; (h) methyl 2-(bis(2, 2, 2-trifluoroethoxy)phosphoryl)acetate, 18-crown-6, KHMDS, THF, −78 ℃, 3 h; (i) DIBAL-H, DCM, −78 ℃, 1 h; (j) Dess-Martin, DCM, 0 ℃, 2 h; (k) ethyl 2-(triphenyl-l5-phosphaneylidene)propanoate, toluene, reflex, 1 h; (l) pyridine/HF-pyridine/MeCN (1:1:2.5, v/v/v), acetonitrile, r.t., 4 h; (m) Pd(PPh3)4, N, N-dimethylbarbituric acid, DCM, reflux, 4 h; (n) LiOH, MeOH/THF/H2O (2:2:1, v/v/v), r.t., 12 h; (o) BOPCl, DIPEA, toluene, 85 ℃, overnight; (p) Cl3CCONCO, DCM, r.t., 20 min; (q) K2CO3, MeOH, r.t., 2.h; (r) TMSBr, DCM, −30 ℃, 3 h; (s) Pd/C, air, EtOAc, r.t., 30 min.

Figure 2 Hsp90 binding affinity and cell viability of synthesized GA derivatives. (A) The competitive binding affinity of compounds on the N-terminal domain of Hsp90α. Different concentrations of compounds 1 and 2 or positive control Hsp90 inhibitors GA and NVP-AUY922 were incubated with VER00051001 and Hsp90α for 48 h and subjected to fluorescence polarization (FP) assay. (B) The antiproliferative inhibition after exposure of compounds 1 and 2 for 72 h was detected by MTT assay.

Figure 3 The effect of compound 1 on cell cycle arrest and apoptosis in human cancer cells. (A) The cell cycle distribution was examined using flow cytometry assay. (B) The proportion of cell apoptosis was measured by flow cytometry assay. (C, D) The protein expressions of G2/M checkpoints and cleaved-PARP were detected by Western blot assay.

Figure 4 The effect of compound 1 on (p)-EGFR, IGFR and downstream signaling molecules in HCT116 (A) and Capan2 (B) cells by Western blot assay.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
 下载:
下载: