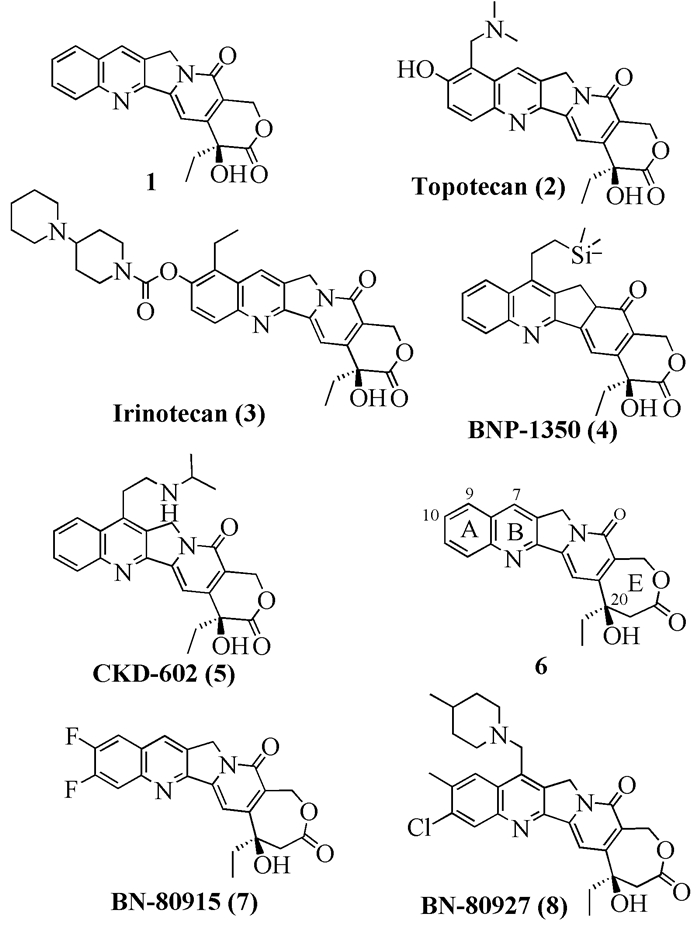
图式 1.
具有生物活性的喜树碱和高喜树碱类化合物
Scheme 1.
Bioactive compounds of different camptothecin and homocamptothecin

喜树碱(Camptothecin,CPT,1)是一种具有显著细胞毒活性的喹啉类生物碱[1],是一个五环的稠环体系。它的(20S)-绝对构型化合物最早报道于1966年,是美国科学家Wall从喜树(Camptotheca acuminata)的树皮中提取出来的。经实验证明,它对骨癌、肝癌、膀胱癌和白血病等多种恶性肿瘤均表现出良好的抑制作用,尤其是对大鼠的白血病细胞(leukemia L1210)有很好的致死性,有显著的抗肿瘤活性[2]。其作用机理主要表现在能有效阻止拓扑异构酶I与DNA直接的相互结合,从而阻碍了病毒细胞的DNA复制而致其凋亡[3, 4]。目前,基于喜树碱的临床诊断技术也在不断研究和发展中[5, 6]。
尽管如此,喜树碱水溶性差、生物利用度低的缺点影响了其应用范围[7]。因此,一些研究机构在喜树碱改造上做了很多工作,已有几个药物(结构式见图式 1)获得美国食品药品监督管理局(FDA)批准上市或处于临床研究阶段,如拓扑替康(Topotecan,2)、伊立替康(Irinotecan,3)[8]和BNP-1350 (4)[9],而倍罗替康(Belotecan,CKD-602,5)亦于2005年在韩国被批准上市。
1
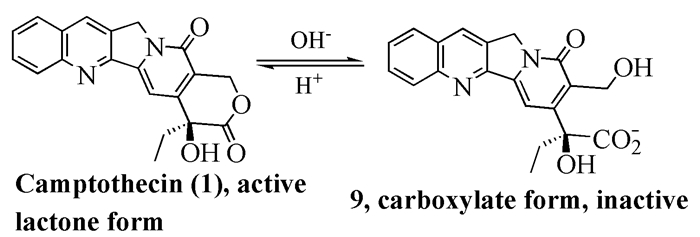
另外,喜树碱还有一个缺陷[10]:在人体组织内喜树碱的内酯很容易开环。在pH=7.4时,喜树碱在1h内迅速水解并达到平衡,内酯结构的浓度仅有20%;当pH=9.1时,水解几乎是立即完成的[11]。开环的产物9(图式 2)是没有任何活性的,并且毒副作用很大。因此,提高内酯环的稳定性,将会在很大程度上提高药物的活性,从而减少药物的使用量,降低药物的血液毒性。
1相比之下,据Bigg小组[4, 12]报道,高喜树碱在pH 7.4的环境下,1h后测定,还保留有84%的内酯结构[11]。喜树碱和高喜树碱处于相同的低浓度时,后者对Ⅰ型拓扑异构酶的抑制能力明显好于前者[13],且其溶解度也稍高。目前,高喜树碱候选药物BN-80915(7)和BN-80927 (8)已处于临床阶段[4, 14]。
基于上述利好因素,研究人员致力于合成高喜树碱化合物,并展开了相关活性研究。所报道的合成方法主要有两种:一种是从喜树碱或10-羟基喜树碱出发,通过半合成方法得到消旋的高喜树碱;另一种是用全合成的方法得到消旋的或(20R)-构型的高喜树碱。
Bigg小组最早通过半合成方法得到了高喜树碱(图式 3)[11]。他们比较了高喜树碱与喜树碱的水解速度[15],在对Topoisomerase I抑制作用上,高喜树碱BN-80245(rac-6)、喜树碱和拓扑替康,处于同一个数量级;而对小鼠白血病癌细胞的活性测试中,BN-80245要高于对照品1个数量级。
1张万年等[8](图式 4)以及李尚丰等[16]用相似的方法,也在开展高喜树碱的合成研究工作。前者将10-羟基喜树碱用亚磷酸三丁酯保护,接着用KBH4还原成半缩醛,再经NaIO4氧化开环以及与溴乙酸叔丁酯进行Reformatsky反应得到化合物17,再用三氟乙酸处理,得到扩环后的高喜树碱磷酸酯18,最后在7位上引入甲基或乙基,得到化合物19a和19b。在此基础上经过拓展,合成了6个磷酸酯类化合物20。这些磷酸酯分别与肿瘤细胞A549、MCF-7和LOVO进行体外测试,其中19a和20c的对3种肿瘤细胞的抑制活性均与对照物伊立替康处在同一个数量级,且略高于对照物。
1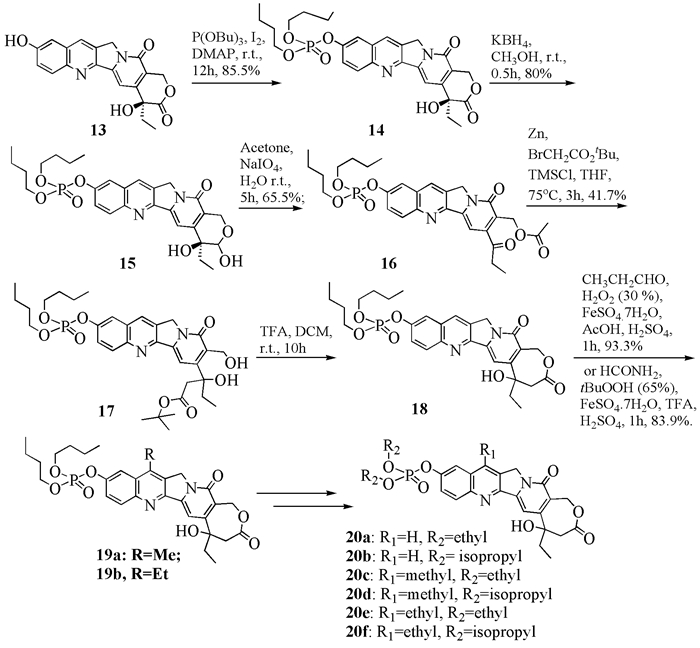
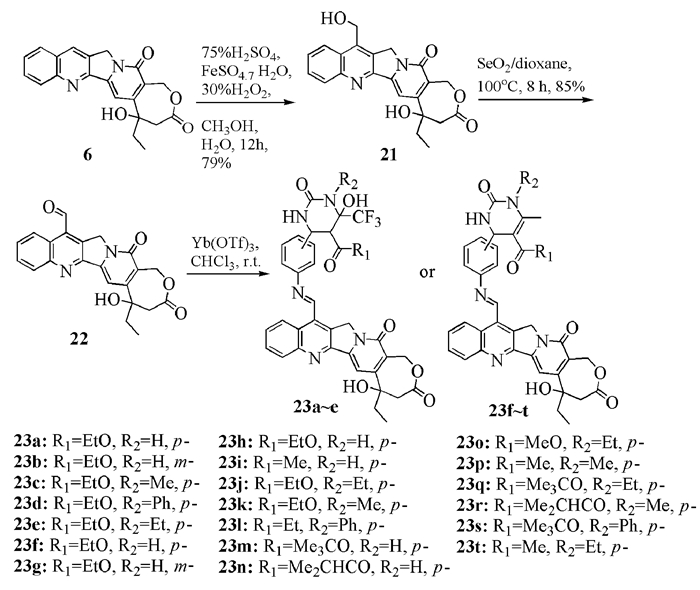
此外,他们在高喜树碱7-位上进行衍生,将另一具有抗癌活性的二氢嘧啶酮片段通过亚胺键的形式连接到7-位上(图式 5)。所合成的大多数化合物对肿瘤细胞A549、MDA-MB-435和HCT116均表现出较好的活性,特别是23b、23k和23m对HCT116的体外抑制活性(分别为78、48和81 nmol/L)要优于拓扑替康[17, 18]。
1Li等[19]用半合成方法得到的高喜树碱,在20-位上引入多种酯,测试了它们对6种人体癌细胞KB、KB/VCR、A549、HCT-8、Bel7402和A2780的体外细胞毒性,结果显示,大部分化合物都表现出与喜树碱相当的抗癌活性。
半合成方法合成快捷,但限制于起始原料的局限性,不能在A环上引入种类多样的取代基,这是该方法的缺憾。
高喜树碱的全合成主要有三种策略,一是最后同时构建B和C环,二是构建B环,三是构建C环(图式 6)。这三种方法分别体现在Curran和Burke、张万年以及Bigg小组的工作中。
1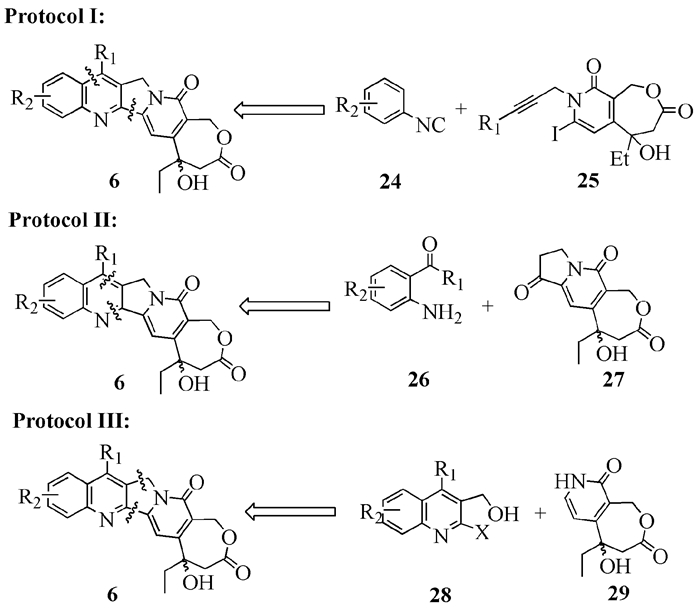
Curran等[20]在7-位上引入硅基合成了一种在血液中和溶液中比较稳定的、亲酯性较好的高喜树碱(见图式 7)。他们沿用制备喜树碱的标准方法,先合成原料30,接着进行扩环,再碘代和N-烷基化引入炔甲基,最后与异腈反应同时构建了B环和C环。随后,他们用Sharpless不对称环氧化方法合成了手性的中间体(R)-30,经过取代反应和关环,制备出了(20R)-高喜树碱以及它在A环上不同位置氟代的衍生物[21]。同年,他们[22]又报道了(20S)-7-TBS-10-羟基高喜树碱的全合成方法。
1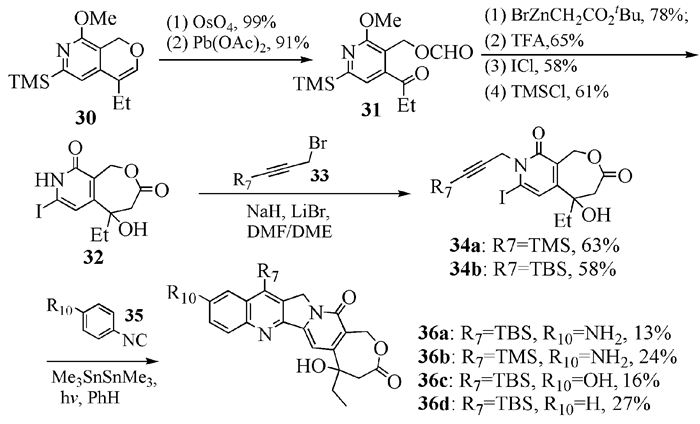
张万年等参考了合成喜树碱的方法[23],先合成出片段37,接着分别经过KBH4还原、NaIO4氧化、Reformatsky反应、分子内酯交换成环得到片段41,然后与42进行关环反应生成BN-80245(图式 8)[24~26]。
1之后,他们[27]用类似的方法,在A环上引入了10-羟基,在B环上引入了7-甲基,合成了一系列10-磷酸酯衍生物(图式 9)。在这些磷酸酯中,45e和45f对3种肿瘤细胞A549、MDA-MB-435和LOVO表现出良好的抑制活性,可以跟伊立替康相媲美,但都比拓扑替康低两个数量级。
1
另外,该小组[28]在化合物44的基础上,合成了12个10-胺烷醚47a~47l以及2个12-胺烷醚48a、48b(图式 10)。其中,化合物47a、47b和48b对肺癌细胞A-549的抑制能力比伊立替康高一个数量级。他们还在A环9-位上引入不同的酰胺官能团(图式 11),研究了各种类似化合物的生物活性。结果表明,50a和51c具有与参照化合物伊立替康相当的乳腺癌细胞(MDA-MB-435)抑制活性。
1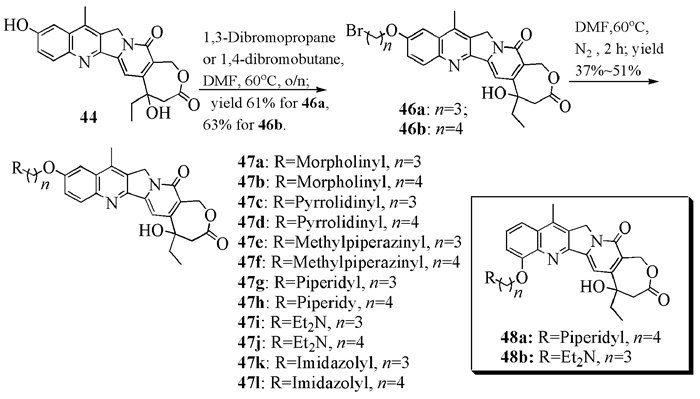
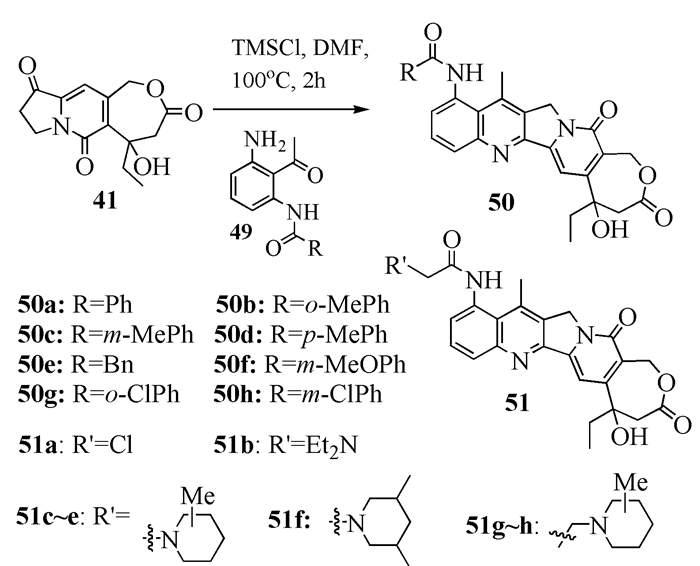
除了研究A环上不同取代基的构效关系之外,张万年等在B环上也进行了改造(图式 12)。研究发现,对乳腺癌细胞(MDA-MB-435)而言,在B环上取代的羰基化合物53d和53e,要比烷基取代产物53a和53b的活性高10倍以上,而与对照物拓扑替康相比,抑制活性高100倍以上。化合物53k与拓扑替康相比,对肿瘤细胞A-549、MDA-MB-435和HCT116的抑制活性分别高出3个、2个和1个数量级[29]。
1
Bigg小组先分别单独合成片段60和61(图式 13),再通过Mitsunobu反应和Heck反应链接上述两个独立的片段构建了封闭的C环,得到多个类似物63。在与肺癌细胞(A427)、前列腺癌细胞(PC-3)、白血病癌细胞(K562adr)和乳腺癌细胞(MCF7mdr)的测试中,化合物63c、63d、63g和63i表现了良好的细胞毒性,特别是63g,即前面提到的化合物BN-80915(7)的消旋体,在对接种了结肠癌细胞(HT-29)的小鼠口服给药跟踪测试中,每天给药0.32mg/kg,可抑制癌细胞繁殖25d,而喜树碱(每天给药0.625mg/kg)只抑制了4d[6, 30]。
1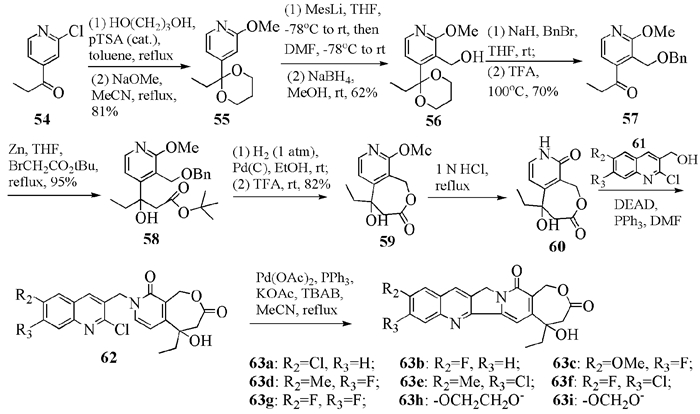
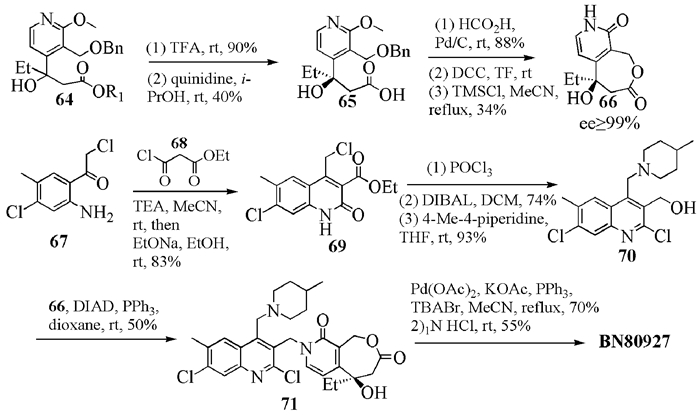
Bigg小组另外一个候选明星药物BN-80927(8)(图式 14)具有单一(20R)-构型,是在构建七元的E环前先用奎尼丁进行初步拆分,得到手性的化合物65,待E环扩环后,再进行重结晶,得到ee值>99%的片段66[13];然后与衍生过的喹啉70关环,得到7-位上取代哌啶的BN-80927。该化合物有较好的生物活性。
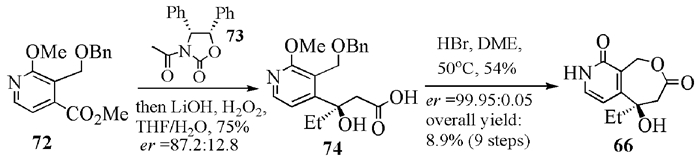
1接着,该小组从手性中间体66出发, 用同样的方法合成了BN-80915(7)以及单氟和双氟在A环上不同位置取代的(20R)-高喜树碱异构体。体外实验表明,BN-80915(7)对HT29、A549和T24r的抗癌活性最优[31]。关于BN-80915 (7)的制备工艺[28],罗氏公司的改进之处是用Evans试剂手性诱导合成了β-羟基丙酸,经过氢溴酸脱苄关环,得到单一构型的66(图式 15)。
1Luo等[32]用手性合成方法得到手性片段79,79与含有不同取代基的80反应,得到双羟基的高喜树碱81(图式 16)。所合成的化合物对结肠癌细胞和肺癌细胞的抑制能力与参照物喜树碱和10-羟基喜树碱相当,有的活性高出1个数量级。
1
总之,全合成方法种类繁多,花式各样,虽然合成繁琐,但可以在不同环上引入需要的官能团,做到药物研发的有的放矢。
高喜树碱的合成方法有半合成和全合成两种,其中全合成法分别可以从B、C环一步生成,最后合成B环以及最后合成C环三种方法来得到消旋的或手性的高喜树碱。另外,高喜树碱的七元E环稳定性大大改善,毒副作用降低,生物利用度提高。其衍生的明星药物分子BN-80915(7)和BN-80927(8)对多种癌细胞有较好的抑制能力,在某方面还优于喜树碱,具有极大的应用前景。目前,高喜树碱手性制备方法也在不断优化,低成本高产率的合成方法以及高活性、低毒性衍生物的制备与应用也将成为该领域的研究热点。
N H Oberlies, D J Kroll. J. Nat. Prod., 2004, 67:129~135. doi: 10.1021/np030498t
M E Wall, M C Wani, C E Cook et al. J. Am. Chem. Soc., 1966, 88:3888~3889. doi: 10.1021/ja00968a057
K Mross, H Richly, N Schleucher et al. Ann. Oncol., 2004, 15:1284~1294. doi: 10.1093/annonc/mdh313
O Lavergne, L L-Ginot, F P Rodas et al. J. Med. Chem., 1998, 41:5410~5419. doi: 10.1021/jm980400l
X Wu, X Sun, Z Guo et al. J. Am. Chem. Soc. 2014, 136:3579~3588. doi: 10.1021/ja412380j
M Ye, X Wang, J Tang et al. Chem. Sci., 2016, 7:4958~4965. doi: 10.1039/C6SC00970K
刘映前, 朱高翔, 张娜 等. CN: 105601641A, 2015.
Y Q Zhang, H J Zhang, J Zhang et al. Aust. J. Chem., 2011, 64:1547~1553. doi: 10.1071/CH11315
H F Hausheer, H Kochat, P Seetharamulu et al. WO: 9835940A1, 1998.
A E Gabarda, W Du, T Isarno et al. Tetrahedron, 2002, 58:6329~6341. doi: 10.1016/S0040-4020(02)00632-4
O Lavergne, L L Ginot, F P Rodas et al. Bioorg. Med. Chem. Lett., 1997, 7:2235~2238. doi: 10.1016/S0960-894X(97)00398-3
C Bailly, A Lansiaux, L Dassonneville et al. Biochemistry, 1999, 38:15556~15563. doi: 10.1021/bi990947h
D Demarquay, M Huchet, H Coulomb et al. Bioorg. Med. Chem. Lett., 1999, 9:2599~2602. doi: 10.1016/S0960-894X(99)00428-X
D Demarquay, M Huchet, H Coulomb. Anti Cancer Drugs, 2001, 12:9~19. doi: 10.1097/00001813-200101000-00003
Z Mi, T G Burke. Biochemistry, 1994, 34:10325~10336.
李尚丰, 马秀光, 何志敏 等. CN106478648A, 2016.
L Zhu, P Cheng, N Lei et al. Arch. Pharm. Chem. Life Sci. 2011, 344:726~734. doi: 10.1002/ardp.v344.11
张万年, 刘文锋, 缪震元 等. CN: 102010418B, 2010.
D Z Li, C Y Wang, R H Liu et al. J. Asian Nat. Prod. Res., 2013, 15(11):1179~1188. doi: 10.1080/10286020.2013.855203
D Bom, D P Curran, A J Chavan et al. J. Med. Chem., 1999, 42:3018~3022. doi: 10.1021/jm9902279
D P Curran, W Du. Org. Lett., 2012, 4:3215~3218.
M C Wani, P E Ronman, J T Lindley et al. J. Med. Chem. 1980, 23:554~560. doi: 10.1021/jm00179a016
Z Miao, C Sheng, W Zhang et al. Bioorg. Med. Chem., 2008, 16:1493~1510. doi: 10.1016/j.bmc.2007.10.046
张万年, 杨松, 姚建忠 等. CN: 1557814A, 2004.
张万年, 杨松, 姚建忠 等. CN: 100408582C, 2004.
Z Miao, J Zhang, L You et al. Bioorg. Med. Chem., 2010, 18:3140~3146. doi: 10.1016/j.bmc.2010.03.039
W Guo, W Liu, L Zhu et al. Chem. Biodiversity, 2011, 8(8):1539~1549. doi: 10.1002/cbdv.201000307
W Liu, L Zhu, W Guo et al. Eur. J. Med. Chem., 2011, 46:2408~2414. doi: 10.1016/j.ejmech.2011.03.024
D Bigg, O Lavergne, F P Rodas et al. USP: 6339091 B1, 2002.
O Lavergne, D Demarquay, C Bailly et al. J. Med. Chem., 2000, 43:2285~2289. doi: 10.1021/jm000129j
R Peters, M Althaus, C Diolez et al. J. Org. Chem., 2006, 71:7583~7595. doi: 10.1021/jo060928v
Y Luo, S Yu, L Tong et al. Eur. J. Med. Chem., 2012, 54:281~286. doi: 10.1016/j.ejmech.2012.05.002

图式 1 具有生物活性的喜树碱和高喜树碱类化合物
Scheme 1 Bioactive compounds of different camptothecin and homocamptothecin

图式 6 高喜树碱的三种逆向合成策略
Scheme 6 Three retro-synthetic protocols for synthesis of homocamptothecin

图式 7 同时构建B和C环合成7-硅基高喜树碱
Scheme 7 Synthesis of 7-silylhomocamptothecin via B&C ring closure

图式 9 B环关环构建10-高喜树碱磷酸酯
Scheme 9 Synthesis of 10-homocamptothecinphosphate via B ring closure

图式 10 B环构建10-和12-胺烷醚高喜树碱
Scheme 10 Synthesis of 10-and 12-aminoalky-loxylhomocamptothecin via B ring closure

图式 11 B环关环构建9-酰胺基高喜树碱
Scheme 11 Synthesis of 9-amidohomocamptothecin via B ring closure

图式 12 B环关环构建10-甲氧基-7-羰基高喜树碱
Scheme 12 Synthesis of 10-methoxy-7-homocamptothecinketone via B ring closure

图式 13 C环关环构建9, 10-取代的高喜树碱
Scheme 13 Synthesis of 9, 10-disubstituted homocamptothecin via C ring closure

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:








 下载:
下载: