引用本文:
董雪, 徐超, 陈靖. 核燃料循环中镅的氧化分离研究进展[J]. 化学通报,
2020, 83(4): 289-295.

Citation: Dong Xue, Xu Chao, Chen Jing. Progress in the Oxidation Separation of Americium in Nuclear Fuel Cycles[J]. Chemistry, 2020, 83(4): 289-295.
Citation: Dong Xue, Xu Chao, Chen Jing. Progress in the Oxidation Separation of Americium in Nuclear Fuel Cycles[J]. Chemistry, 2020, 83(4): 289-295.
核燃料循环中镅的氧化分离研究进展
English
Progress in the Oxidation Separation of Americium in Nuclear Fuel Cycles
Abstract:
Americium mainly exists as trivalent Am(Ⅲ) in aqueous solutions. Due to the similarity in ionic radius and chemical properties of Am(Ⅲ) and trivalent lanthanides (Ln(Ⅲ)), the separation of Am(Ⅲ) from Ln(Ⅲ) is regarded as one of the most challenging tasks in nuclear fuel cycles. Am(Ⅲ) can be oxidized to higher oxidation states such as AmO2+ and AmO22+ through different oxidation methods and then it could be separated from Ln(Ⅲ) by using well-developed solvent extraction or precipitation methods, providing a new route for the separation of Am from Ln. In this paper, the progress on the oxidation separation of Am(Ⅲ) in nuclear fuel cycles have been reviewed, the oxidation principles and relevant mechanisms were described, the advantages and disadvantages of various methods were compared, and the future trends for the oxidation separation of Am(Ⅲ) were also discussed. It hopes to provide a guidance for developing novel techniques for the separation of actinides and lanthanides.
-
Key words:
- Actinides/lanthanides separation
- / Americium
- / Lanthanides
- / Oxidation
-
近年来,为了实现核能的可持续发展,国际上提出了基于“分离-嬗变”策略的先进核燃料循环概念。其特点是不仅对乏燃料中的U、Pu等主要锕系元素进行回收利用,也分离其中的次锕系核素和长寿命裂片核素,再利用核反应或加速器将其嬗变成为短寿命或稳定核素。“分离/嬗变”策略的核心关键之一在于次锕系的分离,目前开发的流程中TRPO流程[1, 2]和DIAMEX流程[3]被认为最有应用前景。
采用TRPO流程等技术处理高放废液时,往往将Am与三价镧系元素(Ln(Ⅲ))分在一组。为了进一步减少α废物体积,需将Am与镧系元素分开。更重要的是,某些镧系元素中子吸收截面积大,其中子毒性会妨碍Am的嬗变,因此Am和镧系元素的分离是将Am成功嬗变为短寿命或稳定核素的关键[4]。另外,对于压水堆来说,乏燃料中的Am主要是241Am核素,Cm主要是短寿命的244Cm核素。241Am由于中子裂变截面大,可以直接进行嬗变,而244Cm则不行。这就要求嬗变前将Am/Cm也进行分离,将分离得到的Cm放置储存,待其衰变为240Pu后再嬗变成为稳定或短寿命核素[5]。
在水溶液中,Am主要以稳定的三价离子Am(Ⅲ)形式存在。长期以来,由于Am(Ⅲ)与Ln(Ⅲ)的半径接近,化学特性极为相似,其相互分离一直是核化学和分离科学领域的重要挑战之一。针对这一挑战,研究人员发展出了不同的分离方法。一种方法是使用含有N、S软供体原子的配体。由于锕系5f轨道比镧系4f轨道更具分散性,N/S-供体配体和锕系元素之间可形成更强的化学键,从而选择性地络合和分离锕系离子。但该方法存在溶剂选择范围窄、酸度范围要求高等缺点。此外,一些配体在强辐射场中会降解产生较多的二次废物,不利于核废物的减容[6]。另一种方法是利用Am的多价态特性, 将Am氧化成与三价镧系离子性质截然不同的高价态AmO2+和AmO22+形式[7, 8],然后再利用成熟的溶剂萃取或沉淀等方法,实现从三价镧系元素中分离出高氧化态Am。
本文将主要介绍国内外学者采用不同氧化方法对水溶液中Am进行氧化分离的研究进展,阐述Am(Ⅲ)的氧化机理及分离机制,比较不同方法的优劣,并对该领域未来的发展趋势进行分析和展望。
1. 不同氧化方法对Am的氧化分离行为
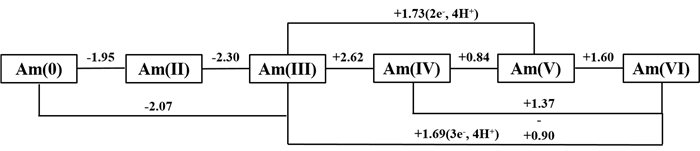
如图 1所示,Am在水溶液中存在Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ等多种氧化态。在没有络合剂时,水溶液中Ⅱ、Ⅲ及Ⅳ价Am以水合离子Am2+、Am3+及Am4+形式存在;而Ⅴ和Ⅵ价Am则以水合的镅酰离子AmO2+和AmO22+形式存在。需要指出的是,许多研究者曾试图在水溶液中确定Am(Ⅱ)的存在并制备其化合物。直到1973年,才制备出了AmCl2、AmBr2和AmI2,但它们在水溶液中极不稳定[9, 10]。此外,也有报道称在冷的强碱溶液中氧化Am(Ⅵ)获得了Am(Ⅶ)[11]。
图 1
1.1 铋酸钠氧化
Bi(Ⅲ)/Bi(Ⅴ)的氧化还原电位为2.0V,而Am(Ⅲ)/Am(Ⅵ)的为1.69V。不难推断,Bi(Ⅲ)/Bi(Ⅴ)电极电势足以支撑Bi(Ⅴ)将Am(Ⅲ)氧化为Am(Ⅵ)。Hara等[13, 14]采用NaBiO3在2mol/L硝酸溶液中将Am3+氧化为AmO22+,证实了NaBiO3氧化得到高价态Am的可能性。他们发现,在较高温度下,酸度越低,Am(Ⅲ)氧化为Am(Ⅵ)速度越快。从过量氧化剂中得到的Am(Ⅵ)仅在0℃条件下较为稳定,在高于10℃条件下则不稳定。Mincher等[15]研究了NaBiO3在0.1mol/L硝酸溶液中对243Am(Ⅲ)的氧化效果。结果表明,在室温下可以氧化生成AmO22+,而在80℃条件下氧化主要生成AmO2+,氟化物沉淀法和光谱吸收均证实了该方法氧化得到高价态Am的可行性。此外,研究者在正十二烷体系下,采用磷酸三丁酯(TBP)对Am(Ⅵ)与其他锕系离子进行了萃取实验,萃取在较短时间内完成,萃取机理与铀酰、镎酰、钚酰离子相似。Richards等[16]在硝酸介质中采用NaBiO3对Am/Cm进行氧化,成功实现了二者之间的分离。在硝酸浓度低于1.0mol/L时,Cm(Ⅲ)比高价态Am更牢固的保留在未溶解的NaBiO3上。特别是在0.1mol/L的硝酸中,分离系数可达90。与大多数正在研究的氧化剂不同,NaBiO3是一种可以大规模工业生产的廉价试剂,使其在Am(Ⅲ)的氧化分离方面具有潜在优势。另外,使用NaBiO3可以实现对Am(Ⅲ)的快速氧化。但这种方法的不足之处在于部分酸性体系(HNO3等)中NaBiO3溶解度较低,溶解在溶液中的氧化剂对Am的氧化容量有限。另外,氧化结束后不溶的物质需要过滤去除,操作较为复杂[15]。
1.2 过硫酸盐氧化
过硫酸盐在水中电离产生过硫酸根离子(S2O82-),其分子中含有过氧基(-O-O-),是一类较强的氧化剂。过硫酸根的自还原过程[17]为式(1):
$ \mathrm{S}_{2} \mathrm{O}_{8}^{2-}+2 e^{-} \rightarrow 2 \mathrm{SO}_{4}^{2-} \quad E^{0}=+2.0 \mathrm{V} $
(1) 其标准氧化还原电位为+2.0V,这一过程证明过硫酸盐体系具有足够的热力学驱动力,可以实现Am(Ⅲ)的氧化。
Asprey等[18]在20世纪50年代首次采用多种过硫酸盐化合物将大量Am(Ⅲ)氧化成Am(Ⅵ)。Newton[19]提出了过硫酸盐氧化Am(Ⅲ)的化学计量关系(式(2)):
$ \begin{array}{c} 3 / 2 \mathrm{S}_{2} \mathrm{O}_{8}^{2-}+\mathrm{Am}^{3+}+2 \mathrm{H}_{2} \mathrm{O} \rightarrow 3 \mathrm{SO}_{4}^{2-}+ \\ \mathrm{AmO}_{2}^{2+}+4 \mathrm{H}^{+} \end{array} $
(2) Elbs等[20]观察到过硫酸盐在水溶液中不稳定,在稀硫酸溶液中会随着氧气释放而分解,而在更浓的硫酸溶液中会形成过氧化氢。Kolthoff等[21]对过硫酸盐的分解动力学做了进一步的研究。过硫酸盐在水溶液中的分解主要遵从以下反应:
$ \mathrm{S}_{2} \mathrm{O}_{8}^{2-}+\mathrm{H}_{2} \mathrm{O} \rightarrow 2 \mathrm{HSO}_{4}^{-}+1 / 2 \mathrm{O}_{2} \\ $
(3) $ \mathrm{H}_{2} \mathrm{S}_{2} \mathrm{O}_{8}+\mathrm{H}_{2} \mathrm{O} \rightarrow \mathrm{H}_{2} \mathrm{SO}_{4}+\mathrm{H}_{2} \mathrm{SO}_{5} $
(4) $ \mathrm{H}_{2} \mathrm{SO}_{5}+\mathrm{H}_{2} \mathrm{O} \rightarrow \mathrm{H}_{2} \mathrm{O}_{2}+\mathrm{H}_{2} \mathrm{SO}_{4} $
(5) 其中,在碱性、中性和稀酸体系中,过硫酸盐通过式(3)分解;而在强酸溶液中,发生的反应为式(4)与式(5)。
Moore等[22]研究表明,0.1~0.2 mol/L过硫酸铵氧化241Am可获得较高生成率(约76%~97%)的高价态Am;进一步提高过硫酸铵浓度,由于过硫酸铵的分解作用,体系中对高价Am具有还原作用的过氧单硫酸和过氧化氢的含量增加,最终导致高价态Am生成率降低。此外,在该体系中加入Ag+可以大大促进Am的氧化。Ag在硝酸中溶解度大,加之其氧化还原电势高,使其具有多电子氧化剂的作用[23]。加入的Ag+被S2O82-迅速氧化到高价态,高价态Ag可以实现241Am的定量氧化。还原后的Ag再被S2O82-氧化,从而起到氧化催化剂的作用。在银催化下,过硫酸盐可以将Am(Ⅲ)直接氧化为Am(Ⅵ),由于Am(Ⅵ)可以在氟化物[24]和磷酸盐体系下稳定存在,且其氟化物或磷酸盐溶解度小,因此可以用一些氟化物和磷酸盐将氧化得到的Am(Ⅵ)沉淀下来实现与镧系元素的分离[25]。最近,Kazi等[26]使用Na2S2O8、Ag(I)和NaOCl在0.01mol/L的HNO3溶液中将Am(Ⅲ)选择性氧化为Am(Ⅵ),再采用次氯酸盐还原为较为稳定的Am(Ⅴ)(价态稳定性超过3d),利用DGA树脂可固定Cm(Ⅲ)而不固定Am(Ⅴ)的特点,实现了Am(Ⅲ)与Cm(Ⅲ)的分离,分离系数可达110±20。
由于银催化的过硫酸盐体系对Am的定量氧化性,这一体系目前被认为是实现Am向高价态转变的可靠方法。但该体系也存在以下两个主要缺点:首先,体系的酸度不能高于0.2mol/L,酸度较高时,过硫酸盐在分解的过程中会产生较多的过氧单硫酸和过氧化氢,这些都会促使Am(Ⅵ)还原;其次,所得到的Am(Ⅵ)溶液会被过量的氧化剂及还原产物污染。这两个缺点会使得研究Am(Ⅵ)的化学性质(例如稳定性、络合和氧化还原反应)较为困难。
1.3 臭氧氧化
尽管O2/O3的标准电极电势为2.07V,远高于Am(Ⅲ)/Am(Ⅵ)的电极电势(1.69V),但即使在酸性介质中加热,也难以将Am(Ⅲ)直接氧化为Am(Ⅵ)[27]。其往往作为辅助氧化剂来实现高价态Am的制备。Tsushima等[28]发现臭氧在加热的HNO3和HClO4溶液中可将Am(Ⅴ)氧化为Am(Ⅵ)。Coleman等[29]先利用K2S2O8在K2CO3溶液中氧化Am(Ⅲ)制备Am(Ⅴ),再使用臭氧将Am(Ⅴ)氧化为Am(Ⅵ)。与酸性溶液相反,Stephanou等[30]报道了臭氧会在25℃甚至更低温度下将碳酸盐溶液中的Am(Ⅲ)和Am(Ⅴ)氧化为Am(Ⅵ)。然而,当温度达到90℃时,Am的价态不会超过+5价。Gogolev等[31]在碳酸氢盐溶液中使用臭氧将Am(Ⅲ)氧化,以此观察Am(Ⅵ)/Am(Ⅳ)的动力学过程。结果表明,采用臭氧对碳酸氢盐溶液中的Am(OH)3悬浮液进行处理,Am(Ⅵ)的产率会随初始Am含量的增加而降低。此外,值得注意的是,在实验过程中采用低比放射性的243Am代替241Am不会改变高价态Am的产率及其氧化动力学。由此可推测该体系中241Am导致的辐解产物对高价Am的还原影响不大。臭氧作为一种气态且不会遗留大量二次废物的氧化剂,在Am氧化方面具有操作简便且洁净的优势,但其较弱的氧化能力限制了其实际应用。
1.4 电化学氧化
Asprey等[32]在20世纪50年代首次采用铂电极在电流密度为0.025A/cm2条件下研究了不同浓度高氯酸中Am(Ⅲ)的阳极氧化过程,并且给出了Am(Ⅴ)和Am(Ⅵ)的氧化还原电位。他们发现在6mol/L高氯酸中电解1h后获得了产率为80%的Am(Ⅵ),而在较低的酸度下,未观察到明显氧化现象。Myasoedov等[33]发现在3mol/L KHCO3-K2CO3中电解氧化243Am(Ⅲ)的效果取决于溶液的pH和阳极电位。在+1.25V电势下,可在pH=8.2~9.7范围内获得Am(Ⅳ),在pH=11~12.5范围内获得Am(Ⅵ),在pH>13.5范围内获得Am(Ⅴ)。Hobart等[34]采用电化学方法在多种碳酸盐((NH4)2CO3、Na2CO3、K2CO3、Cs2CO3)体系中将243Am(Ⅲ)电解氧化为243Am(Ⅳ),并采用循环伏安法确定在pH=9、2mol/L Na2CO3-NaHCO3中存在准可逆的Am(Ⅳ)/Am(Ⅲ)氧化还原过程。与标准氢电极相比,该介质中对应的氧化还原电势约为0.92±0.01 V。
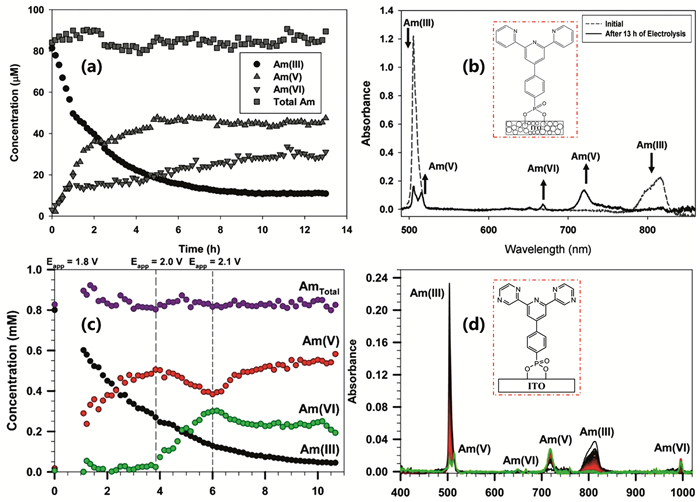
Dares等[6]首次采用三联吡啶(p-tpy)作为表面结合配体修饰介观金属氧化物纳米颗粒电极(图 2),证明了在相对较低电势(低至1.8V)条件下可将Am(Ⅲ)氧化为Am(Ⅵ),为研究没有络合剂存在的无机酸溶液中高氧化态Am的氧化还原过程提供了思路。该方法的显著特点在于非极性有机配体的附着改变了电极材料的表面疏水性,相对于单一的纳米电极,增加了水氧化的过电位,从而能够降低生成Am(Ⅵ)所需电势,同时可有效地避免因电势过高而带来的负面效果,如过氧化氢及亚硝酸的大量生成等。在这一研究基础上,Lopez等[35]使用共价连接二吡嗪吡啶(dpp)配体修饰的介孔锡掺杂氧化铟电极在pH=1的硝酸体系下实现了Am(Ⅲ)到AmO2+和AmO22+的电化学氧化(图 2)。对比之前的研究,dpp配体与Am(Ⅲ)的配位能力比p-tpy衍生配体更强,可能是实现电化学氧化生成Am(Ⅵ)更好的选择。研究表明,施加的电势会影响Am的最终氧化态。在电位为1.8V时,观察到Am(Ⅴ)。将电位提高到2.0V时,增加了Am(Ⅲ)和Am(Ⅴ)电化学氧化成Am(Ⅵ)的速率,但同时也增加了可还原Am(Ⅵ)的过氧化氢的生成速率。因此,采用电化学氧化Am时,不能仅考虑如何开发用于新型Am氧化的配体修饰电极,还需要考虑如何提高不必要反应的过电位,避免水氧化成过氧化氢以及辐解反应产生的还原剂(例如:亚硝酸)导致的电化学失活。另外,电化学氧化技术对电极材料的要求较高,不同材质的电极材料对Am的氧化效率差异很大。考虑到电极材料与强放射性废液直接接触,还需考虑电极材料的辐照稳定性。
图 2

1.5 光化学氧化
光化学氧化基本原理是将光能转化为化学反应所需的能量,使体系中的特殊成分激发成极具氧化力的自由基或离子,进而实现金属离子的氧化调价。大量研究表明,光化学方法(激光或紫外光诱导)可以实现金属离子的价态调节[36, 37]。与传统的直接分离方法和电化学氧化法等方法相比,光化学氧化分离是一种新型的、绿色环保的过程。光化学调价技术操作简单,最显著的优势在于过程中光子可以代替化学试剂参与反应。这一功能在核燃料后处理中显得尤为重要,可以减少放射性二次废物量的产生,实现废物的减容[38]。此外,光化学氧化调价具有传统调价方式所不具有的特性,特别是在混合体系下,通过调控光源波长、强度、体系种类及添加剂种类及用量可以实现金属离子的选择性氧化还原调价[39]。
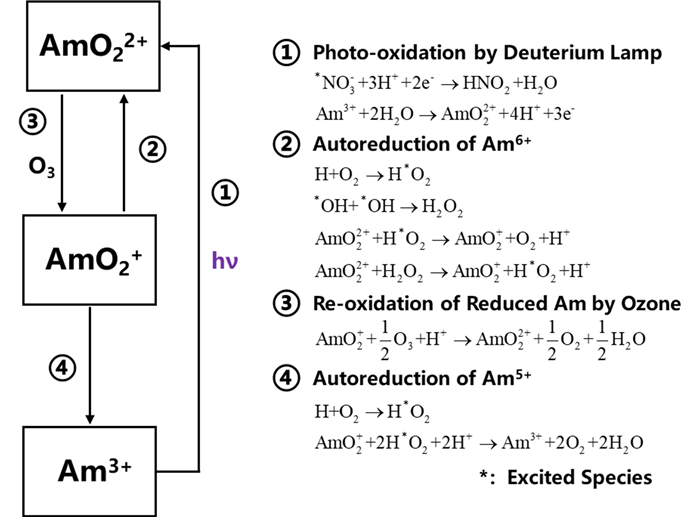
Wada等[40]在含有Pu(Ⅲ)和Np(Ⅴ)的3mol/L硝酸溶液中,采用汞灯(0.015W)进行光化学氧化。研究表明,在照射10min内,超过95%的Pu(Ⅲ)被氧化为Pu(Ⅳ),而Np(Ⅴ)价态并未改变。由于Am的氧化还原电位(Am(Ⅲ)/Am(Ⅵ)=1.69V)较Np(Np(Ⅳ)/Np(Ⅵ)=0.74V)和Pu(Pu(Ⅳ)/Pu(Ⅵ)=1.04V)的更高,使Am的光氧化条件比Np、Pu更为苛刻。Tsushima等[41]证明了通过光解将硝酸溶液中Am(Ⅲ)氧化为更高价态的可能性,并将其用于Am与镧系元素的分离。在氧化过程中,研究者发现高价态Am的分布比例虽然随光照时间的增加而略有增加,但远小于实现有效分离所需比例,推测可能是水的α辐解产物降低了氧化度,并使高价态Am逐渐变为Am(Ⅲ)[38]。在后续研究中,为了减少及避免Am的还原,他们将臭氧引入溶液中以提高氧化效率(图 3)。通过30W氘灯发出的紫外光以及向溶液中通入臭氧,在0.1mol/L的稀硝酸溶液中成功将Am(Ⅲ)光化学氧化为Am(Ⅵ)。65℃下的氧化速率为5%/h,但该速率随硝酸浓度的增加而降低。通过以上研究可知,除非氧化速率超过由α辐解形成的自由基和离子引起的Am自还原速率,否则难以进行有效的光氧化(图 4)。此外,有研究对氧化过程中酸度及温度对光化学氧化Am(Ⅲ)的影响进行了探究,发现酸度越低,温度越高,越有利于光化学氧化Am(Ⅲ)至Am(Ⅵ)。近来,还有研究者考察了室温条件下在KHCO3+K2CO3溶液中XeO3光氧化Am(Ⅲ)的可能性[42]。结果表明,将Am(Ⅲ)和XeO3暴露在波长小于420nm的紫外光和可见光下,Am(Ⅲ)的特征吸收峰显著下降,实现了Am(Ⅲ)的氧化。在XeO3存在的情况下,Am(Ⅲ)的光氧化速率加快了数十倍。
图 3

图 4

采用光化学方法对目标金属离子进行调价时,光源不与含放射性金属离子的溶液直接接触,既避免了二次废物的产生,又不影响实验仪器的二次使用,同时便于远程控制,对材料的辐照稳定性要求也不高[43]。目前采用光化学氧化Am(Ⅲ)的研究相对较少,氧化过程中不同影响因素(光源波长、强度、氧化剂类型等)对Am(Ⅲ)氧化的影响尚不明确,还需更深层次的探讨。
1.6 其他氧化方法
由于硝酸可在超声波辐照下产生自由基及活性分子,王辉等[44]采用声化学的方法对核燃料后处理流程中的锕系元素(铀、镎、钚、镅)进行调价。其中在镅的调价过程中发现,在有超声波辐照的条件下,臭氧在硝酸介质中氧化Am(Ⅴ)生成Am(Ⅵ)的速率明显加快[45]。这一方法在调价过程中无需加入任何试剂,有望实现锕系元素调价的无盐化,但还需要更多研究证明其实际应用的可行性。
2. 结语
在过去的数十年中,不同氧化态Am的化学性质已被广泛研究。采用合适的氧化方法将Am(Ⅲ)氧化至高价态,进而通过溶剂萃取或沉淀法选择性地实现高价态Am与Ln(Ⅲ)之间及Am与其他锕系元素之间的分离,为Am分离研究提供了一种新思路。目前,可直接将Am(Ⅲ)氧化至高价态Am的化学试剂/方法非常有限,即使生成了高价态Am,也很容易通过自还原和辐解作用回到Am(Ⅲ)。一些氧化方法在调价过程中无需加入其他试剂(如电/光化学氧化方法),符合核燃料后处理工艺无盐化的发展潮流。此外,虽然一些氧化方法在核燃料循环领域展现了其应用的可能性与优异性,但目前大多数研究还处于基础理论研究阶段,要想实现工业应用,还需重点解决以下两个问题:(1)反应机理的深入研究。尽管已报道的氧化Am(Ⅲ)的方法较多,但不同氧化方法的作用机理、不同反应参数(体系、浓度等)的影响规律等尚不明确,特别是一些新型氧化方法(如电化学氧化和光化学氧化)的氧化过程复杂,涉及氧化、自还原和歧化等多个物理化学过程,体系条件和参数的微小改变可能导致氧化效果的较大差异。此外,Am的各种氧化还原反应的动力学也有待进一步研究。(2)相关工艺过程的进一步优化研究。可考虑将两种以上的氧化技术联合使用,从而减少还原性物质的产生并提供充足的氧化环境,同时尽量降低氧化过程中二次废物的产生。另外,应加强与其他分离技术(萃取、吸附)的合理耦合,以更好地发挥协同效应,进一步提高效率,实现Am的高效分离。
#中国化学会“中国青年化学家元素周期表”活动特约稿件
-
-
[1]
陈靖, 王建晨.化学进展, 2011, 7:1366~1371.
-
[2]
Zhu Y J, Song C L. Recovery of neptunium, plutonium, and americium from highly active waste. Proceedings of Transuranium Elements, 1992.
-
[3]
Cuillerdier C, Musikas C, Hoel C, et al. Sep. Sci. Technol., 1991, 26(9):1229~1244. doi: 10.1080/01496399108050526
-
[4]
Potential Benefits and Impacts of Advanced Nuclear Fuel Cycles with Actinide Partitioning and Transmutation. NEA No. 6894; OECD, Nuclear Energy Agency (NEA): Paris, 2011
-
[5]
Ekberg C, Fermvik A, Retegan T, et al. Radiochim. Acta., 2008, (96):225~233.
-
[6]
Dares C J, Lapides A M, Mincher B J, et al. Science, 2015, 350(6261):652~655. doi: 10.1126/science.aac9217
-
[7]
Runde W H, Mincher B J. Chem. Rev., 2011, 111(9):5723~5741. doi: 10.1021/cr100181f
-
[8]
Mincher B J, Schmitt N C, Tillotson R D, et al. Solvent. Extr. Ion. Exc., 2014, 32(2):153~166. doi: 10.1080/07366299.2013.850288
-
[9]
Baybarz R D. J. Inorg. Nucl. Chem., 1973, 35(2):483~487. doi: 10.1016/0022-1902(73)80560-3
-
[10]
Baybarz R D, Aspery L B, Strouse C E, et al. J. Inorg. Nucl. Chem., 1972, 34(11):3427~3431. doi: 10.1016/0022-1902(72)80237-9
-
[11]
Krot N N, Shilov V P, Nikolaevskii V B, et al. Preparation of americium in heptavalent state (No. ORNL-tr-2828). AN SSSR, Moscow (USSR). Inst. Fizicheskoj Khimii., 1974.
-
[12]
Morss L R. The Chemistry of the Actinide and Transactinide Elements. Dordrecht: Springer, 2006.
-
[13]
Hara M, Suzuki S. Bull. Chem. Soc. Jpn, 1979, 52(4):1041~1045. doi: 10.1246/bcsj.52.1041
-
[14]
Hara M, Suzuki S. J. Radioanal. Nucl. Chem., 1977, 36(1):95~104.
-
[15]
Mincher B J, Martin L R, Schmitt N C. Inorg. Chem., 2008, 47(15):6984~6989. doi: 10.1021/ic800667h
-
[16]
Richards J M, Sudowe R. Anal. Chem., 2016, 88(9):4605~4608. doi: 10.1021/acs.analchem.6b01026
-
[17]
Reed W A, Garnov A Y, Rao L et al. Sep. Sci. Technol., 2005, 40(5):1029~1046. doi: 10.1081/SS-200049860
-
[18]
Asprey L B, Stephanou S E, Penneman R A. J. Am. Chem. Soc., 1950, 72(3):1425~1426. doi: 10.1021/ja01159a528
-
[19]
Newton T W. Kinetics of the Oxidation-Reduction Reactions of Uranium, Neptunium, Plutonium, and Americium in Aqueous Solutions. No. TID-26506. Los Alamos Scientific Lab., N. Mex.(USA), 1975.
-
[20]
Elbs K, Schönherr O. Zeitschrift für Elektrochemie, 1895, 2(12):245~252. doi: 10.1002/bbpc.18950021202
-
[21]
Kolthoff I M, Miller I K. J. Am. Chem. Soc., 1951, 73(7):1~30.
-
[22]
Moore F L. Anal. Chem., 1963, 35(6):715~719. doi: 10.1021/ac60199a010
-
[23]
Mincher B J, Law J D, Goff G S et al. Higher Americium Oxidation State Research Roadmap. No. INL/EXT-15-37534. Idaho National Lab.(INL), Idaho Falls, ID (United States), 2015.
-
[24]
Thompson R C, Appelman E H. Inorg. Chem., 1981, 7(20):2114~2115.
-
[25]
Yanir E, Givon M, Marcus Y. Inorg. Nucl. Chem. Lett., 1969, 5(5):369~372. doi: 10.1016/0020-1650(69)80082-6
-
[26]
Kazi Z, Nicolas G, Christl M et al. J. Radioanal. Nucl. Chem., 2019, 321(5):227~233.
-
[27]
Shultz W W. The Chemistry of Americium, ERDA Critical Review Series, TID-26971, 1976.
-
[28]
Tsushima S, Nagasaki S, Suzuki A. Sep. Sci. Technol., 1996, 31(17):2443~2453. doi: 10.1080/01496399608001058
-
[29]
Coleman J S, Armstrong D E, Asprey L B, et al. Purification of gram amounts of americium. No. LA-1975. Los Alamos Scientific Lab., N. Mex., 1955.
-
[30]
Stephanou S E, Nigon J P, Penneman R A. J. Chem. Phys., 1953, 21(1):42~45.
-
[31]
Gogolev A V, Tananaev I G, Myasoedov B F. Radiochemistry, 2004, 46(3):246~248. doi: 10.1023/B:RACH.0000031680.78106.98
-
[32]
Asprey L B, Stephanou S E, Penneman R A. J. Am. Chem. Soc., 1951, 73(12):5715~5717. doi: 10.1021/ja01156a065
-
[33]
Myasoedov B F, Lebedev I A, Khizhnyak P L, et al. J. Less Common Metals, 1986, 122(86):189~193.
-
[34]
Hobart D E, Samhoun K, Peterson J R. Radiochim. Acta., 1982, 31(3/4):139~146.
-
[35]
Lopez M J, Sheridan M V, Mclachlan J R, et al. Chem. Commun., 2019, 55(28):4035~4038. doi: 10.1039/C9CC00837C
-
[36]
Wada Y, Morimoto K, Goibuchi T, et al. J. Nucl. Sci. Technol., 1995, 32(10):1018~1026. doi: 10.1080/18811248.1995.9731810
-
[37]
Fukasawa T, Kawamura F. J. Nucl. Sci. Technol., 1991, 28(1):27~32. doi: 10.1080/18811248.1991.9731318
-
[38]
Tsushima S, Nagasaki S, Suzuki A. J. Nucl. Sci. Technol., 1995, 32(2):154~156. doi: 10.1080/18811248.1995.9731685
-
[39]
Sasaki S, Wada Y, Tomiyasu H. Prog. Nucl. Energ., 1998, 32(3/4):403~410.
-
[40]
Wada Y, Wada K, Goibuchi T, et al. J. Nucl. Sci. Technol., 1994, 31.(7):700~710. doi: 10.1080/18811248.1994.9735212
-
[41]
Tsushima S, Nagasaki S, Suzuki A. J. Nucl. Sci. Technol., 1995, 32(2):154~156. doi: 10.1080/18811248.1995.9731685
-
[42]
Shilov V P, Gogolev A V, Fedosseev A M. Russ. Chem. Bull., 2019, 68(7):1458~1459. doi: 10.1007/s11172-019-2578-0
-
[43]
Enokida Y, Suzuki A. J. Nucl. Sci. Technol., 1989, 26(8):770~776. doi: 10.1080/18811248.1989.9734381
-
[44]
王辉, 刘方, 魏艳等.应用声学, 2013, 32(1):61~65. http://www.cnki.com.cn/Article/CJFDTotal-YYSN201301014.htm
-
[45]
Nikonov M V, Shilov V P, Krot N N. Radiokhimiya, 1989, 31(5):23~26.
-
[1]
-
-

 下载:
下载:

 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 11
- 文章访问数: 805
- HTML全文浏览量: 251
 下载:
下载:
