CAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
b.
University of Chinese Academy of Sciences, Beijing 100049, China
c.
Zhang Dayu School of Chemistry, Dalian University of Technology, Dalian 116024, China
* Corresponding author. E-mail address: qinggy@dicp.ac.cn (G. Qing).
Received Date:
07 September 2022 Accepted Date:
25 December 2022 Revised Date:
18 December 2022 Available Online:
15 August 2023
Abstract:
Nature chooses phosphorylation as a key modification to modulate and program the functions of proteins. Various phosphorylated peptides (PPs) have been widely identified and investigated by biologists, but the possibility that PPs could become a building unit for artificial materials is neglected. Here we report for the first time a supramolecular assembly of PPs with the assistance of dysprosium ions (Dy3+). Dy3+ bridges multiple phosphate groups in double-phosphorylated peptides (di-PPs), and braid these peptide chains into nanofibers. The assembly occurs inside nanochannels and blocks the channels, leading to prominent "ON–OFF" switching in transmembrane ionic current. The di-PPs' assembling process could be dynamically regulated by the addition or deletion of phosphate groups under the control of kinases or phosphatases. This study proves the huge potential of PPs being utilized as materials via self-assembling, which will promote the design of novel bio-inspired artificial materials and devices.
Self-assembly of biological macromolecules such as molecular motors, enzymes, and subcellular structures is critical in life activities [1]. In the past decade, researchers have developed various promising applications from the fabrication of biomolecule self-assemblies [2–4]. Research of self-assembling peptides (e.g., Aβ peptides [5]) has been covering many applications, including healing materials [6,7], biosensors [8], enzyme substrates [9], drug delivery [10] and nanoelectronics devices [11]. Nevertheless, the further development of self-assembling peptides is still hampered by the relatively unpredictable conformation of peptides for now [12]. Till now, the development of self-assembling peptides has mainly contributed to the studying and mimicking of natural peptide sequences. Nature has evolved complex and various protein structures, but most of these structures are based on a few elementary binding modes: mainly α-helices and β-sheets [13]. The relatively conservative binding modes in nature may also limit the exploration of novel self-assembling peptides.
Phosphorylation is one of the most fundamental, common, and important mechanisms of protein functioning [14]. The high negative charge, the large hydrated shell of the phosphate group, and its relatively balanced energetics make phosphorylation a facile and reversible modification that brings substantial conformational and functional change to biomolecules [15]. The process of phosphorylation fulfils the necessity of easily and selectively regulating the function of proteins, and endows such process with specificity and controllability [16]. PPs are considered crucial regulatory modules in organisms, and current research on phosphorylation concentrates on investigating the regulatory mechanisms of phosphorylation in signal pathways and their influence on various diseases, particularly cancers [17]. Nevertheless, the research using phosphorylated peptides as self-assembly materials has been rare [18,19], and in the limited studies, the phosphate groups did not govern the binding interaction but rather acted mainly as hydrophilic groups. The lack of phosphorylation-governed self-assembly examples in nature may prevent the researchers from exploiting the appealing properties of phosphate groups as stated above. Since phosphorylation offers highly specific and flexible sites of interaction and recognition for proteins in nature [20], we prelude that, a self-assembling system of phosphorylated peptides built on the selective interaction of phosphate groups could circumvent the disadvantages of current self-assembling peptides and possess superior programmability and specificity [21].
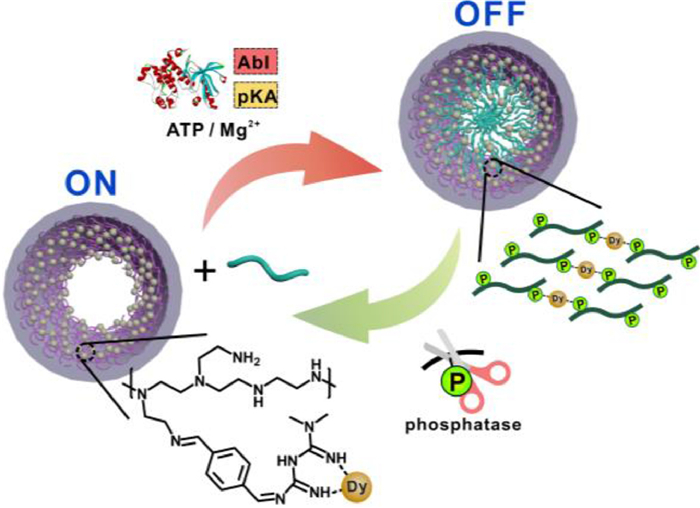
Here we report a supramolecular assembly of PPs based on the interaction between phosphate groups and Dy3+ (Scheme 1). This phenomenon was observed unexpectedly in nanochannels in the form of transmembrane ionic current blockages [22,23]. Initially, nanochannels were used as a detection platform for PPs. Polyethylene terephthalate (PET) foils with conical nanochannels were prepared according to the method developed by Apel [24]. In short, the PET foil was irradiated with heavy metal ions beforehand and treated with UV, and then etched with alkaline from one side, generating conical nanochannels with an average pore base diameter of ∼600 nm and an average pore tip diameter of 22 nm. To construct a PP recognition system, metformin (with its dual-guanidine group capable of binding PPs) was grafted onto polyethyleneimine (PEI) chain using 1, 4-phthalaldehyde as a linker, abbreviated as PEI-g-Met (Figs. S1 and S2 in Supporting information), the grafting ratio of Met in PEI was 5.1%), then the polymer was immobilized on the inner walls of the nanochannels via an amine-carboxyl coupling reaction (Figs. S3-S5 in Supporting information). Subsequently, a small piece of modified PET membrane was mounted between two Teflon cells for the measurement of transmembrane ionic current (inset of Fig. 1A) [25].
Scheme 1
Scheme 1.
Illustration of the assembly of Dy3+ and PPs inside nanochannels. A layer of Dy3+ was absorbed on the walls of the nanochannels via the modified PEI-g-Met polymers. Then, the addition of PPs caused assembly between Dy3+ and PPs, leading to the blockage of the nanochannels. Phosphatase or kinases can control the degree of phosphorylation on the peptides, and in turn modulate the assembly process.
Figure 1.
(A) I-V plots of PEI-g-Met modified nanochannels in response to different concentrations of Dy3+ and 2pS. The modules were filled with 0.01 mol/L NaCl solution before adding the analytes. The experiments were carried out at 25 ℃ unless specifically mentioned. (B) Comparison of ionic current decrease (I/I0 at + 2 V) of the nanochannels after additions of Dy3+, 2pS, or both analytes (5 × 10−5 mol/L), respectively. Concentration-dependent ionic current decreases of the nanochannels in response to the additions of Dy3+ with three di-PPs (C) or with the corresponding single-PPs (D). Insets show the peptide sequences of PPs, the phosphate-modified amino acids are indicated by red characters. (E) Concentration-dependent current decreases of the nanochannels in response to the additions of 2pS and other metal ions replacing Dy3+. SEM images capturing the base pore sides of the nanochannels before (F) and after (G) the assembly of Dy3+ and 2pS.
A series of PPs, including single PPs and di-PPs, were tested for the nanochannels. The di-PPs showed approximately 30% decreases in ionic current at the concentrations of 10–5 mol/L, and the single PPs showed about 10% decreases in current (Fig. S6 in Supporting information). In attempting to improve the performance, Dy3+ entered our scope of interest. It had been proved to bind selectively with metformin [26], and we anticipated that the incorporation of Dy3+ could provide an additional binding with PPs and possibly amplify the response. Fig. 1A displays the current-voltage (I-V) curves of the nanochannel upon the additions of 2pS (i.e., HpSPIAPSSPpSPK) and Dy3+. In this work, the concentrations of Dy3+ and PPs were kept equimolar unless specifically mentioned (Fig. S8 in Supporting information). The decrease of the ionic current was neglectable when the concentrations of 2pS and Dy3+ were lower than 10–7 mol/L, but the current started to decrease rapidly when their concentrations reached 10–6 mol/L. Finally, the ionic current decreased by 94.4% at the concentration of 5 × 10–5 mol/L. The addition of Dy3+ or 2pS alone into the polymer-modified nanochannel cut down about 28% of the original current at the concentration of 5 × 10–5 mol/L (Fig. 1B). However, at the same concentration, the addition of both Dy3+ and 2pS caused a much more drastic decrease to the ionic current. Interestingly, the decrease of the current was not proportional to the concentration of the analytes, but rather an abrupt change when a certain critical concentration was reached.
Phosphorylation could occur on three kinds of amino acid sites: Serine (pS), Threonine (pY), and Tyrosine (pT) [27]. To investigate how the type of phosphorylation sites and the number of phosphate groups influence the assembly of PPs with Dy3+, several other peptides were tested. As shown in Fig. 1C, PEI-g-Met modified nanochannel devices display evidential ionic current response to the additions of both di-PPs and Dy3+. Differently, the assembly between Dy3+ and 2pT or 2pY caused a relatively smaller decrease in the current, reaching 78.9% for 2pT and 80.7% for 2pY at the concentrations of 5 × 10–5 mol/L. Regarding the number of the phosphate group, di-PPs could aggregate with Dy3+ and block the nanochannel, by comparison, single PPs only resulted in slight decreases in the ionic current (Fig. 1D). We presumed that di-PPs tended to bind with multiple Dy3+ and form cross-linked polymer network; by comparison, single PPs could only bind with one Dy3+ per molecule, and thus failed to form polymer aggregates. This selectivity of the co-assembling effect towards di-PPs has alluring potential in phosphorylation proteomics, since double-site and multi-site phosphorylation plays a critical role in the modulation of signal pathways.
Some other metal ions were tested in replacement of Dy3+, as shown in Fig. 1E. Most other ions including Fe3+ and Ca2+ showed much less responses than Dy3+, while Tb3+, another lanthanide, showed comparable co-assembling performance (Fig. S7 in Supporting information). Rare earth metals have larger ionic radii, and among the list of rare earth metals, Tb3+ and Dy3+ are special in that they are relatively "harder" ions. We speculated that both chemical hardness and ionic radius are crucial to the interaction. Besides, Dy3+ also had strong binding with metformin groups in the grafted polymers [26], provoking the 2pS–Dy3+ aggregates to grow on the polymer layers and block the nanochannels. We also tested nanochannels with different pore diameters, and the results showed that the nanochannels with either too-large or too-small tip diameters exhibited less current response (Fig. S9 in Supporting information). When the tip diameter increased from 22 nm to 34 nm, the ionic current change became smaller, which indicated that the 2pS–Dy3+ aggregates could not easily block the large nanochannels. By contrast, the response of the nanochannels with smaller tip sizes of 14 nm was also less obvious, which could be reasonably attributed to the increased difficulty of the polymer to be functionalized inside the smaller nanochannels, as well as the difficulty of the diffusion of 2pS.
Scanning electron microscopy (SEM) images directly capturing the morphology of the nanochannels' base sides (Figs. 1F and G) displayed that the nanochannels were blocked after the addition of Dy3+ and 2pS. Therefore, we speculated that 2pS might aggregate with Dy3+ in the inner space of the nanochannels. A layer of Dy3+ was absorbed on the walls of the nanochannels via the modified PEI-g-Met polymer firstly, which could interact with the phosphate groups on 2pS. As the concentration gradually increased, the aggregation between Dy3+ and 2pS started from the wall and expanded to the whole channel, leading to a sudden decrease in the ionic current response [28].
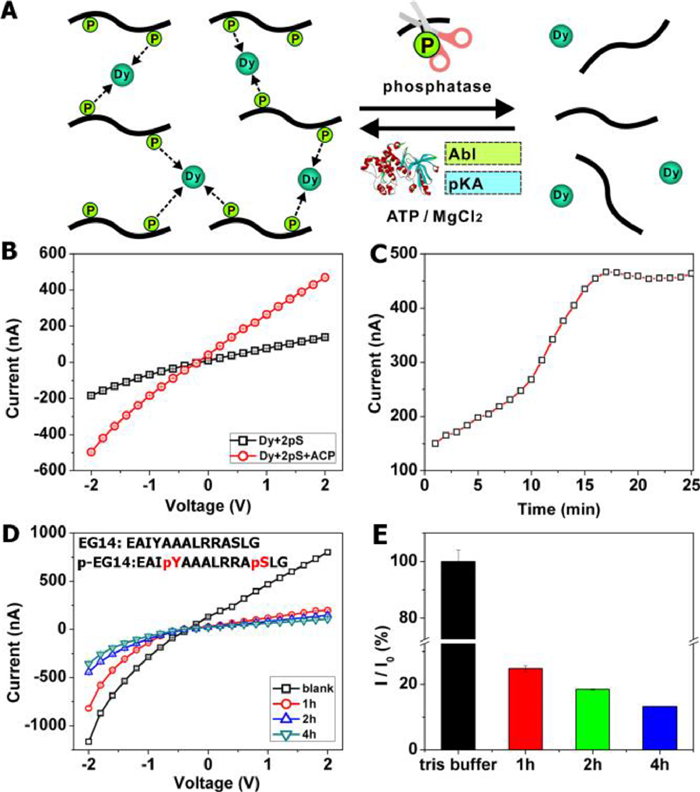
The assembly process between di-PPs and Dy3+ could be dynamically controlled by phosphatases and kinases, as illustrated in Fig. 2A. Acid phosphatase (ACP) is an enzyme that specifically catalyzes the hydrolysis of phosphate groups on PPs [29]. If the assembling was determined by the interactions between phosphates and Dy3+, the addition of ACP would cut off the phosphate groups in PPs and thus lead to the breakage of di-PP–Dy3+ assembly. To prove this assumption, the nanochannels were first blocked by 2pS and Dy3+, then ACP was added to the solution. The ionic current exhibited a substantial increase, as shown in Fig. 2B. The current gradually recovered from 150 nA to 467 nA in about 15 min and kept stable afterward (Fig. 2C), which inferred that the assembly was disintegrated after the addition of ACP, and proved our speculation.
Figure 2
Figure 2.
(A) Schematic illustration of the phosphatase or kinase catalysis experiment. ACP cuts off the phosphate groups in di-PPs and leads to the breakage of di-PPs–Dy3+ assembly, while kinases provoke the assembly of Dy3+ and the peptide by adding phosphate groups to it. (B) I-V plots of 2pS–Dy3+ assembled nanochannels before (black) and after (red) addition of ACP. The nanochannels were blocked with 2pS–Dy3+ assembly, then 40 U/mL of ACP was added to the solution. (C) Time-dependent ionic current increases after the addition of ACP. (D) I-V plots of the nanochannels responding to Dy3+ and the mixture of EG-14 with two kinases (Abl and PKA) at different reaction times. The mixture solution also contained ATP (1 mmol/L), DTT (2 mmol/L), MgCl2 (10 mmol/L), and NaCl (10 mmol/L) in Tris-HCl buffer. In both experiments, the reaction temperature was kept at 37 ℃. (E) Relative current decreases for different reaction times in (D).
On the other hand, kinases can catalyze the phosphorylation of peptides with specific amino acid sequences, and it is expected that adding kinase to the corresponding peptide would provoke the assembling of Dy3+ and the peptide. To test the effect of kinases, a peptide EAIYAAALRRASLG (EG14) was introduced to this experiment. EG14 contains two active sites at the 4th Tyr (Y) and 12th Ser (S), which could be phosphorylated by two kinds of kinases, namely Abl (Tyrosine-protein kinase ABL1) and PKA (protein kinase A), respectively [30]. The function of kinases is the reverse of phosphatases; thus, it was expected that the kinases could facilitate the assembly of EG14. Initially, the nanochannels maintained an "open" state (black line in Fig. 2D) when EG14 and Dy3+ were added. Upon the addition of Abl and PKA simultaneously, the ionic current gradually decreased (Fig. 2D). After 4 h, the current fell to 11.8% of the original value (Fig. 2E). High-resolution mass spectrum (peak of [2p-EG14+Mg]2+, Fig. S10 in Supporting informtion) confirmed the formation of double phosphorylated EG14 product (pEG14). Therefore, the sharp decrease of the ionic current could be reasonably attributed to the emergence of pEG14–Dy3+ assembly inside the nanochannels. However, the reversibility of the off-regulation and on-regulation was limited due to the damage caused by the decomposition process of the assemblies to the polymer layer. To conclude, by introducing phosphatase or kinases into the system, the degree of phosphorylation of peptides could be controlled, providing a powerful tool to modulate the supramolecular assembling.
The co-assembled capacities of PPs with Dy3+ were evaluated on the surface of a biosensor using a label-free Corning Epic technology assay, which could effectively detect the aggregation in solution via the variation of refraction light (Fig. S11 in Supporting information) [31]. The addition of equimolar mixtures of 2pS and Dy3+ led to a change in the refractive index, which displayed a clear concentration-dependent response at 30 min (Fig. 3A). The signal spiked up when the concentration reached 5 × 10−6 mol/L (Fig. 3B), which indicated the formation of the assembly in the solution. In addition, the Epic response change induced by 2pS and Dy3+ (140 ppm) was remarkably larger than that induced by 1pS and Dy3+ (20 ppm), demonstrating a stronger co-assembling capacity of 2pS with Dy3+. Atom force microscopy (AFM) three-dimensional image described the morphology of the 2pS–Dy3+ assemblies in bulk solution. A few thin nanofibers with an average length of 800 nm, a width of 20 nm, and a height of 8 nm could be clearly observed (Fig. 3C).
Figure 3
Figure 3.
(A) EPIC optical responses for equimolar mixture solutions of Dy3+ and 2pS at different concentrations in H2O, from 0.1 µmol/L to 1 mmol/L. (B) Concentration-dependent EPIC responses for 2pS–Dy3+ and 1pS–Dy3+ assembly. (C) AFM 3D image of the 2pS–Dy3+ assembly at 10−3 mol/L concentration. (D) The 31P NMR spectra of pSer and its equimolar mixture with Dy3+ in D2O at room temperature. (E) EPR spectra of Dy3+, its mixture with PEI-g-Met, or with both the polymer and 2pS. The samples were analyzed at 77 K. (F) Possible interaction model between Dy3+ and 2pS obtained from quantum chemistry calculation. The distances between Dy3+ and oxygen atoms on the phosphate groups are 2.197, 2.173, 2.269 and 2.163 angstroms, respectively.
The influence of Dy3+ on the phosphorus atom of PPs was characterized by 31P NMR (Fig. 3D). O-phosphorylated serine (pSer), a core unit in 2pS, was used in this experiment to strengthen the 31P signal. The phosphorus atom in pSer had a sharp peak at –0.4 ppm, which shifted to –12 ppm and broadened after interaction with Dy3+. The Dy3+ ion caused a decrease in the electron density of the phosphorus atom, leading to deshielding effect and the shift of the coordinated phosphate resonances towards downfield. The magnitude of the shift was consistent with previous reports (10-14 ppm) [32]. The results of the 31P NMR spectra provided direct evidence for the coordination effect of Dy3+ on the phosphate group. On the other hand, Dy3+ possesses a strong paramagnetic effect due to its half-filled 4f7 orbit, therefore, electron paramagnetic resonance (EPR) could specifically define the interaction between Dy3+ and the surrounding molecules by monitoring the electron spin of Dy3+. Fig. 3E shows the EPR spectra of Dy3+, its complex with PEI-g-Met, and with both PEI-g-Met and 2pS. The peak at ∼3090G (corresponding to a g-value of 2.177) could be attributed to the resonance peak of Dy3+. After the addition of PEI-g-Met, the peak decreased prominently, and when 2pS was added to the mixture, the positive peak of Dy3+ nearly vanished, indicating a stronger interaction between Dy3+ and the phosphate groups.
Theoretical simulation of the interaction mechanism of Dy3+ and pSer was further conducted (Fig. 3F). The calculated distances between Dy3+ and oxygen atoms on the phosphate groups were 2.20, 2.17, 2.27 and 2.16 angstroms respectively. The results of the calculation indicated the formation of stable coordination bonds between Dy3+ and 2pS, which further verified the experimental data.
In summary, an assembly of di-PPs with Dy3+ by the coordination between pSer and Dy3+ was reported. This work raises the possibility of PPs being potential artificial materials, and expands the realm of artificial biomolecule self-assembly into PPs, which vastly increased the information loaded and the possible conformations of the self-assembly [33]. The high precision and controllability endowed by kinases or phosphatases open up a broad space for imagination about the applications of PPs assembly. Besides, the paramagnetic properties of Dy3+ also allow potential applications in the construction of novel nanoscale magnetic devices [34], and corresponding works are being carried out in our group.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 21922411 and 22174138), DICP Innovation Funding (No. DICP-RC201801 and I202008), and the Dalian Outstanding Young Scientific Talent (No. 2020RJ01).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.108106.
T. Pugh, F. Tuna, L. Ungur, et al., Nat. Commun. 6 (2015) 7492–7499. doi: 10.1038/ncomms8492
Scheme 1
Illustration of the assembly of Dy3+ and PPs inside nanochannels. A layer of Dy3+ was absorbed on the walls of the nanochannels via the modified PEI-g-Met polymers. Then, the addition of PPs caused assembly between Dy3+ and PPs, leading to the blockage of the nanochannels. Phosphatase or kinases can control the degree of phosphorylation on the peptides, and in turn modulate the assembly process.
Figure 1
(A) I-V plots of PEI-g-Met modified nanochannels in response to different concentrations of Dy3+ and 2pS. The modules were filled with 0.01 mol/L NaCl solution before adding the analytes. The experiments were carried out at 25 ℃ unless specifically mentioned. (B) Comparison of ionic current decrease (I/I0 at + 2 V) of the nanochannels after additions of Dy3+, 2pS, or both analytes (5 × 10−5 mol/L), respectively. Concentration-dependent ionic current decreases of the nanochannels in response to the additions of Dy3+ with three di-PPs (C) or with the corresponding single-PPs (D). Insets show the peptide sequences of PPs, the phosphate-modified amino acids are indicated by red characters. (E) Concentration-dependent current decreases of the nanochannels in response to the additions of 2pS and other metal ions replacing Dy3+. SEM images capturing the base pore sides of the nanochannels before (F) and after (G) the assembly of Dy3+ and 2pS.
Figure 2
(A) Schematic illustration of the phosphatase or kinase catalysis experiment. ACP cuts off the phosphate groups in di-PPs and leads to the breakage of di-PPs–Dy3+ assembly, while kinases provoke the assembly of Dy3+ and the peptide by adding phosphate groups to it. (B) I-V plots of 2pS–Dy3+ assembled nanochannels before (black) and after (red) addition of ACP. The nanochannels were blocked with 2pS–Dy3+ assembly, then 40 U/mL of ACP was added to the solution. (C) Time-dependent ionic current increases after the addition of ACP. (D) I-V plots of the nanochannels responding to Dy3+ and the mixture of EG-14 with two kinases (Abl and PKA) at different reaction times. The mixture solution also contained ATP (1 mmol/L), DTT (2 mmol/L), MgCl2 (10 mmol/L), and NaCl (10 mmol/L) in Tris-HCl buffer. In both experiments, the reaction temperature was kept at 37 ℃. (E) Relative current decreases for different reaction times in (D).
Figure 3
(A) EPIC optical responses for equimolar mixture solutions of Dy3+ and 2pS at different concentrations in H2O, from 0.1 µmol/L to 1 mmol/L. (B) Concentration-dependent EPIC responses for 2pS–Dy3+ and 1pS–Dy3+ assembly. (C) AFM 3D image of the 2pS–Dy3+ assembly at 10−3 mol/L concentration. (D) The 31P NMR spectra of pSer and its equimolar mixture with Dy3+ in D2O at room temperature. (E) EPR spectra of Dy3+, its mixture with PEI-g-Met, or with both the polymer and 2pS. The samples were analyzed at 77 K. (F) Possible interaction model between Dy3+ and 2pS obtained from quantum chemistry calculation. The distances between Dy3+ and oxygen atoms on the phosphate groups are 2.197, 2.173, 2.269 and 2.163 angstroms, respectively.

 DownLoad:
DownLoad:



 下载:
下载:





 下载:
下载:
