引用本文:
陈婷, 朱志良. 铁基水处理材料除砷技术的研究进展[J]. 化学通报,
2018, 81(10): 880-889.

Citation: Chen Ting, Zhu Zhiliang. Research Progress in Arsenic Removal Technology of Iron-Based Water Treatment Materials[J]. Chemistry, 2018, 81(10): 880-889.
Citation: Chen Ting, Zhu Zhiliang. Research Progress in Arsenic Removal Technology of Iron-Based Water Treatment Materials[J]. Chemistry, 2018, 81(10): 880-889.
铁基水处理材料除砷技术的研究进展
摘要:
自然环境中的砷污染问题被认为是全球最严重的环境威胁之一,人类长期暴露于含砷饮用水环境中会引起各种疾病的发生,因此,开发经济有效的除砷技术一直是砷污染治理领域的研究热点。铁基水处理材料由于其对砷的良好亲和力、表面反应活性强、价廉易制、便于回收等特点,一直备受关注。本文综述了近年来不同铁基水处理材料如铁(氢)氧化物、纳米零价铁、铁基多金属氧化物复合材料除砷技术的研究进展,论述了铁基水处理材料对水相中砷去除的影响因素及机理;同时,对影响铁基水处理材料砷解吸的因素和毒性评估研究进行了总结;指出了目前铁基水处理材料砷污染去除技术研究中存在的主要问题,并对水相砷污染去除技术研究中值得关注的重要发展方向进行了展望。
English
Research Progress in Arsenic Removal Technology of Iron-Based Water Treatment Materials
Abstract:
Arsenic in the natural environment is considered as one of the most serious environmental threats in the world. Long-term exposure to arsenic from drinking water can cause various diseases. Therefore, there is an urgent need to develop economically effective arsenic removal technology. Iron-based water treatment materials have attracted more attention on arsenic removal due to its good affinity to arsenic, strong reactivity on the surface, low cost, easy preparation and recycling. This paper summarizes the progress in arsenic removal technology by iron-based materials, such as ferric (hydr) oxide, zero-valent iron, iron-based multi-mental oxide and their composites. Possible influence factors and mechanisms of arsenic removal in aqueous solution by different iron-based water treatment materials were discussed in details. At the same time, the influence factors of arsenic desorption and the toxicity assessment of the above mentioned materials were summarized. Finally, the main problems about arsenic removal existed in present research are put forward. The prospects for the further development of iron-based water treatment materials for arsenic removal are also discussed.
-
Key words:
- Iron-based materials
- / Arsenic removal technology
- / Water treatment
- / Toxicity assessment
-
砷污染是目前全球饮用水安全的主要威胁之一[1]。在印度、中国、孟加拉国、越南、尼泊尔和柬埔寨等地砷污染情况十分严峻[2~5]。据估计,目前全世界有超过一亿的人口涉及饮用受砷污染地下水的问题[6, 7]。由于砷的毒性风险,世界卫生组织(WHO)、美国环境保护署(EPA)将饮用水中砷的最大污染物水平限制为10μg/L。我国《生活饮用水卫生标准》(GB 5749-2006)也将砷的最高允许浓度规定为10μg/L。
在自然环境中,砷主要以+3价和+5价的无机形态存在于地下水中[8]。在富氧环境中,砷以稳定的五价砷酸分子H3AsO4、砷酸根含氧阴离子H2AsO4-、HAsO42-和AsO43-等形式存在。然而在缺氧环境中,三价砷(如H3AsO3)占主导地位[9]。经毒理学研究表明,As(Ⅲ)比As(Ⅴ)物种的毒性更大[10, 11]。一旦进入人体,大部分砷会积累在头发、指甲、骨骼、肝脏和肾脏等器官中[12],从而影响细胞及机体的正常代谢,引起组织损伤和机体功能紊乱,甚至直接导致死亡[13]。
由于世界范围内砷污染问题依然十分严峻,人们一直在不断开发各类除砷技术。其中吸附法由于其成本及能耗低、操作简单等特点而备受推崇[14]。因此,活性炭[15]、氧化铝[16]、树脂[17]、分子筛[18, 19]、二氧化硅[20, 21]及生物吸附剂[22]等各类吸附材料不断被开发及改性,以期通过合成经济高效的吸附材料去除水相中的砷,将饮用水中的砷浓度降低到WHO规定的水平以下。其中,铁基水处理材料由于其表面反应活性强、价廉易制、便于回收及对砷的亲和力高等特点受到研究者们特别的关注,常被用作消除砷污染的主要吸附材料[23~25]。此外,某些铁基材料还可将As(Ⅲ)氧化为As(Ⅴ),利用砷的形态转变在降低砷毒性的同时,借助自身对五价砷的强吸附特性实现对水相中砷的有效去除[26~28]。
因此,本文聚焦于近年来铁基水处理材料除砷技术的研究进展,通过对不同铁基除砷材料如铁(氢)氧化物、纳米零价铁(nZVI)、铁基双金属氧化物及其复合材料的制备方法、影响因素及去除机理等的主要进展回顾,对不同铁基水处理材料除砷的条件及影响因素进行对比分析,结合对生物的毒性作用,讨论不同铁基水处理材料的构效关系、实用性及发展前景,进一步分析了现有除砷技术研究中存在的主要问题,对今后铁基水处理材料除砷技术的重点发展方向提出了相应建议。
1. 铁基水处理材料对水相中砷的去除
1.1 铁(氢)氧化物除砷
近年来,铁氧化物由于其丰富的孔隙结构、巨大的比表面积和超顺磁性等优点成为国内外研究者关注的热点。目前,常见的应用于砷污染处理的铁氧化物材料包含水合(氢)氧化铁、磁性Fe2O3、磁性Fe3O4等。
1.1.1 水合(氢)氧化铁
水合(氢)氧化铁由于其成本低廉、性质稳定以及良好的环境相容性常用于吸附溶液中的重金属及阴离子[29, 30]。例如,Zhang等[31]用介质溶液法合成了超小且分散均匀的β-FeOOH纳米棒,研究了其对As(Ⅴ)的吸附性能。结果表明,该材料对As(Ⅴ)的吸附容量达到29.03mg/g,远高于Fe3O4、α-Fe2O3和α-FeOOH。
水合(氢)氧化铁的颗粒细小,在水溶液中的团聚作用限制了材料的表面活性,从而使材料的吸附性能显著降低。因此研究人员通过对其表面改性来提高材料的稳定性和分散性,促进对水溶液中污染物的吸附。Jia等[32]研究发现,向利用溶液法合成的α-FeOOH中加入十六烷基三甲基溴化铵(CTAB)可有效避免材料的聚集,从而使单分散的α-FeOOH表面拥有大量的羟基位点,通过离子交换机理实现对水溶液中As的吸附。CTAB作为表面活性剂包裹了α-FeOOH,由于其分子之间的强排斥力可减少α-FeOOH的聚集并提高其稳定性。Andjelkovic等[33]用简单的方法在三维石墨烯气凝胶网络中掺入氧化铁纳米颗粒显著增强了材料对As的吸附性能,由于α-FeOOH纳米石墨烯气凝胶粒子具有随机分散的3D多孔结构,对As(Ⅴ)的吸附量为81.3mg/g,对As(Ⅲ)的吸附量为13.4mg/g。
随着研究的不断深入,δ-FeOOH也被开发出来用于水相中砷的去除。Faria等[34]通过化学沉淀法合成了具有高比表面积及小颗粒尺寸的δ-FeOOH晶体。TEM图像及BET测试表明,该材料平均孔径为18nm,比表面积高达135m2/g。在中性环境中,该吸附剂对砷的吸附容量可达37.3mg/g。
制备工艺以及吸附条件是影响水合(氢)氧化铁吸附性能的主要因素。但在实际水处理过程中,由于面临的环境条件的复杂性和不确定性,需要不断优化水合(氢)氧化铁的制备方法以提高不同环境条件下的吸附能力。
1.1.2 磁性Fe2O3
磁性Fe2O3常作为良好的吸附剂吸附去除水溶液中的砷。Lin等[35]在室温下通过共沉淀法合成磁性γ-Fe2O3纳米颗粒用于对As(Ⅲ)和As(Ⅴ)的去除,并探究了pH和竞争离子对As(Ⅲ)和As(Ⅴ)吸附作用的影响。结果表明,合成的磁性γ-Fe2O3纳米颗粒对As(Ⅲ)或As(Ⅴ)的吸附在30min内可达到饱和,在10、30、50 ℃对As(Ⅲ)和As(Ⅴ)的吸附量分别为59.25、67.02、74.83 mg/g和88.44、95.37、105.25 mg/g,溶液pH以及Cl-、SO42-和NO3-对吸附没有显著影响,但是PO43-由于具有与砷相似的外部电子结构产生竞争作用,可导致材料的吸附能力显著降低。反应后,通过施加强度大于0.35T的磁场可以回收吸附饱和的磁性γ-Fe2O3纳米颗粒。
为比较磁性γ-Fe2O3和磁性Fe3O4对砷的去除效果,Mou等[23]通过溶剂热反应和N2氛围下的热处理,成功制备了具有栗状分层纳米结构(CNHs)的磁性γ-Fe2O3和磁性Fe3O4。结果表明,γ-Fe2O3 CHNs对As(Ⅴ)有非常高的去除能力,且吸附速度比Fe3O4快,其吸附容量高达101.4mg/g,能很好地与被处理水溶液快速分离,可作为理想的除砷磁性吸附剂。
同时,不同的制备方法对材料的吸附性能有很大影响,合成方法的改进可使铁氧化物材料的吸附性能不断提高,从而更高效率地去除水相中的砷。Wei等[36]通过微波加热以及甘油介导的水热过程成功合成了具有空心结构的α-Fe2O3,合成的材料对水中As、Cr及刚果红的去除表现出优异性能。其中,甘油含量对空心结构的形成有很大影响,通过不断调整甘油的投加量形成适当大小的空心结构可有效地提高材料对As的吸附能力。
1.1.3 磁性Fe3O4
磁性Fe3O4由于其具有较高的比表面积和超顺磁性的优点,常用于处理重金属污染废水[37~39]。Yavuz等[40]研究表明,在低场强下可从溶液中去除临界尺寸为20nm的Fe3O4颗粒。因此,在外加磁场的控制下,使用后的Fe3O4很容易从溶液中快速分离出来,可避免对环境的二次污染。由于裸露的Fe3O4对砷的吸附能力有一定的限度,吸附能力并不强[41],因此,研究者们将各种材料包裹在Fe3O4表面或通过制备Fe3O4复合材料[42~44]等方法对Fe3O4进行改性,通过增加其表面缺陷和羟基数量,从而提高对砷的吸附能力。
Feng等[45]利用水热法在没有任何模板的情况下成功合成抗坏血酸涂覆的Fe3O4纳米颗粒。通过BET测定,该材料比表面积为180m2/g,其较高的比表面积展现出对砷的强吸附能力。这是由于体系内的抗坏血酸改善了Fe3O4纳米粒子在悬浮液中的分散性。同时,该材料有效地抑制了铁离子在溶液中的浸出,降低了对环境的二次污染。超顺磁性的优点也利于该材料在使用后的成功分离。
Guo等[46]在碱性条件下通过聚多巴胺改性合成了具有三维结构的Fe3O4-石墨烯凝胶用于对水相中含量10-9级别的砷的去除。该方法简单易行、成本低廉,聚多巴胺的加入可有效增强石墨烯的宏观结构同时也可增强Fe3O4纳米颗粒对砷的结合能力和承载能力。
提高Fe3O4除砷效率还有一种办法是在其组成中掺杂其他金属元素,可使纳米颗粒的反应活性增加。Wang等[37]在磁性Fe3O4中掺杂Cu2+用于对砷的吸附。结果表明,随着Cu2+的增加,该材料对As(Ⅲ)和As(Ⅴ)的吸附量分别从8.12和7.32 mg/g增加到37.97和42.90 mg/g。原因是Cu2+的加入可以催化As(Ⅲ)氧化为As(Ⅴ),同时减小了材料粒径,增加材料的表面积、孔隙率和ζ电位,使得材料表面的吸附位点对As(Ⅴ)的亲和力增加。
Luo等[44]用氧化石墨烯(GO)、Fe3O4和MnO2纳米颗粒通过两步共沉淀反应合成Fe3O4-rGO-MnO2。此纳米复合材料具有高吸附容量和优异的磁性,在pH为7.0时,Langmuir吸附模型计算的单层吸附量分别为14.04和12.22 mg/g(As(Ⅲ)和As(Ⅴ))。高吸附容量归因于rGO的高比表面积。同时,吸附剂表面的MnO2可将As(Ⅲ)氧化成As(Ⅴ),然后通过减少Fe3+的聚集为As(Ⅴ)提供更多的吸附位点。此外,吸附作用在pH 2~10范围内都保持稳定。这种吸附剂在处理砷污染水的治理中具有较好潜在价值。
表 1总结了水合(氢)氧化铁、磁性Fe2O3、磁性Fe3O4及其复合材料对水中污染物砷的吸附条件及最大吸附量。由表可见,改性后的铁氧化物对As的吸附能力明显增强;同时,大多数铁氧化物对砷酸盐的吸附能力比对亚砷酸盐的强;不同的吸附条件,例如吸附剂的剂量、砷初始浓度、pH等对吸附效果有很大影响。因此,如何提高吸附剂的吸附能力、如何有效改善吸附最佳条件同时加强对毒性较大的亚砷酸盐的吸附是值得关注的重点。
表 1

 下载:
导出CSV
下载:
导出CSV
吸附剂 吸附剂剂量
/(g/L)砷初始浓度
/(mg/L)T/℃ 平衡时间/h 平衡浓度/(mg/L) pH 最大吸附量
/(mg/g)β-FeOOH 1 5.2 25 24 70(Ⅴ) 7 29.03(Ⅴ) CTAB-α-FeOOH 1 5.21 25 24 150~200(Ⅴ) - 30.64(Ⅴ) GN-α-FeOOH 0.05 1~16 20±2.0 - 8.5(Ⅲ),13.5~14(Ⅴ) 8~9 13.4(Ⅲ),81.3(Ⅴ) δ-FeOOH 0.25 2.5~20 25±1.0 6 9.5~10(Ⅴ) 7 37.3(Ⅴ) γ-Fe2O3 0.8 10~150(Ⅲ),10~200(Ⅴ) 30 1.5 45(Ⅲ),45(Ⅴ) 6(Ⅲ), 3(Ⅴ) 67.02(Ⅲ),95.37(Ⅴ) γ-Fe2O3 CHNs 0.4 6.96~200 25±1.0 3 152(Ⅴ) 4 101.4(Ⅴ) Fe3O4 CHNs 0.4 6.96~200 25±1.0 3 4.52(Ⅴ) 4 6.07(Ⅴ) α-Fe2O3 0.5 10~500 - 2 450~500(Ⅴ) - 75.3(Ⅴ) 抗坏血酸-Fe3O4 0.06 1 - 24 65~70(Ⅲ),45~50(Ⅴ) 7 46.06(Ⅲ),16.56(Ⅴ) Fe3O4-石墨烯 2 0.1~1 30 12(Ⅲ), 24(Ⅴ) 0.075Ⅲ),0.25~0.3(Ⅴ) 7 0.452(Ⅲ),0.373(Ⅴ) Cu-Fe3O4 0.5 1~85(Ⅲ),1~45(Ⅴ) 25 4 60~70(Ⅲ),10~15Ⅴ) 5±0.2 37.97(Ⅲ),42.9(Ⅴ) Fe3O4-RGO-MnO2 0.5 0.01~10 25.5±0.2 24 2.0~2.5(Ⅲ),3.5~4.0(Ⅴ) 7±0.2 14.04(Ⅲ),12.22(Ⅴ) 1.2 纳米零价铁除砷
纳米零价铁(nZVI)是降低水溶液中As浓度的有效材料之一。由于其无毒、价廉易制且还原过程几乎不需维护,近年来受到国内外学者广泛关注[47~50]。然而,由于nZVI抗氧化性较弱,其稳定性和重复利用性较差,在水溶液中活性较低、易团聚,容易受到各种环境条件的影响,且反应后不易从溶液中分离。因此,为了克服这些缺点,研究者们使用各种方法对nZVI进行改性,以期实现材料的高效利用价值。
Zhu等[51]利用活性炭优异的机械强度和多孔结构性质将nZVI负载到活性炭上得到nZVI/AC复合材料,评估了吸附剂用量、pH、常见共存离子等各种参数对砷去除的影响。结果表明,大部分形如针状的零价铁颗粒被装载到了活性炭的孔隙和裂缝中,保证在吸附过程中复合材料的重复使用,极大程度地降低了nZVI的丢失。实验表明,nZVI/AC对As(Ⅲ)和As(Ⅴ)的吸附容量分别为18.2和12.0 mg/g。同时,在磷酸盐或硅酸盐存在时,砷的去除效率显著降低。常见金属阳离子Ca2+、Mg2+的存在增强了As(Ⅴ)的去除,但是Fe2+会抑制As(Ⅲ)的吸附。吸附-再生循环8次后材料的吸附容量并没有明显降低。表明通过活性炭改性后的nZVI虽然可有效降低流失率,提高材料的稳定性和重复利用性,但在除砷的吸附效果方面还有待提高。
具有多孔结构的MnO2作为维持反应活性的物种,可避免零价铁表面氧化沉淀的聚集,从而有效维持零价铁的反应活性。因此,Bui等[52]通过超声化学合成法制备了具有核壳结构的nZVI/Mn氧化物,并与nZVI进行了不同pH条件下对溶液中As(Ⅲ)和As(Ⅴ)的去除效果的比较。结果表明,nZVI/Mn复合物对As的去除高度依赖于pH。在酸性条件下,nZVI/Mn氧化物除砷能力比nZVI强。这主要是由于酸性条件可促进FeO氧化(腐蚀)生成铁(氢)氧化物从而达到对As的吸附和共沉淀作用。同时,nZVI/Mn复合物对As(Ⅴ)和As(Ⅲ)的最大吸附容量比nZVI的分别高2和3倍。在4次循环实验后,nZVI/Mn材料对As(Ⅴ)去除效率仍保持较高水平,而nZVI的去除效率则明显降低。这主要是由于nZVI核和锰氧化物壳的协同作用增强了材料的活性,避免了材料表面沉淀的聚集,从而为As提供了更多表面吸附位点。
nZVI由于粒径较小,不适用于连续流动的系统。如果将其加载到适当的载体上进行反应可提高其除砷效率。还原态氧化石墨烯(rGO)由于具有特殊的电子传输结构、良好的机械性能以及高比表面积等优点,可作为优良的载体来去除水溶液中的砷。Wang等[53]将rGO负载到nZVI上制备成rGO-nZVI复合材料,并评价其对As(Ⅲ)和As(Ⅴ)的去除效果。批量实验表明,该材料在低剂量情况下对As(Ⅲ)和As(Ⅴ)表现出较强的亲和力,吸附能力分别可达到35.83和29.04 mg/g。Bhowmick等[54]利用蒙脱石负载的nZVI去除水溶液中的砷。这些改性方法都在不同程度上提高了对水溶液中砷的去除效率,避免了nZVI在水中易凝聚等缺点。
表 2总结了多种nZVI材料在其实验最佳条件下对水中砷的吸附效果。从表中可以发现,不同的nZVI的最优吸附条件以及对砷吸附能力差别很大,改性后的nZVI除砷能力均有一定程度的提高。因此,在实际水处理过程中,应根据受砷污染的特点去设计nZVI复合材料,优化材料的吸附条件从而提高材料对污染物的吸附性能。
表 2
下载:
导出CSV
吸附剂 吸附剂量
/(g/L)砷初始浓度
/(mg/L)T/℃ 平衡时间/h 平衡浓度/(mg/L) pH 最大吸附量
/(mg/g)NZVI/AC 1 2 25 72 0.20~0.23(Ⅲ),0.22~0.23(Ⅴ) 6.5 18.2(Ⅲ),12.0(Ⅴ) nZVI/Mn oxide 0.3 1~20 25 2 12~15(Ⅲ),9~10(Ⅴ) 4.8 29.4(Ⅲ),35.7(Ⅴ) nZVI 0.3 1~20 25 2 9(Ⅲ),9~10(Ⅴ) 4.8 9.5(Ⅲ),21.7(Ⅴ) nZVI-RGO 0.4 1~10 25±0.5 2 0.7~0.8(Ⅲ),1.2~1.3(Ⅴ) 7 35.83(Ⅲ),29.04(Ⅴ) Mt-nZVI 1 5 22±1.0 4 250~300(Ⅲ),150~200(Ⅴ) 7 59.9(Ⅲ),45.5(Ⅴ) 1.3 铁基多金属氧化物复合材料除砷
铁基材料除砷的不足在于在水溶液中适应的pH条件范围较窄;而两种或更多种金属氧化物的复合材料不仅继承了母体氧化物的优点,而且有明显的协同效应,同时还能拓宽铁基材料应用的pH范围。例如,Zhang等[25]通过简便的共沉淀法合成了一种新型具有纳米结构的Fe-Cu二元氧化物。当Fe:Cu摩尔比为2:1时,Fe-Cu二元氧化物对水中As(Ⅴ)和As(Ⅲ)的去除表现出优异的性能。当pH=7.0时,对As(Ⅴ)和As(Ⅲ)的吸附量分别达到了82.7和122.3 mg/g, 实现了通过简单而低成本的合成材料在中性pH条件下对As(Ⅴ)和As(Ⅲ)的有效去除。
除此之外,研究者通常还将Ce、Al、Zn、Zr、Mn等金属加入到铁氧化物中,以改善铁氧化物的相关功能并用于对砷的去除。例如,Liu等[55]合成的FeAlOxHy对水相中砷和氟离子的去除效果明显好于FeOxHy和AlOxHy。Zhang等[56]采用氧化共沉淀法低成本开发了一种结合MnO2氧化性能和铁氧化物的高吸附性能的新型Fe-Mn二元氧化物材料,并将其用于去除水中的砷。结果表明,该吸附剂能将As(Ⅲ)完全氧化为As(Ⅴ),去除效果显著。
笔者所在课题组近年开展了有关层状双金属氢氧化物(LDH)功能材料除砷的系列研究[57, 58]。首先,通过共沉淀法合成了Zn-Fe-LDH材料,并以此为前体在不同的煅烧温度下转换合成了一系列不同Zn-Fe摩尔比的双金属混合氧化物(Zn-Fe-MMO)。吸附砷的实验结果表明,煅烧温度为300℃,Zn-Fe摩尔比为4时,材料吸附性能最好[57]。我们还采用共沉淀法制备了三金属LDH:Cu-Zn-Fe-LDH,利用其类Fenton反应活性开展了同步去除水中对乙酰氨基酚和砷的研究。在H2O2存在下,毒性较强的As(Ⅲ)可以同步被氧化成As(Ⅴ),然后被Cu-Zn-Fe-LDH吸附去除[58]。
为了将As(Ⅲ)原位氧化并实现在水中同时除去As(Ⅲ)和As(Ⅴ)的目的,Lu等[59]还利用LDH的“记忆效应”通过煅烧重建方法制备了一种新颖的过二硫酸根插层的LDH:Mg-Fe-S2O8-LDH,并对其吸附砷的性能和去除机理进行了研究。实验结果表明,LDH中插层的S2O82-组分可将水中的As(Ⅲ)几乎完全氧化成As(Ⅴ),同时进一步吸附在Mg-Fe-S2O8-LDH上,是可同时去除水中不同价态砷污染物的高效吸附剂材料。
由于As(Ⅲ)比As(Ⅴ)的毒性更大且环境中流动性较强,相对而言在溶液中较难去除。Hong等[60]为了获得去除As(Ⅲ)的高效功能材料,采用共沉淀法合成了α-丙氨酸插层的铁基LDH:Mg-Fe-Ala-LDH,并探究了Mg-Fe-Ala-LDH材料对亚砷酸根和砷酸根的吸附性能。结果发现,对As(Ⅲ)的最大吸附容量为49.8mg/g,明显高于对As(Ⅴ)的23.6mg/g。解吸实验也表明,Mg-Fe-Ala-LDH对As(Ⅲ)显示出比对As(Ⅴ)更强的亲和力。机理研究表明,这与层状双氢氧化物材料中的α-丙氨酸插层组分的特性密切相关。这表明所合成的Mg-Fe-Ala-LDH材料是去除水中As(Ⅲ)潜在的高效吸附剂。
表 3总结了不同铁基多金属(氢)氧化物材料对As(Ⅲ)或As(Ⅴ)的吸附条件和吸附能力,不同种类的金属与铁所形成的铁基金属(氢)氧化物对砷的吸附能力差异较大。对层状双金属氢氧化物来说,不同插层阴离子对As(Ⅲ)或As(Ⅴ)吸附能力也有显著差别。因此,在实际水处理中,应根据水质特征选择合适的吸附材料用于水中砷去除。
表 3
下载:
导出CSV
吸附剂 吸附剂剂量
/(g/L)砷初始浓度
/(mg/L)T/℃ 平衡时间/h 平衡浓度
/(mg/L)pH 最大吸附量/(mg/g) Fe-Cu oxide 0.2 5~60 24±1.0 24 35~40(Ⅲ),30~35(Ⅴ) 7 122.3(Ⅲ),82.7(Ⅴ) FeAlOxHy 0.067(Fe), 0.054(Al) 50 25±1.0 4 - 7.5 362.7(Ⅴ) Zn-Fe-MMO 0.2 1 25 24 45~50(Ⅲ),35~40(Ⅴ) 6 36.0(Ⅲ),176.3 (Ⅴ) Cu-Zn-Fe-LDH 0.1~0.4 1 25 24 75~80(Ⅲ),70(Ⅴ) 7 126.13(Ⅴ) Mg-Fe-S2O8-LDH 0.2 1~50 25 24 35~38(Ⅲ),40~45(Ⅴ) 3~10 75.0(Ⅲ),75.63(Ⅴ) Mg-Fe-Ala-LDH 0.2 1~15 25 24 8~9(Ⅲ),5~6(Ⅴ) 6 49.8(Ⅲ),23.6(Ⅴ) 2. 铁基水处理材料除砷机理
2.1 铁(氢)氧化物除砷机理
由于不同铁氧化物吸附材料结构的不同,其除砷时的吸附条件也有很大区别,吸附机理也随吸附条件的改变而有所不同。一般而言,铁氧化物除砷可通过氧化吸附、静电吸引、离子交换、配位等多种吸附机理去除水溶液中的砷污染物。
水合氧化铁在不同pH时由于表面羟基化作用会发生质子化或去质子化,使材料表面带上正电荷或负电荷,以不同功能团的形式存在于水中,从而对水中的砷酸根离子等物种有很好的吸附作用[61]。
磁性Fe3O4由于其表面大量的活性位点可作为砷的吸附位点,使用后的Fe3O4由于其超顺磁性可进行简单的回收分离。磁铁矿纳米粒子(MNPs)与As形成表面络合物通过促进As和Fe之间的电子转移过程从而达到除砷的目的[62]。
同时,为规避铁氧化物低pH条件下除砷效率较低、易团聚等缺点常对材料进行表面改性。改性后的铁氧化物增强了其分散性和稳定性,除砷效率明显提高,但同时除砷机理也变得复杂。例如,在pH=7时,As主要以HAsO42-存在于溶液中,生物炭改性后的Fe2O3表面的羟基通过质子化作用带正电,因此,HAsO42-可通过静电吸引与材料表面带正电的官能团相互作用,从而达到除砷目的[63]。负载珠状纤维素的β-FeOOH的二齿-双核共角结构具有强吸附中心,可实现对As(Ⅲ)和As(Ⅴ)特异性吸附[64]。
2.2 纳米零价铁除砷机理
nZVI的除砷机理相当复杂。已有研究认为nZVI除砷机理包括吸附、表面沉淀以及与铁(氢)氧化物等铁的腐蚀产物的共沉淀[65]等。
nZVI的反应活性可通过在nZVI表面上发生的一系列化学反应来解释[66, 67]。如式(1)~(3)所示,nZVI在酸性条件下被腐蚀生成Fe2+,Fe2+进一步被氧化成Fe3+。这些腐蚀产物以铁氢氧化物或铁氧化物(式(4)~(6))的形式在nZVI表面沉淀以便和As(Ⅴ)结合。同时,过程中产生的·OH可以将As(Ⅲ)氧化成As(Ⅴ)再进行吸附。相较于铁(氢)氧化物而言,nZVI的腐蚀过程可以加强对As(Ⅲ)的氧化吸附。
$ \text{Fe}\left( 0 \right)+{{\text{O}}_{\text{2}}}+\text{2}{{\text{H}}^{\text{+}}}\to \text{F}{{\text{e}}^{\text{2+}}}+{{\text{H}}_{\text{2}}}{{\text{O}}_{\text{2}}} $
(1) $ \text{Fe}\left( \text{0} \right)\text{+}{{\text{H}}_{\text{2}}}{{\text{O}}_{\text{2}}}\text{+2}{{\text{H}}^{\text{+}}}\to \text{F}{{\text{e}}^{\text{2+}}}\text{+2}{{\text{H}}_{\text{2}}}\text{O} $
(2) $ \text{F}{{\text{e}}^{\text{2+}}}+{{\text{H}}_{\text{2}}}{{\text{O}}_{\text{2}}}\to \text{F}{{\text{e}}^{\text{3+}}}+\cdot \text{OH+O}{{\text{H}}^{\text{-}}} $
(3) $ \text{F}{{\text{e}}^{\text{2+}}}+\text{2O}{{\text{H}}^{\text{-}}}\to \text{Fe}{{\left( \text{OH} \right)}_{\text{2}}} $
(4) $ \text{F}{{\text{e}}^{\text{3+}}}\text{+3O}{{\text{H}}^{\text{-}}}\to \text{Fe}{{\left( \text{OH} \right)}_{\text{3}}} $
(5) $ \text{6Fe}{{\left( \text{OH} \right)}_{\text{2}}}\text{+}{{\text{O}}_{\text{2}}}\to \text{2F}{{\text{e}}_{\text{3}}}{{\text{O}}_{\text{4}}}\text{+6}{{\text{H}}_{\text{2}}}\text{O} $
(6) 然而在高pH条件下,nZVI的腐蚀产物发生了变化,砷的去除机制无法用上述机理进行解释[68]。为研究pH对nZVI除砷机理的影响,Wu等[69]研究了不同pH条件下nZVI对As(Ⅴ)去除效率,深入分析了nZVI在不同pH下的吸附机理。结果表明,在初始pH=4时,nZVI的腐蚀产物由赤铁矿(γ-Fe2O3)、磁铁矿(Fe3O4)和纤铁矿(γ-FeOOH)组成。然而在pH=10时,赤铁矿(γ-Fe2O3)和磁铁矿(Fe3O4)成为腐蚀产物的主要成分。同时,随着初始pH增加到7,再到10,nZVI对As(Ⅴ)吸附容量分别下降了0.80%和7.57%。这可以从两个方面进行解释,一是nZVI的远距离静电排斥,减少了As(Ⅴ)的去除;另一个是nZVI腐蚀产物的变化,导致nZVI与As(Ⅴ)的配位能力改变。
同时,在有氧条件下和无氧条件下nZVI除砷机理也各不相同。Sun等[70]在有氧和相对厌氧条件下对含有砷酸盐和亚砷酸盐的水溶液分别进行了批量实验。结果表明,在相对厌氧条件下,亚砷酸盐的去除速度比砷酸盐的快,而有氧条件下则相反。这主要是由于有氧条件下ZVI的腐蚀产物对As的吸附共沉淀作用占主导地位,同时As(Ⅲ)可被氧化成As(Ⅴ)再去除,而在厌氧条件下认为As主要通过沉淀作用去除。
由此可见,环境条件的不同对nZVI除砷机理有很大影响,需根据特定的环境条件进行具体分析研究。
2.3 铁基多金属复合氧化物除砷机理
单纯的铁(氢)氧化物水处理材料虽然对重金属有较强的吸附功能,但同时也存在不足。通过在铁氧化物中掺杂一种或多种金属元素形成铁基多金属氧化物可有效规避铁氧化物的缺点,从而达到高效除砷的目的。在铁基多金属氧化物中,铁氧化物常作为吸附基础材料,掺杂的金属元素氧化物有不少是作为氧化剂可将As(Ⅲ)氧化成As(Ⅴ)后再对As(Ⅴ)进行表面吸附,通过转化砷的价态提高了对毒性更大的As(Ⅲ)的吸附去除能力。典型吸附机理如式(7)~(9)所示,其中X代表掺杂的金属元素,XO2代表金属氧化物,-SFe-X代表Fe-X氧化物表面上的吸附位点。
$ \text{As}\left( \text{III} \right)\text{+}\left( \text{-SFe-X} \right)\to \text{As}\left( \text{III} \right)\text{-SFe-X} $
(7) $ \text{As}\left( \text{III} \right)\text{+X}{{\text{O}}_{\text{2}}}\text{+4}{{\text{H}}^{\text{+}}}\to \text{As}\left( \text{V} \right)\text{+}{{\text{X}}^{\text{2+}}}+\text{2}{{\text{H}}_{\text{2}}}\text{O} $
(8) $ \text{As}\left( \text{V} \right)\text{+}\left( \text{-SFe-X} \right)\to \text{As}\left( \text{V} \right)\text{-SFe-X} $
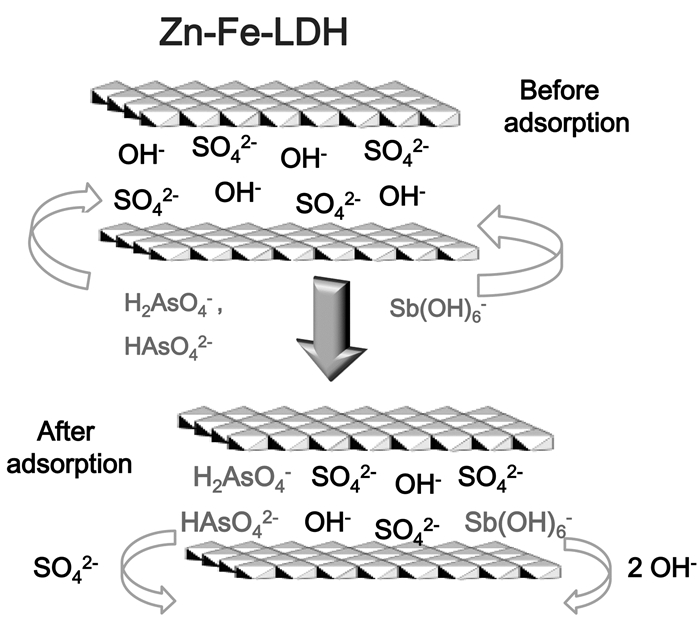
(9) 但对于LDH来讲,由于层间阴离子具有交换能力,可与溶液中的砷酸根阴离子交换从而有效吸附砷物种。Lu等[71]通过XPS测定了Zn-Fe-LDH处理含As(Ⅴ)和Sb(Ⅴ)废水前后表面元素分布特征,As 3d和Sb 3d核心峰的出现表明吸附后Zn-Fe-LDH表面存在As(Ⅴ)和Sb(Ⅴ)物种,从而推测是由于层间SO42-和OH-同砷酸根离子和锑酸根离子发生了离子交换。
图 1

3. 影响铁基水处理材料砷解吸的因素
尽管铁基水处理材料在除砷方面表现出优异的性能,但由于环境的复杂性,吸附后的砷在一定条件下会被解吸释放到环境中。例如,受到环境中共存阴离子的影响[72],铁(氢)氧化物的还原溶解[73]等都会导致砷的再次释放。
Caporale等[74]研究了竞争配体对Mg-Fe-LDH吸附及解吸亚砷酸盐的影响。结果表明,有机和无机配体显示出与As(Ⅲ)竞争Mg-Fe-LDH吸附位点的不同能力。其中,硫酸盐是竞争配体中作用最弱的。柠檬酸盐由于具有高电荷密度和形成强内层复合物的能力,其抑制As(Ⅲ)的效率明显高于草酸盐;而PO43-由于与砷具有相似的外层电子结构,是竞争抑制As(Ⅲ)吸附的强配体。解吸实验结果表明,停留时间的长短会显著影响砷的解吸效果。这主要是因为停留时间短意味着吸附主要发生在外表面,从而导致较快的解吸[75]。随着As(Ⅲ)在LDH表面的停留时间的增加,这些阴离子对As(Ⅲ)的解吸影响会减少。除PO43-外,所有配体抑制As(Ⅲ)吸附并解吸As(Ⅲ)的能力均较对As(Ⅴ)的低。
氢氧化铁通常被认为是优越的砷吸附剂。然而,由于铁在水环境中会发生氧化还原反应,致使氢氧化铁在吸附砷后可转化为溶解形式,从而释放出砷[76]。Masue等[77]为了抑制砷的再释放,采用共沉淀法合成了双金属铝铁氢氧化物用于对砷的吸附,研究表明,在氢氧化铁中加入氢氧化铝可使氢氧化铁的溶解速率降低,从而有效抑制砷的解吸。
4. 铁基水处理材料的毒性评估
不同铁基水处理材料除砷技术在实验室阶段已取得了不少成果。由于其价廉易制、便于回收等优点在实际水处理工程中也得到了很好应用。但铁基材料的实际应用也会导致环境中纳米颗粒的不断增加,吸附的有毒有害污染物也有可能二次释放,给生态环境安全带来了或多或少的威胁。因此,有效评估铁基水处理材料对生物的毒性作用才能更好的优化铁基材料在实际工程中的应用条件,避免给环境带来不利影响。
Wang等[78]评价了α-Fe2O3纳米粒子对卤虫卵囊及其三个幼虫时期的毒性。结果表明,卤虫一龄,二龄和三龄阶段的幼虫的体长和个体干重在α-Fe2O3纳米粒子暴露下均下降,其卵囊的孵化率明显降低。二龄和三龄阶段幼虫的LC50分别为177.424和235.495 mg/L。由此可见,高剂量α-Fe2O3纳米粒子对环境中的微生物有潜在毒性影响。
nZVI在土壤修复以及水处理过程中的使用越来越普遍。研究表明,nZVI可以吸附在细菌的细胞膜上或穿透细胞膜,从而引起细胞的功能紊乱甚至导致细菌死亡[79]。El-Temsah等[80]将蚯蚓暴露于含0~2000mg/kg nZVI的土壤中,评价其对蚯蚓的影响。实验证明,当nZVI浓度大于500mg/kg时,蚯蚓的繁殖受到严重影响,并对有机体造成了急性副作用。Auffan等[81]选用nZVI、γ-Fe2O3、Fe3O4比较其氧化还原状态对大肠杆菌的毒性作用。结果表明,化学稳定的γ-Fe2O3对细胞没有明显的毒性作用。但含有Fe2+的Fe3O4以及nZVI由于氧化作用对细胞表现出极强的毒性。此外,铁基水处理材料作为矿物类材料吸附的污染物在环境中可以二次释放,也会造成对生物的毒性作用,但这方面的研究还十分有限。
因此,在制备过程中,应选用安全环保的材料以高效、经济的方法合成稳定性好的铁基材料。当铁基材料用于实际水处理过程时,应注意这些材料潜在的生态毒理学影响,也应关注吸附材料的二次环境污染问题,对各类材料进行全过程毒性评估。投加时应严格把控投加剂量,优化吸附条件,尽量规避对环境的二次伤害。
5. 结语
铁基水处理材料由于对砷的亲和力强、吸附容量大、价廉易制、易于回收、环境友好等优点成为国内外在砷污染去除领域的研究热点。各种铁基水处理材料的制备、功能化改性、除砷机理研究等方面均取得了一定的成果。但是,该类材料的实际运用过程中,由于材料自身的特点及环境条件的复杂性,仍存在许多亟待解决的问题。值得关注的主要问题包括:(1)某些铁基水处理材料作用时的适用pH范围很窄,吸附条件严苛,且大多数铁基水处理材料对As(Ⅴ)的去除效果优于毒性更大的As(Ⅲ),导致除总砷效率不高,很难应用于实际水处理过程;(2)功能化改进后的复合材料,往往存在制备成本高、耗时较长、材料稳定性较差,增加了二次污染的可能性;(3)对除砷机理的研究仍不够深入和清晰,缺乏规律性的除砷机理认识;(4)缺少对铁基水处理材料的毒性评估以及对人体的健康风险评价研究;(5)实际环境中的共存阴离子、腐殖酸等有机质对吸附影响的研究方面还不够深入系统。
因此,基于上述分析,对今后铁基水处理材料除砷技术研究,重点应关注以下几个方面:(1)根据构效关系原理,寻找更为高效、安全、经济适用的铁基除砷材料合成方法;(2)针对不同的砷污染实际环境,优化不同铁基水处理材料的吸附条件和过程,深入考察实际共存离子及污染物对铁基水处理材料除砷效率的影响,扩大其适应pH范围,提高对总砷尤其是As(Ⅲ)的去除效果;(3)通过合理的改性、复合和加工,改善铁基水处理材料易团聚、不易分离、金属离子溶出等缺点;(4)采用先进的表征和分析技术,探究除砷的机理及规律,在分子、原子水平明晰不同铁基水处理材料除砷的详细机理和长效去除机制,使获得的结论更具有理论指导意义;(5)加强对使用后的铁基水处理材料对环境的毒性评估,采用科学的风险评价方法探明其对环境生物及人体健康的影响。
-
-
[1]
P Ravenscroft, H Brammer, K Richards. Arsenic Pollution: A Global Synthesis. Chichester, UK: Wiley-Blackwell, 2009.
-
[2]
T Pal, P K Mukherjee, S Sengupta et al. Gondwana Res., 2002, 5(2): 501~512. doi: 10.1016/S1342-937X(05)70738-3
-
[3]
M Berg, C Stengel, T K Pham et al. Sci. Total Environ., 2007, 372(2~3): 413~425. http://www.sciencedirect.com/science/article/pii/S0048969706006978
-
[4]
H Zhang, D S Ma, X X Hu. Environ. Geol., 2002, 41(6): 638~643. doi: 10.1007/s002540100442
-
[5]
J Gurung, H Ishiga, M S Khadka. Environ. Geol., 2005, 49(1): 98~113. doi: 10.1007/s00254-005-0063-6
-
[6]
S Fendorf, H A Michael, A Geen. Science, 2010, 328(5982): 1123~1127. doi: 10.1126/science.1172974
-
[7]
P Kay. Area, 2011, 43(1): 118~119. doi: 10.1111/j.1475-4762.2010.00983_4.x
-
[8]
龚仓, 徐殿斗, 马玲玲.化学通报, 2014, 77(6): 502~509. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20140122001&flag=1
-
[9]
B K Mandal, K T Suzuki. Talanta, 2002, 58(1): 201~235. doi: 10.1016/S0039-9140(02)00268-0
-
[10]
X P Liu, W F Zhang, Y N Hu et al. Chemosphere, 2015, 119: 273~281. doi: 10.1016/j.chemosphere.2014.06.067
-
[11]
T V Nguyen, S Vigneswaran, H H Ngo et al. Sep. Purif. Technol., 2008, 61(1): 44~50. doi: 10.1016/j.seppur.2007.09.015
-
[12]
Y X Jiang, X C Zeng, X T Fan et al. Ecotoxicol. Environ. Safety, 2015, 122: 198~204. doi: 10.1016/j.ecoenv.2015.07.018
-
[13]
M M Rahman, J C Ng, R Naidu. Environ. Geochem. Health, 2009, 31: 189~200. doi: 10.1007/s10653-008-9235-0
-
[14]
D Mohan, C U Pittman. J. Hazard. Mater., 2007, 142(1~2): 1~53. http://www.sciencedirect.com/science/article/pii/S0304389410012021
-
[15]
T Budinova, D Savova, B Tsyntsarski et al. Appl. Surf. Sci., 2009, 255(8): 4650~4657. doi: 10.1016/j.apsusc.2008.12.013
-
[16]
Y H Kim, C M Kim, I H Choi et al. Environ. Sci. Technol., 2004, 38(3): 924~931. doi: 10.1021/es0346431
-
[17]
T M Suzuki, J O Bomani, H Matsunaga et al. React. Funct. Polym., 2000, 43(1~2): 165~172. http://www.sciencedirect.com/science/article/pii/S1381514899000383
-
[18]
K R Sharma. Chem. Res. Toxicol., 2003, 16(12): 1684.
-
[19]
Y L Wang, S F Wang, X Wang et al. Water Air Soil Pollut., 2016, 227(8): 257. doi: 10.1007/s11270-016-2955-3
-
[20]
M Hasanzadeh, F Farajbakhsh, N Shadjou et al. Environ. Technol., 2015, 36(1): 36~44. doi: 10.1080/09593330.2014.934744
-
[21]
M Salim, Y Munekage. Int. J. Environ. Res., 2009, 3(1): 13~22. https: //www.cabdirect.org/?target=%2fcabdirect%2fabstract%2f20093065376
-
[22]
V M Boddu, K Abburi, J L Talbott et al. Water Res., 2008, 42(3): 633~642. doi: 10.1016/j.watres.2007.08.014
-
[23]
F Z Mou, J G Guan, H R Ma et al. ACS Appl. Mater. Interf., 2012, 4(8): 3987~3993. doi: 10.1021/am300814q
-
[24]
Z M Ren, G S Zhang, J P Chen. J. Colloid Interf. Sci., 2011, 358(1): 230~237. doi: 10.1016/j.jcis.2011.01.013
-
[25]
G S Zhang, Z M Ren, X W Zhang et al. Water Res., 2013, 47(12): 4022~4031. doi: 10.1016/j.watres.2012.11.059
-
[26]
A Gupta, M Yunus, N Sankararamakrishnan. Chemosphere, 2012, 86(2): 150~155. doi: 10.1016/j.chemosphere.2011.10.003
-
[27]
V Chandra, J Park, Y Chun et al. ACS Nano, 2010, 4(7): 3979~3986. doi: 10.1021/nn1008897
-
[28]
L C Roberts, S J Hug, T Ruettimann et al. Environ. Sci. Technol., 2004, 38(1): 307~315. doi: 10.1021/es0343205
-
[29]
G Z Nie, J Wang, B C Pan et al. Sci. China Chem., 2015, 58(4): 722~730. doi: 10.1007/s11426-014-5285-6
-
[30]
X L You, D Guay, A Farran et al. J. Chem. Technol. Biotechnol., 2016, 91(3): 693~704. doi: 10.1002/jctb.4629
-
[31]
Y X Zhang, Y Jia. Appl. Surf. Sci., 2014, 290: 102~106. doi: 10.1016/j.apsusc.2013.11.007
-
[32]
Y Jia, T Luo, X Y Yu et al. RSC Adv., 2013, 3(36): 15805~15811. doi: 10.1039/c3ra40980e
-
[33]
I Andjelkovic, D N H Tran, S Kabiri et al. ACS Appl. Mater. Interf., 2015, 7(18): 9758~9766. doi: 10.1021/acsami.5b01624
-
[34]
M C S Faria, R S Rosemberg, C A Bomfeti et al. Chem. Eng. J., 2014, 237: 47~54. doi: 10.1016/j.cej.2013.10.006
-
[35]
S Lin, D N Lu, Z Liu. Chem. Eng. J., 2012, 211: 46~52.
-
[36]
Z H Wei, R G Xing, X Zhang et al. ACS Appl. Mater. Interf., 2013, 5(3): 598~604. doi: 10.1021/am301950k
-
[37]
T Wang, W C Yang, T T Song et al. RSC Adv., 2015, 5(62): 50011~50018. doi: 10.1039/C5RA03951G
-
[38]
Y Jia, H M Yu, L Wu et al. Anal. Chem., 2015, 87(12): 5866~5871. doi: 10.1021/acs.analchem.5b00712
-
[39]
S A Baig, T T Sheng, C Sun et al. PloS One, 2014, 9(6): e100704. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM24967645
-
[40]
C T Yavuz, J T Mayo, W W Yu et al. Science, 2006, 314(5801): 964~967. doi: 10.1126/science.1131475
-
[41]
T Wang, L Y Zhang, H Y Wang et al. ACS Appl. Mater. Interf., 2013, 5(23): 12449~12459. doi: 10.1021/am403533v
-
[42]
R Z Chen, C Y Zhi, H Yang et al. J. Colloid Interf. Sci., 2011, 359(1): 261~268. doi: 10.1016/j.jcis.2011.02.071
-
[43]
Z G Liu, F S Zhang, R Sasai. Chem. Eng. J., 2010, 160(1): 57~62. doi: 10.1016/j.cej.2010.03.003
-
[44]
X B Luo, C C Wang, S L Luo et al. Chem. Eng. J., 2012, 187: 45~52. doi: 10.1016/j.cej.2012.01.073
-
[45]
L Y Feng, M H Cao, X Y Ma et al. J. Hazard. Mater., 2012, 217: 439~446. http://www.sciencedirect.com/science/article/pii/S0304389412003676
-
[46]
L Q Guo, P R Ye, J Wang et al. J. Hazard. Mater., 2015, 298: 28~35. doi: 10.1016/j.jhazmat.2015.05.011
-
[47]
H Alijani, Z Shariatinia. Chemosphere, 2017, 171: 502~511. doi: 10.1016/j.chemosphere.2016.12.106
-
[48]
M Baikousi, Y Georgiou, C Daikopoulos et al. Carbon, 2015, 93: 636~647. doi: 10.1016/j.carbon.2015.05.081
-
[49]
F C Su, H J Zhou, Y X Zhang et al. J. Colloid Interf. Sci., 2016, 478: 421~429. doi: 10.1016/j.jcis.2016.06.035
-
[50]
C Trois, A Cibati. J. Environ. Chem. Eng., 2015, 3(1): 488~498. doi: 10.1016/j.jece.2014.12.019
-
[51]
H J Zhu, Y F Jia, X Wu et al. J. Hazard. Mater., 2009, 172(2~3): 1591~1596. http://www.sciencedirect.com/science/article/pii/S0304389409013119
-
[52]
T H Bui, C Kim, S P Hong et al. Environ. Sci. Pollut. Res. Int., 2017, 24(31): 24235~24242. doi: 10.1007/s11356-017-0036-9
-
[53]
C Wang, H J Luo, Z L Zhang et al. J. Hazard. Mater., 2014, 268: 124~131. doi: 10.1016/j.jhazmat.2014.01.009
-
[54]
S Bhowmick, S Chakraborty, P Mondal et al. Chem. Eng. J., 2014, 243: 14~23. doi: 10.1016/j.cej.2013.12.049
-
[55]
R P Liu, W X Gong, H C Lan et al. Sep. Purif. Technol., 2012, 92: 100~105. doi: 10.1016/j.seppur.2012.03.020
-
[56]
G S Zhang, J H Qu, H J Liu et al. Water Res., 2007, 41(9): 1921~1928. doi: 10.1016/j.watres.2007.02.009
-
[57]
G L Di, Z L Zhu, H Zhang et al. Chem. Eng. J., 2017, 328: 141~151. doi: 10.1016/j.cej.2017.06.112
-
[58]
H T Lu, Z L Zhu, H Zhang et al. ACS Appl. Mater. Interf., 2016, 8(38): 25343~25352. doi: 10.1021/acsami.6b08933
-
[59]
H T Lu, Z L Zhu, H Zhang et al. Water Air Soil Pollut., 2016, 227(4): 125. doi: 10.1007/s11270-016-2828-9
-
[60]
J Hong, Z L Zhu, H T Lu et al. Chem. Eng. J., 2014, 252: 267~274. doi: 10.1016/j.cej.2014.05.019
-
[61]
许建红, 高乃云, 唐玉霖等.水处理技术, 2011, 37(8): 22~25+34. http://www.cnki.com.cn/Article/CJFDTotal-SCLJ201108007.htm
-
[62]
C H Liu, Y H Chuang, T Y Chen et al. Environ. Sci. Technol., 2015, 49(13): 7726~7734. doi: 10.1021/acs.est.5b00381
-
[63]
S S Wang, B Gao, A R Zimmerman et al. Bioresour. Technol., 2015, 175: 391~395. doi: 10.1016/j.biortech.2014.10.104
-
[64]
X J Guo, Y H Du, F H Chen et al. J. Colloid Interf. Sci., 2007, 314(2): 427~433. doi: 10.1016/j.jcis.2007.05.071
-
[65]
M S H Mak, P Rao, I M C Lo. Water Res., 2009, 43(17): 4296~4304. doi: 10.1016/j.watres.2009.06.022
-
[66]
F Fu, D D Dionysiou, H Liu. J Hazard. Mater., 2014, 267: 194~205. doi: 10.1016/j.jhazmat.2013.12.062
-
[67]
V Tanboonchuy, J C Hsu, N Grisdanurak et al. J Hazard. Mater., 2011, 186(2~3): 2123~2128. http://www.sciencedirect.com/science/article/pii/S030438941100015X
-
[68]
赵雅光, 万俊锋, 王杰等.化工学报, 2015, 66(2): 730~737. http://www.cnki.com.cn/article/cjfdtotal-hgsz201502033.htm
-
[69]
C Wu, J W Tu, W Z Liu et al. Environ. Sci.-Nano, 2017, 4(7): 1544~1552. doi: 10.1039/C7EN00240H
-
[70]
H W Sun, L Wang, R H Zhang et al. J. Hazard. Mater., 2006, 129(1~3): 297~303. http://www.sciencedirect.com/science/article/pii/S1381116906008065
-
[71]
H T Lu, Z L Zhu, H Zhang et al. Chem. Eng. J., 2015, 276: 365~375. doi: 10.1016/j.cej.2015.04.095
-
[72]
A G Caporale, M Pigna, J J Dynes et al. J. Hazard. Mater., 2011, 198: 291~298. doi: 10.1016/j.jhazmat.2011.10.044
-
[73]
M L Polizzotto, C F Harvey, G C Li et al. Chem. Geol., 2006, 228(1~3): 97~111. http://www.sciencedirect.com/science/article/pii/S0009254106000349
-
[74]
A G Caporale, M Pigna, S M G G Azam et al. Chem. Eng. J., 2013, 225: 704~709. doi: 10.1016/j.cej.2013.03.111
-
[75]
C Luengo, V Puccia, M Avena. J. Hazard. Mater., 2011, 186(2~3): 1713~1719. https: //www.sciencedirect.com/science/article/pii/S0304389410016547
-
[76]
X G Meng, G P Korfiatis, C Y Jing et al. Environ. Sci. Technol., 2001, 35(17): 3476~3481. doi: 10.1021/es010645e
-
[77]
Y Masue, R H Loeppert, T Kramer et al. Environ. Sci. Technol., 2007, 41(3): 837~842. doi: 10.1021/es061160z
-
[78]
C Wang, H Jia, L Zhu et al. Sci. Total Environ., 2017, 598: 847~855. doi: 10.1016/j.scitotenv.2017.04.183
-
[79]
M Stefaniuk, P Oleszczuk, Y S Ok. Chem. Eng. J., 2016, 287: 618~632. doi: 10.1016/j.cej.2015.11.046
-
[80]
Y S El-Temsah, E J Joner. Chemosphere, 2012, 89(1): 76~82. doi: 10.1016/j.chemosphere.2012.04.020
-
[81]
M Auffan, W Achouak, J Rose et al. Environ. Sci. Technol., 2008, 42(17): 6730~6735. doi: 10.1021/es800086f
-
[1]
-
表 1 铁(氢)氧化物及其复合材料对水相中砷的去除条件和效果的对比[23, 31~37, 44~46]
Table 1. Comparison of arsenic removal conditions and efficiency in aqueous by ferric (hydr) oxide and related composite materials[23, 31~37, 44~46]
吸附剂 吸附剂剂量
/(g/L)砷初始浓度
/(mg/L)T/℃ 平衡时间/h 平衡浓度/(mg/L) pH 最大吸附量
/(mg/g)β-FeOOH 1 5.2 25 24 70(Ⅴ) 7 29.03(Ⅴ) CTAB-α-FeOOH 1 5.21 25 24 150~200(Ⅴ) - 30.64(Ⅴ) GN-α-FeOOH 0.05 1~16 20±2.0 - 8.5(Ⅲ),13.5~14(Ⅴ) 8~9 13.4(Ⅲ),81.3(Ⅴ) δ-FeOOH 0.25 2.5~20 25±1.0 6 9.5~10(Ⅴ) 7 37.3(Ⅴ) γ-Fe2O3 0.8 10~150(Ⅲ),10~200(Ⅴ) 30 1.5 45(Ⅲ),45(Ⅴ) 6(Ⅲ), 3(Ⅴ) 67.02(Ⅲ),95.37(Ⅴ) γ-Fe2O3 CHNs 0.4 6.96~200 25±1.0 3 152(Ⅴ) 4 101.4(Ⅴ) Fe3O4 CHNs 0.4 6.96~200 25±1.0 3 4.52(Ⅴ) 4 6.07(Ⅴ) α-Fe2O3 0.5 10~500 - 2 450~500(Ⅴ) - 75.3(Ⅴ) 抗坏血酸-Fe3O4 0.06 1 - 24 65~70(Ⅲ),45~50(Ⅴ) 7 46.06(Ⅲ),16.56(Ⅴ) Fe3O4-石墨烯 2 0.1~1 30 12(Ⅲ), 24(Ⅴ) 0.075Ⅲ),0.25~0.3(Ⅴ) 7 0.452(Ⅲ),0.373(Ⅴ) Cu-Fe3O4 0.5 1~85(Ⅲ),1~45(Ⅴ) 25 4 60~70(Ⅲ),10~15Ⅴ) 5±0.2 37.97(Ⅲ),42.9(Ⅴ) Fe3O4-RGO-MnO2 0.5 0.01~10 25.5±0.2 24 2.0~2.5(Ⅲ),3.5~4.0(Ⅴ) 7±0.2 14.04(Ⅲ),12.22(Ⅴ)  下载: 导出CSV
下载: 导出CSV
表 2 纳米零价铁及其复合材料对水相中砷的去除条件及效果的对比[51~54]
Table 2. Comparison of arsenic removal conditions and efficiency in water by nZVI and composite materials[51~54]
吸附剂 吸附剂量
/(g/L)砷初始浓度
/(mg/L)T/℃ 平衡时间/h 平衡浓度/(mg/L) pH 最大吸附量
/(mg/g)NZVI/AC 1 2 25 72 0.20~0.23(Ⅲ),0.22~0.23(Ⅴ) 6.5 18.2(Ⅲ),12.0(Ⅴ) nZVI/Mn oxide 0.3 1~20 25 2 12~15(Ⅲ),9~10(Ⅴ) 4.8 29.4(Ⅲ),35.7(Ⅴ) nZVI 0.3 1~20 25 2 9(Ⅲ),9~10(Ⅴ) 4.8 9.5(Ⅲ),21.7(Ⅴ) nZVI-RGO 0.4 1~10 25±0.5 2 0.7~0.8(Ⅲ),1.2~1.3(Ⅴ) 7 35.83(Ⅲ),29.04(Ⅴ) Mt-nZVI 1 5 22±1.0 4 250~300(Ⅲ),150~200(Ⅴ) 7 59.9(Ⅲ),45.5(Ⅴ)
下载: 导出CSV
表 3 铁基多金属氧化物对水相中砷的去除条件及效果的对比[25, 55, 57~60]
Table 3. Comparison of arsenic removal conditions and efficiency in water by iron-based multi-metal oxides[25, 55, 57~60]
吸附剂 吸附剂剂量
/(g/L)砷初始浓度
/(mg/L)T/℃ 平衡时间/h 平衡浓度
/(mg/L)pH 最大吸附量/(mg/g) Fe-Cu oxide 0.2 5~60 24±1.0 24 35~40(Ⅲ),30~35(Ⅴ) 7 122.3(Ⅲ),82.7(Ⅴ) FeAlOxHy 0.067(Fe), 0.054(Al) 50 25±1.0 4 - 7.5 362.7(Ⅴ) Zn-Fe-MMO 0.2 1 25 24 45~50(Ⅲ),35~40(Ⅴ) 6 36.0(Ⅲ),176.3 (Ⅴ) Cu-Zn-Fe-LDH 0.1~0.4 1 25 24 75~80(Ⅲ),70(Ⅴ) 7 126.13(Ⅴ) Mg-Fe-S2O8-LDH 0.2 1~50 25 24 35~38(Ⅲ),40~45(Ⅴ) 3~10 75.0(Ⅲ),75.63(Ⅴ) Mg-Fe-Ala-LDH 0.2 1~15 25 24 8~9(Ⅲ),5~6(Ⅴ) 6 49.8(Ⅲ),23.6(Ⅴ)
下载: 导出CSV
-


 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 10
- 文章访问数: 222
- HTML全文浏览量: 34
 下载:
下载:
