
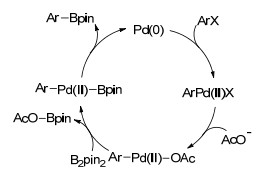
Scheme 1.
卤代芳烃钯催化硼基化机理
Scheme 1.
Mechanism of Pd-catalyzed borylation of aryl halides

芳基硼酸(酯)是碳-碳键及碳-杂原子键构建的重要试剂, 特别是芳基硼酸酯具有高稳定性、低毒性、易操作等优点, 被广泛应用于药物和活性天然产物合成中.此外, 在生物学、材料科学及医学研究中, 芳基硼酸已被广泛应用于糖类的传感器、酶的抑制剂及核苷和糖类的运输载体等[1].因此, 开发高效、温和的芳基硼酸(酯)合成方法一直都是化学家们关注的重点.
芳香硼酸(酯)的经典合成方法是通过格氏试剂或锂试剂和硼酸酯反应[2].然而对于分子中带有酯基、氰基、硝基、羰基等官能团的芳香卤代物来说, 无法通过有机金属试剂来直接制备芳香硼酸.而过渡金属催化硼基化反应因具有反应条件温和、选择性单一、官能团耐受性好等优点, 受到化学工作者的青睐.
1995年, Miyaura等[3]率先通过联硼酸频哪酯和卤代芳烃发生钯催化偶联反应制备了相应的芳基硼酸酯.随后发现除卤代芳烃外, 芳基三氟甲磺酸酯、芳基重氮盐等底物均可通过过渡金属催化的硼基化反应制得芳基硼酸酯.由于该反应选择性高、定位性强, 底物适用范围广, 因此已被广泛应用于有机硼酸类化合物的制备.后来科学家们还发展了包括镍、铜、锌、铁等过渡金属催化剂在内的芳基硼基化反应; 近五年来, 关于非金属催化、光诱导的芳基硼基化反应见诸报端. Murata[4]在2012年和Kwong小组[5]在2013年分别对芳香卤代物和芳基三氟甲磺酸酯过渡金属催化的硼基化反应进行了综述, 2017年Ito小组[6]综述了芳香卤代物金属及非金属催化的硼基化反应, 上述文章对芳香含氮化合物和羧酸类化合物硼基化反应及近期发展并未涉及. 2017年, 李鹏飞等[7]综述了非过渡金属参与的芳基硼基化反应, 同年, 严国兵课题组[8]总结了经自由基反应历程的芳基硼基化反应.本文从催化策略的角度出发, 综述了近二十年来金属、非金属催化及光诱导合成芳基硼酸类化合物的研究进展.鉴于碳氢活化硼化反应内容广泛且在最近已有一些重要综述文章[9], 故不在本文讨论范围之内.
1995年, Miyaura等[3]第一次报道了卤代芳烃(碘和溴)与联硼酸频那醇酯通过钯催化偶联反应来制备相应有机硼酸酯(Eq. 1).该方法化学环境温和, 有较强的底物适应性及官能团耐受性, 并且可通过进一步的Suzuki-Miyaura反应合成联芳化合物, 广泛应用于药学及材料科学[6].
|
|
(1) |
Miyaura提出了卤代芳烃的硼基化机理(Scheme 1), 催化循环过程经历氧化加成、金属转移和还原消除三个阶段, 首先是卤代芳烃与Pd(0)发生氧化加成反应生成Pd(Ⅱ)配合物Ar—Pd(Ⅱ)—X.然后Pd(Ⅱ)配合物与碱KOAc作用形成有机钯醋酸根化合物, 该中间体与联硼化合物发生金属转移反应生成硼和钯的配合物, 随后发生还原消除反应, 生成相应的芳基硼化合物及Pd(0).
选择合适的碱及溶剂对于硼基化反应来说至关重要.尽管KOAc等弱碱通常并不作为偶联反应的促进剂, 但KOAc却是该反应最合适的碱, 其他碱性略强的碱如K3PO4或K2CO3会进一步使ArB(OR)2和ArX发生Suzuki偶联反应.此外, 极性溶剂也有利于反应的进行[3].
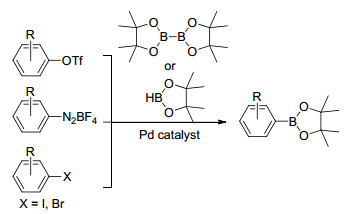
除卤代芳烃, 芳基三氟甲磺酸酯、芳基重氮盐也可作为底物制备芳基硼酸酯, 此外, 原子经济性较高的频哪醇硼烷也可以作为钯催化硼基化反应的硼源[10] (Scheme 2).
近些年, 化学家们主要通过开发新的配体, 使得氯代芳烃及含有大位阻取代的卤代芳烃的硼基化反应成为可能.
与溴代芳烃和碘代芳烃相比, 氯代芳烃虽然反应活性较低, 但更加经济易得.因此, 氯代芳烃代替溴代和碘代芳烃进行硼基化反应, 将可以大大降低反应成本, 有着更高的工业价值和经济价值.
2001年, Ishiyama等[11]采用富电子配体三环己基膦(PCy3), 钯催化剂Pd(dba)2, 以醋酸钾作碱, 1, 4-二氧六环作溶剂成功实现了氯代芳烃的硼基化.值得一提的是, 该催化体系还可以缩短溴代芳烃和芳基三氟甲磺酸酯的硼基化反应时间; 但是对于有邻位取代或者具有富电子基团的氯代芳烃, 该反应需要更高载量的催化剂和更长的反应时间(Eq. 2).
|
|
(2) |
2007年, Buchwald小组[12]报道了Pd2(dba)3与配体2-二环己基膦-2', 4', 6'-三异丙基联苯(XPhos)或2-双环己基膦-2', 6'-二甲氧基联苯(SPhos)组成的催化体系, 该体系高度稳定且催化活性较高, 可在2 h内以高产率完成氯代芳烃的硼基化, 而且使用配体SPhos可以在室温下完成反应.该方法底物适应性较广, 芳杂环氯代物也能以较高产率制备芳基硼酸酯(Eq. 3).随后, 该课题组[13]又采用PdCl2(CH3CN)2/SPhos催化体系, 利用更加经济的频哪醇硼烷作为硼酸化试剂成功实现氯代芳烃硼基化.
|
|
(3) |
已知的钯催化硼基化反应一般采用联硼酸频哪醇酯、频哪醇硼烷等作为硼基化试剂, 经过脱保护才能得到相应的芳基硼酸, 因此往往会引入二醇类化合物等副产物难以除去.基于该问题, 2010年, Molander等[14]使用配体XPhos, 首次实现四羟基二硼与氯代芳烃的钯催化偶联反应, 直接制备相应芳基硼酸; 该策略使用了原子经济性更高的四羟基二硼, 并且无须使用手套箱和无水溶剂, 操作简单, 且上述芳基硼酸可进一步转化为相应的硼酸酯或三氟硼酸盐(Scheme 3). Molander还采用该方法进行了一锅两步Suzuki偶联反应, 产率在55%~90%.
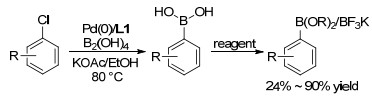
随后, Kwong小组[15]报道了吲哚基配体PPh2-Andole-phos参与的氯代芳烃硼基化反应, 该配体含有廉价的PPh2部分, 且活性较高, 能够高产率的完成多种氯代芳烃底物的硼基化, 最高收率可达97% (Eq. 4).
|
|
(4) |
2016年, Yorimitsu等[16]报道了氯代芳烃的钯催化硼基化反应, 在催化剂Pd(dba)3、配体SPhos、溶剂甲苯, 无碱参与的条件下, 可得到一系列芳基硼酸酯, 该方法可应用于碱性敏感基团(硅氧基、笏甲氧羰基等)取代的芳香氯代物的硼基化. Yamamoto课题组针对硼基转移这一关键步骤做了DFT计算研究, 结果表明硼原子与氯原子间强烈的相互作用促进了钯金属转移过程的进行, 使该反应能够发生(Eq. 5).
|
|
(5) |
由于卤代芳烃邻位取代基的位阻效应会阻碍Miyaura硼基化反应的发生, 含有较大位阻取代基的芳基硼酸类化合物往往难以合成. 2000年, Baudoin等[17]以2-(二环己基膦)联苯(Cy-JohnPhos)为配体, 合成了一系列的邻位取代的芳基硼酸酯, 并将该反应应用于一锅两步Suzuki反应, 合成了2, 2'-取代的联苯化合物(Eq. 6).
|
|
(6) |
2005年, Wang题组[18]发现在Pd(dppf)Cl2催化的作用下, 采用位阻相对较小的联硼酸新戊二醇酯代替联硼酸频那醇酯与邻位取代的卤代芳烃反应可以避免脱卤素产物的生成, 从而得到相应的芳基硼酸酯, 产率较高, 2-甲氧基-5-硝基溴代苯的硼基化反应产率由20%提升到72% (Eq. 7).
2011年, Tang小组[19]报道了联苯型单膦配体L5, 将它与低催化量的Bedford钯前驱物(0.5%~1% Pd)联用, 可以实现对大位阻取代溴代芳烃的硼基化, 得到了一系列邻位双取代的芳基硼酸酯, 产率可达59%~96% (Eq. 8).但是, 由于Bedford钯前驱物中的膦配体具有π-酸性, 氯代芳烃与钯催化剂的氧化加成难以进行, 该催化体系不能应用于氯代芳烃的硼基化反应.同年, Sawamura等[20]使用基于硅的单膦配体Silica-SMAP, 实现了多取代氯代芳烃的硼基化反应, 扩展了大位阻取代芳烃硼基化反应底物的范围(Eq. 9).
|
|
(7) |
|
|
(8) |
|
|
(9) |
由于钯催化剂价格昂贵, 价格相对便宜的镍催化剂便成为钯催化剂较好的替代品.因此, 镍催化的芳基硼基化反应研究有着重要的意义.
2000年, Tour等[21]报道了以三乙胺作碱, 在Ni(dppp)Cl2的催化下, 1, 3, 5-三溴苯与频哪醇硼烷发生偶联反应得到相应硼酸酯, 并随之经过酸解生成芳基硼酸(Eq. 10). 2008年, Percec小组[22]发现该反应体系可实现多种取代的碘代芳烃及溴代芳烃的硼基化反应.
随后Percec等[23]又采用Ni(dppp)Cl2和dppf的催化体系, 成功实现了氯代芳烃的硼基化, dppp和dppf混合磷配体显著提高了反应产率, 而且富电子及缺电子的氯代芳烃均可通过该法以较好产率得到相应硼酸酯产物, 该研究也证明了混合配体在催化反应中的潜力(Eq. 11).
|
|
(10) |
|
|
(11) |
2011年, Yamakawa小组[24]报道了氯代芳烃与联硼酸频哪醇酯镍催化偶联反应, 以NiCl2(PMe3)2作催化剂, CsF作碱, 四氢呋喃为溶剂, 在三甲基(2, 2, 2-三氟乙氧基)硅烷(TMSOCH2CF3)的参与下可以高产率地得到一系列相应的芳基硼酸酯.该催化体系官能团耐受性较好, 并且以邻位取代的氯代芳烃为底物时也有着良好催化效果, 产率高达78%~93% (Eq. 12).
|
|
(12) |
2013年, Molander小组[25]报道了卤代芳烃与四羟基二硼的镍催化偶联反应, 在以Ni(dppp)Cl2/PPh3为催化剂, N, N-二异丙基乙胺(DIPEA)作碱, 乙醇作溶剂的条件下, 多种官能团取代的氯代或溴代芳烃均可通过该法以较高产率得到相应的芳基硼酸, 而且多数反应可以在室温下完成(Eq. 13).
|
|
(13) |
二甲氨基是芳香环上重要的定位基团, 实现二甲氨基向其他官能团的转化在有机合成中有重要意义. 2015年, Itami小组[26]以N, N-二甲基芳胺为原料, 与三氟甲磺酸甲酯反应合成相应季铵盐, 随后在Ni(COD)2作催化剂, 正三丁基膦为配体, 叔丁醇钠作碱, 二氧六环作溶剂, 70 ℃条件下与联硼酸频哪醇酯偶联得到一系列相应的硼酸酯(Eq. 14).同年, 史壮志小组[27]采用N-杂卡宾配合物ICy为配体, Ni(COD)2为催化剂实现了N, N-二甲基芳胺到芳香硼酸酯的转化(Eq. 15).
|
|
(14) |
|
|
(15) |
尽管大量碳卤键(C—Cl、C—Br、C—I)的硼基化反应被纷纷报道, 但是由于C—F键键能较强, 氟代芳烃的硼基化反应难以实现. 2015年, Martin等[28]第一次报道了氟代芳烃镍催化的硼基化反应, 通过优化反应条件:以Ni(COD)2作催化剂, PCy3为配体, NaOPh作碱, 联硼酸新戊二醇酯为硼基化试剂, 四氢呋喃作溶剂, 110 ℃回流12 h可以取得最大收率(Eq. 16).
|
|
(16) |
2016年, Tobisu等[29]报道了芳基-2-吡啶基醚与联硼酸新戊二醇酯的镍催化偶联反应, 在催化剂NiCl2(DME)、配体PCy3•HBF4、K3PO4作碱、溶剂乙二醇二甲醚的条件下, 合成了一系列不同基团取代的芳基硼酸酯, 对于邻位取代芳烃该方法也有着较高的产率, 2-氟苯基硼酸酯的产率高达71% (Eq. 17).值得一提的是, 由于OPy是碳氢活化邻位定位基且在氧化条件下具有稳定性, 因此可以在进行邻位碳氢氧化反应后进一步制备相应的硼酸酯.
|
|
(17) |
芳酸及其衍生物广泛存在于自然界中, 是重要的有机合成中间体, 在农业、医药及化工等领域都有着广泛的应用, 近年来, 羧酸衍生物通过失去CO2引发的偶联反应在合成领域已显示出广阔的前景. 2016年, 史壮志课题组[30]报道了N-叔丁氧羰基活化的二级芳香酰胺在Ni(OAc)2•4H2O催化下的硼基化反应, 以ICy作为配体, K3PO4作碱, 甲苯和环己烷作为混合溶剂, 150 ℃条件下发生反应, 收率最高可达91%, 且该催化体系对氮氧杂环取代的底物也有良好的适应性.他们通过对反应关键中间体B、C的分离及结构确证, 提出了活化酰胺C—N实现脱羧偶联的反应机理(Scheme 4).
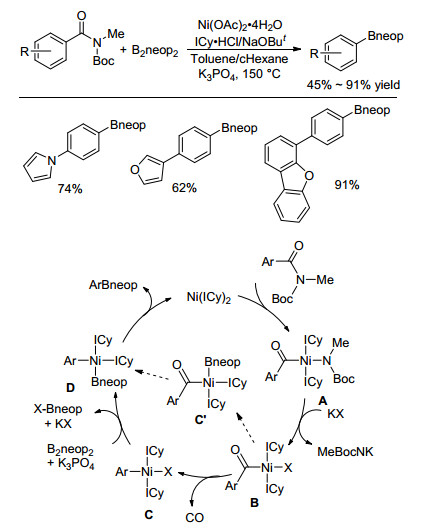
同年, 史壮志小组[31]又以N-杂环类卡宾ICy为配体, 使用催化剂Ni(COD)2成功实现了芳香羧酸酯脱羧硼化反应, 经条件筛选发现, Na2CO3/NaCl (V:V=1:1)作为添加物, 二氧六环为溶剂, 160 ℃条件下收率最高(Eq. 18).几乎同一时间, Rueping小组[32]报道了膦配体三正丁基膦参与的镍催化芳香羧酸酯的脱羧硼化反应, 该催化体系有着好的官能团耐受性, 高选择性(Eq. 19).
|
|
(18) |
|
|
(19) |
由于铜具有廉价、低毒的特点, 而且其反应条件温和, 反应配体也相对简单, 因此铜催化的硼基化反应有着巨大的工业潜力, 成为构建芳基硼酸酯的一条重要思路.
2006年, 马大为课题组[33]第一次报道了碘代芳烃铜催化的硼基化反应, 采用CuI作为催化剂, NaH作碱, 室温条件下完成了碘代芳烃与频哪醇硼烷的偶联反应.该方法对于取代基位于邻位、间位、对位以及是否为供电子或吸电子均无明显的选择性, 产率能达61%以上, 然而该方法并不适用于溴代芳烃的偶联反应.此外由于频哪醇硼烷与NaH具有强的还原性, 含有羰基取代的碘代芳烃会转变为相应的还原副产物(Eq. 20).
|
|
(20) |
2009年, Marder等[34]将催化量CuI与三正丁基膦配体应用于卤代芳烃的硼基化反应, 芳香碘代物及芳香溴代物均能发生反应, 得到相应的硼酸酯化合物.值得一提的是, 对于具有空间位阻的芳香碘代物及芳香溴代物, 该体系催化的硼基化反应也有着较高的产率(Eq. 21).
|
|
(21) |
2012年, 严国兵等[35]报道了无配体的卤代芳烃与联硼酸频那醇酯铜催化偶联反应. t-BuONa作碱, DMF为溶剂, 在CuBr的催化下, 得到了一系列芳基硼酸酯. DMF被认为充当了配体的角色, 对关键中间体硼铜复合物起稳定作用(Eq. 22).
同年, 于海涛小组[36]报道了铜催化的芳香重氮盐与联硼酸频哪醇酯的偶联反应, 经条件筛选, CuBr作催化剂, 乙腈和水作为混合溶剂[V(CH3CN)/V(H2O)=2/1]时, 室温条件下产率最高.该催化体系对多数官能团都有较好的容忍性, 但是对于具有位阻的芳香重氮盐, 反应产率较低, 2-硝基苯四氟硼酸重氮盐硼基化反应产率仅有24% (Eq. 23).
|
|
(22) |
|
|
(23) |
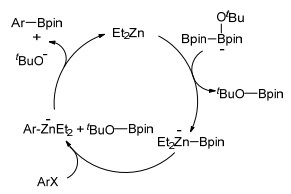
有机锌化合物的出现为有机化学及有机金属化学开辟了新的领域, 2013年, Takita等[37]报道了有机锌催化的卤代芳烃的硼基化反应.经过条件筛选, Et2Zn作催化剂, 四氢呋喃为溶剂, 在叔丁醇钠的参与下, 75 ℃回流碘代芳烃与联硼酸频哪醇酯反应得到相应芳基硼酸酯.溴代芳烃在相同催化体系, 更高温度(120 ℃)下也能得到相应产物.对于吸电子及给电子和大位阻官能团取代的卤代芳烃, 该方法均能取得较高的产率(Eq. 24). Takita等又通DFT模型计算给出了合理的反应机理(Scheme 5).
|
|
(24) |
2014年, Marder小组[38]发现卤代芳烃在ZnBr2及N-杂环卡宾(NHC)类配体催化下可与联硼酸频哪醇酯反应得到相应的芳基硼酸酯.经条件筛选, MeOK作碱, IMes为配体, 甲基叔丁基醚作溶剂时产率最高.该催化体系对溴代及碘代芳烃均有较好的活性, 并对于取代基位于邻位、间位、对位以及是否为供电子或吸电子均无明显的选择性(Eq. 25).随后, Marder及其同事[39]发现ZnCl2/4, 4'-二叔丁基-2, 2'-联吡啶(L9)或ZnBr2/5, 5'-二甲基-2, 2'-联吡啶(L10), 在MeOK作碱, 甲基叔丁基醚作溶剂的条件下, 可以活化碘代或溴代芳烃中的C—X键及邻位C—H键得到相应的1, 2-芳基二硼酸酯(Eq. 26).
|
|
(25) |
|
|
(26) |
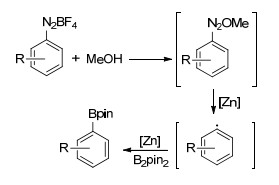
2017年, Wu小组[40]报道了锌催化的芳基重氮盐的硼基化反应, 在催化剂Zn(ClO4)2、溶剂甲醇, 40 ℃加热的条件下, 四氟硼酸芳基重氮盐与联硼酸频哪醇酯反应得到相应的芳基硼酸酯.该催化体系下, 供电子及吸电子取代的四氟硼酸芳基重氮盐为底物时均能获得较高的收率, 4-硝基苯四氟硼酸重氮盐的硼基化产率高达94% (Eq. 27).该反应被认为是自由基反应历程, 四氟硼酸芳基重氮盐首先与甲醇反应得到中间体1-甲氧基-2芳基二氮烯, 该中间体不稳定, 在锌催化剂的参与下可分解为相应的芳基自由基, 芳基自由基再与联硼酸频哪醇酯反应得到相应的芳基硼酸酯(Scheme 6).
|
|
(27) |
2012年, Chatani小组[41]报道了[RhCl(COD)]2和配体4, 5-双(二苯基膦)-9, 9-二甲氧基氧杂蒽(xantphos)催化芳腈与联硼酸新戊二醇酯的交叉偶联反应, 该反应以1, 4-二氮杂二环[2.2.2.]辛烷(DABCO)为碱, 甲苯为溶剂100 ℃条件下加热15 h.该催化体系有着较好的官能团耐受性及底物适应性, 邻位官能团取代芳腈和杂环芳腈仍以较高产率转化为相应芳香硼酸酯(Eq. 28). 2015年, Chatani等[42]又报道了[RhCl(COD)]2催化下芳基-2-吡啶基醚的硼基化反应, 这对OPy在碳氢活化领域中的广泛应用有着重要的意义(Eq. 29).
|
|
(28) |
|
|
(29) |
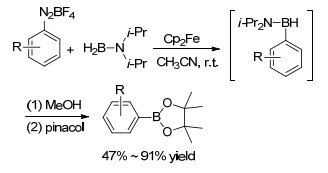
2013年, Pucheault课题组[43]报道了二茂铁(Cp2Fe)催化的芳基重氮盐与N, N-二异丙基氨基硼烷偶联反应, 乙腈是反应最适合的溶剂, 芳基重氮盐在低催化量的Cp2Fe (0.1 mol%)的催化下即可以较高产率得到相应的芳基硼烷类化合物, 该产物可以转变为应用更为广泛的硼酸酯、硼酸或硼酸盐的化合物(Scheme 7).
2014年, Bedford等[44]采用[FeCl2(dppe)] (5 mol%)、t-BuLi (1.4 equiv.)、MgBr (20 mol%)体系, 实现了卤代芳烃与联硼酸频哪醇酯的偶联反应, 但是产率较低. 2017年, Yoshida等[45]报道了1-氯代萘铁催化的硼基化反应, 以三乙酰丙酮铁[Fe(acac)3]为催化剂, t-BuOK作碱, 甲苯为溶剂, 110 ℃回流条件下氯代芳烃与联硼酸频那醇酯发生偶联反应得到相应硼酸酯; 但是反应的底物适应性较差, 硝基、氰基和羧基取代的氯代苯并不能转化为相应的产物(Eq. 30).
|
|
(30) |
2016年, 胡爱国、黄正等[46]首次报道了金属钴催化的芳香卤代物硼基化反应, 他们合成了两个Co催化剂前驱物A、B, 经条件优化, 当以B作催化剂, 甲基锂作催化剂活化剂, 甲醇钾作碱, 甲基叔丁基醚为溶剂时该反应可取得最高收率.该催化体系有着较好官能团耐受性和底物适应性, 值得一提的是, 芳香氯代物和类芳香卤代物(磺酸酯、重氮盐)也可在该体系下得到相应的芳香硼酸酯(Eq. 31).
|
|
(31) |
同年, Komeyama小组[47]报道了金属钴及铬联合催化的芳香有机锌化合物硼基化反应, 他们发现以甲氧基硼酸频哪醇酯为硼源, CoBr2和CrCl3(THF)3为催化剂, xantphos为配体, 四氢呋喃为溶剂, 在锌粉和三甲基氯硅烷的参与下收率最佳.由于芳香卤代物可以在CoBr2/ xantphos的催化下转变为相应的芳香锌化合物, 因此来源更广、使用更加方便的芳香卤代物也可直接在该催化体系下反应制备相应的芳香硼酸酯.值得一提的是, 当使用空间位阻相对较小的甲氧基硼酸新戊二醇酯代替甲氧基硼酸频哪醇酯时, 该反应的产率有了大幅提升(Eq. 32).
|
|
(32) |
在过去二十多年间, 过渡金属催化的硼基化反应得到了迅速发展, 但是过渡金属催化存在价格昂贵、污染环境及过渡金属残留等问题, 在制药工业中的应用受到了限制[6].因此, 廉价易得、毒性更小的非金属催化反应具有很高的优越性, 更具有吸引力.
芳胺类化合物可由相应的硝基芳香化合物经还原得到, 是一种价格低廉、种类丰富的化工及制药原料. 2010年王剑波等[48]报道了第一个非金属催化的芳胺类化合物的硼基化反应.起始原料芳胺与亚硝酸特丁酯(t-BuONO)反应制得相应芳香重氮盐, 该重氮盐再与联硼酸频哪醇酯反应得到产物芳基硼酸酯, 芳基硼酸酯不经纯化可直接进一步进行钯催化的Suzuki偶联反应.该反应对多数官能团都有着较好的容忍性, 间位或对位有吸电子基团时, 产率相比较高, 而当邻位有取代基团时, 反应产率有所降低.此外, 该反应可在室温下进行, 并且无需进行无氧操作(Eq. 33).
|
|
(33) |
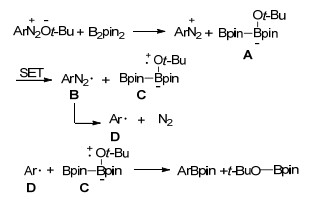
2012年, 莫凡洋和王剑波等[49]又对上述硼基化反应条件进行了优化, 并且扩大了反应底物的范围.他们通过条件优化发现, 当反应温度提升到80 ℃时, 无须添加BPO, 反应产率有大幅度提升, 而且一些杂环芳胺也可以转化为相应的硼酸酯, 该类硼酸酯在药物发现和制药工业中有着广泛的应用.缺电子的杂环芳胺硼基化反应产率较高, 而富电子杂环芳胺在t-BuONO的存在下因易被氧化而导致硼基化反应产率降低. Mo等基于反应, 提出了合理的反应机理:叔丁氧基负离子首先与联硼酸频哪醇酯相互作用得到四配位的硼中间体A, 之后中间体A与芳香重氮盐之间发生单电子转移生成自由基B, 然后释放一分子N2得到芳基自由基D, D与中间体C反应得到硼基化产物(Scheme 8).
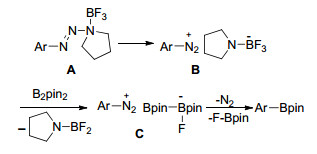
同年, Yamane等[50]报道了三氟化硼乙醚(BF3•Et2O)介导的芳基三氮烯的硼基化反应.芳基三氮烯可以看作是芳基重氮盐的前驱体, 它可以由相应的芳胺经反应以较高的产率制得.该反应的官能团容忍性较好, 但是对于有强吸电子基团取代的芳基三氮烯硼基化反应产率较低或不反应(Eq. 34). Yamane认为芳基三氮烯的硼基化为亲核取代反应历程:芳基三氮烯在BF3的存在下生成芳基重氮盐B, 然后氟负离子由三氟化硼负离子转移到联硼酸频那醇酯得到C.最后发生亲核取代得到目标产物芳基硼酸酯(Scheme 9).
|
|
(34) |
2014年, Blanchet小组[51]报道了芳香重氮盐与四羟基二硼直接制备芳基硼酸的反应.以四氟硼酸芳香重氮盐为原料, 室温下与四羟基二硼在DMF中反应得到相应芳基硼酸, 反应速率快, 15 min即可反应完成(Eq. 35). Blanchet等也直接由芳胺在NaNO2/HCl的条件下制备芳香重氮盐, 不经后处理直接在水相中加入2 equiv.的四羟基二硼和NaOAc, 室温下反应20 min即得到相应芳基硼酸, 与四氟硼酸芳香重氮盐相比产率有所提高(Eq. 36).
同年, 薛东等[52]报道了甲醇促进的芳胺的硼基化反应, 该方法直接由芳胺在甲醇和水混合溶剂中通过NaNO2/HCl制备芳香重氮盐, 不经后处理与联硼酸频哪醇酯或四羟基二硼在甲醇的参与下经芳香环亲核取代反应(SNAr)得到相应芳基硼酸酯或芳基硼酸(Eq. 37).
|
|
(35) |
|
|
(36) |
|
|
(37) |
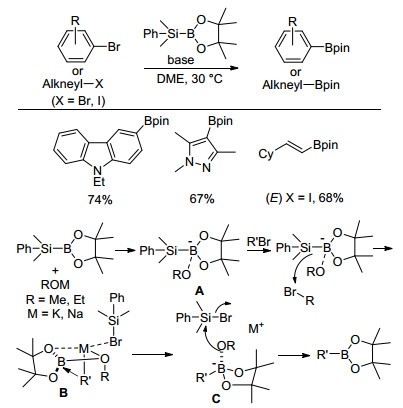
2012年, Ito及其同事[53]报道了烷氧基碱参与下硅硼烷(PhMe2Si—Bpin)与卤代芳烃生成相应芳基硼酸酯的反应.经过条件筛选, 溴代芳烃作为反应底物, KOMe作碱, 乙二醇二甲醚为溶剂时, 硼基化反应的产率最高.该反应可以在室温下进行, 而且反应速率很快, 反应一般在1 h内即反应完全.该方法对于取代基位于邻位、间位、对位以及是否为供电子或吸电子均无明显的选择性, 位阻较大的卤代芳烃也能以较高的产率转化为相应的芳基硼酸酯, 其中2, 6-二甲基溴苯的硼基化产率可达85% (Eq. 38).
|
|
(38) |
2015年, Ito等[54]对上述反应进行了进一步的底物扩展及机理研究, 多种芳杂环硼酸酯都可以通过该方法制得.值得一提的是, 使用NaOEt代替KOMe, (E)-和(Z)-卤代烯烃进行硼基化反应后仍保持原烯烃的构型.他们基于实验数据提出了合理的反应机理.首先硅硼烷与碱形成硅硼烷/烷氧基复合物A, 随后A进攻溴代物的溴原子生成含有阴离子的复合物B, 阴离子部分亲核进攻硼原子生成相应有机硼中间体C, 最后中间体C与溴硅烷反应生成相应芳基硼酸酯.此后, Ito等[55]又通过一些实验数据及DFT计算研究对该机理进行深刻阐明(Scheme 10).
2013年, Zhang等[56]报道了碘代芳烃在Cs2CO3的参与下与联硼酸频哪醇酯的硼基化反应, 经条件筛选, MeOH是该反应的最佳溶剂.这是第一个没有过渡金属参与的碘代芳烃与联硼酸频哪醇酯的反应, 该反应对大多数官能团都有着较好的耐受性, 并且能够以较高的产率得到相应的芳基硼酸酯.在自由基捕获剂TEMPO的存在下, 反应仍能取得正常收率, 表明该反应过程并不是一个自由基历程(Eq. 39).
|
|
(39) |
2017年, Cho等[57]报道了卤代芳烃与1, 1-二[(频哪醇)硼基]烷烃制备芳基硼酸酯的反应.叔丁醇钠作碱, 甲苯/四氢呋喃(V/V=1/1)为溶剂, 50~120 ℃下碘代芳烃与双[(频哪醇)硼基]甲烷反应得到相应硼酸酯, 产率最高, 反应具有良好的化学选择性和官能团耐受性.当采用溴代芳烃参与反应时, 1, 1-二[(频哪醇)硼基]乙烷作为硼源的产率高于双[(频哪醇)硼基]甲烷.经过理论计算及实验研究, 他们提出该反应经过卤代芳烃与α-硼碳负离子加合物生成芳基负离子, 然后与1, 1-二[(频哪醇)硼基]烷烃或叔丁醇频哪醇硼酸酯反应生成芳基硼酸酯(Scheme 11).
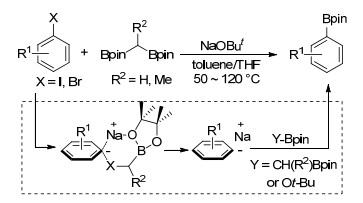
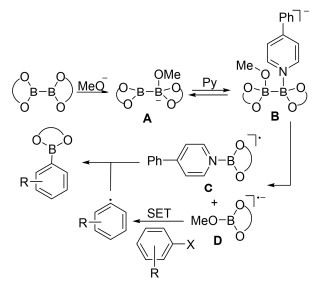
同年, 焦雷课题组[58]报道了吡啶催化的卤代芳烃的硼基化反应, 4-苯基吡啶作为催化剂, 甲醇钾作碱, 甲基叔丁基醚(MTBE)为溶剂, 85 ℃回流的条件下, 碘代芳烃或者溴代芳烃可以与联硼酸频哪醇酯反应得到相应的芳基硼酸酯.该反应有着良好的官能团耐受性, 而且产率较高, 但是吸电子基团取代的卤代芳烃反应产率要低于给电子基团取代的卤代芳烃(Eq. 40).
与之前报道的自由基机理不同的是, 该反应是由两种不同类型的自由基偶联得到硼酸酯产物.首先, 甲氧基负离子与联硼酸频哪醇酯反应得到复合物A, A再与吡啶作用得到中间体B. B发生均裂得到稳定的硼自由基C和自由基负离子D, D作为电子供体与卤代芳烃发生单电子转移(SET)形成芳基自由基, 最后硼自由基C捕获芳基自由基得到目标产物芳基硼酸酯(Scheme 12).
|
|
(40) |
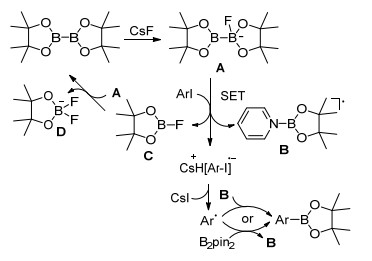
随后, Pucheault小组[59]报道了氟离子介导的碘代芳烃的硼基化反应, 经过条件优化, CsF作碱, DMSO为溶剂, 105 ℃加热, 在吡啶的参与下反应收率最佳(Eq. 41).他们基于实验数据提出了合理反应机理, 首先氟离子与联硼酸频哪醇酯形成复合物A, 碘代芳烃与A发生单电子转移生成自由基阴离子, 同时, 吡啶与联硼酸频哪醇酯的复合物B和频哪醇氟硼烷C随之生成, C可与A反应生成联硼酸频哪醇酯和D.自由基与复合物B或联硼酸频哪醇酯反应得到目标产物芳香硼酸酯(Scheme 13).
|
|
(41) |
二芳基碘鎓盐是一种比较稳定的高价碘化合物, 在有机合成中, 它们可以作为许多无机及有机亲核试剂的芳化试剂. 2015年, Fernández等[60]采用二芳基乙酰碘鎓盐与联硼酸频哪醇酯反应实现了非金属催化的芳基硼基化, 该反应的最佳溶剂为甲醇, 甲醇与乙酰根负离子可以作为路易斯碱与联硼酸频哪醇酯配位, 令其发生极性反转得到具有亲核性的二硼复合物, 与二芳基碘鎓盐发生亲核取代反应获得芳基硼酸酯.该方法对于取代基位于邻位、间位、对位以及是否为供电子或吸电子均能取得较好的收率, 而且位阻较大的二芳基碘鎓盐也能反应得到相应硼酸酯产物(Eq. 42).
|
|
(42) |
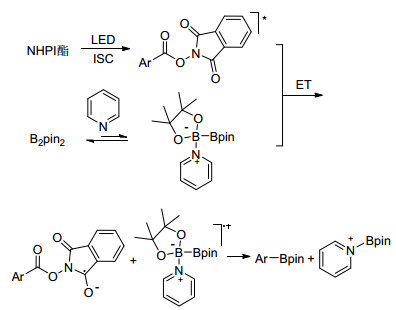
2017年, 傅尧等[61]报道了芳香羧酸无金属参与的脱羧硼化反应.通过条件优化, 异烟酸叔丁酯作为催化剂, 三氟甲苯作溶剂时, 芳香羧酸的N-羟基邻苯二甲酰亚胺酯(NHPI酯)与联硼酸频哪醇酯反应以较高的收率得到目标硼化产物.该方法反应条件简单, 催化剂价格低廉, 反应可以轻松放大到克级规模, 具有应用于工业生产的巨大潜力.反应通过形成吡啶稳定的硼自由基, 经历了一个自由基偶联的过程, NHPI酯与联硼酸频哪醇酯络合物促进了分子内的电子转移, 进而活化活性酯产生自由基脱羧作用(Eq. 43).
|
|
(43) |
寻求绿色、高效、高选择性、安全的合成策略一直是化学工作者孜孜不倦的追求.由于光能具有清洁、可再生的优点, 光诱导的反应自然受到了化学家们的广泛关注.
2016年, 李鹏飞小组[62]发现碘代芳烃在紫外光(UV)诱导下可以与联硼酸酯反应得到相应的芳基硼酸酯, 经过条件筛选, 乙腈、丙酮和水的作为混合溶剂, 0.5当量的N, N, N', N'-四甲基甲二胺(TMDAM)作碱时, 反应的产率最高; 此外, 溴代芳烃也能转化为相应硼酸产物, 产率较碘代芳烃稍低.该方法对于取代基位于邻位、间位、对位以及是否为供电子或吸电子均无明显的选择性, 当采用连续流动反应系统时, 反应产率有明显提高(Eq. 44). 2016年Li小组[63]报道了连续流动系统下富电子氯代芳烃UV诱导的硼基化反应, 并将反应底物进一步扩展到富电子的氟代芳烃及酚类衍生物.
|
|
(44) |
同年, Larionov小组[64]报道了UV诱导的卤代芳烃的硼基化反应.使用甲醇作为反应溶剂, 溴代芳烃在UV的诱导下与四羟基二硼发生反应得到相应的芳基硼酸, 该反应对多种官能团均有较好的耐受性.此外采用该方法, 碘代芳烃及氯代芳烃也可以获得相应的芳基硼酸.由于碳卤键的键解离能不同(C—I 272, C—Br 336, C—Cl 400 kJ•mol−1), 反应速率:碘代芳烃>溴代芳烃>氯代芳烃. Larionov等还拓展了底物的范围, 富电子的氟代芳烃及芳香类季铵盐也能在UV的诱导下进行硼基化反应, 另外在UV诱导下卤代芳烃在乙腈中可以与联硼酸频哪醇酯反应得到相应芳基硼酸酯, 产率较高(Eq. 45).
2017年, Li课题组[65]以NaI作为电子供体, 在UV的作用下与芳基三氟甲磺酸酯发生电子转移后经历一个自由基的过程得到相应芳基硼酸酯(Eq. 46).
|
|
(45) |
|
|
(46) |
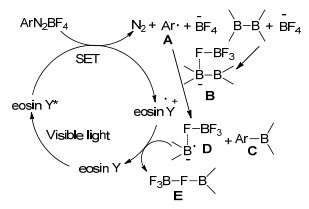
2012年, 严国兵小组[66]报道了首例可见光诱导的芳基硼基化反应.以曙红Y (eosin Y)为光敏剂, 乙腈作为溶剂, 四氟硼酸芳基重氮盐在可见光的诱导下成功合成了相应的芳基硼酸酯.无论富电子或缺电子的芳香氮盐都能以较高的产率得到目标产物硼酸酯(Eq. 47).
|
|
(47) |
在可见光的作用下, eosin Y变成有较高还原能力的激发态eosin Y*, 与四氟硼酸芳香重氮盐发生一个单电子转移(SET)过程得到芳香自由基A.四氟硼酸根负离子与联硼酸频哪醇酯配位得到一个四配位的硼复合物B, 然后与芳香自由基A反应得到目标产物芳香硼酸酯C和自由基阴离子D, 最后阴离子D被eosin Y*氧化得到E完成催化循环过程(Scheme 14).
2016年, Ranu小组[67]报道了可见光催化的芳胺类化合物的硼基化反应, 以eosin Y为光敏剂, 乙腈作为溶剂, 芳胺在t-BuONO的存在下与联硼酸频哪醇酯反应得到芳基硼酸酯.该方法与Wang小组[49]报道的反应相比, 反应条件更温和, 产率更高(Eq. 48).
同年, 付华小组[68]采用fac-Ir(ppy)3为光催化剂, 以紧凑型荧光灯为光源, 实现了卤代芳烃的硼基化反应.该方法不需要特殊的实验装置, 易于操作, 条件温和, 而且具有良好的底物适应性(Eq. 49).
|
|
(48) |
|
|
(49) |
2017年, Glorius小组[69]基于发光淬灭筛选方法发现苯并三氮唑衍生物在可见光催化下生成生成邻位苯酰胺取代的芳基硼酸酯, 经过条件筛选, [Ir(ppy)2(NHC-F2)]作为催化剂, 乙腈为溶剂, 在苯甲酸酐和N, N-二异丙基乙胺的参与下, 收率最高.他们又通过鲁棒性测试检测了该方法的官能团耐受性, 结果表明, 多种官能团都可以在该条件下存在且不影响反应(Eq. 50).
|
|
(50) |
随着一些脱羧硼化反应的报道, 2017年, 李鹏飞课题组[70]报道了铱光催化剂催化下的脂肪羧酸酯的脱羧硼化反应, 然而芳香羧酸酯在该条件下的硼基化产率很低.同年Glorius小组[71]报道了无金属催化剂参与可见光诱导的芳基羧酸的脱羧硼化反应, 芳基羧酸转化为相应的N-羟基邻苯二甲酰亚胺酯(NHPI酯), NHPI酯与联硼酸频哪醇酯、吡啶、Cs2CO3在400 nm LED灯的激发下得到芳基硼酸酯.该反应条件温和、普适性好, 可以应用于已商品化的药物分子合成中(Eq. 51).同时, 在文中探讨了该反应的机理, NHPI酯在光激发下得到激发态, 与吡啶复合物发生电子转移, 随后经过一个自由基历程得到目标产物硼酸酯(Scheme 15).随后, Aggarwal小组[72]报道了光诱导脂肪羧酸的脱羧硼化反应, 大大缩减了上述反应所需添加剂, 该反应仅需要两种底物及溶剂即可在光诱导下完成转化.
|
|
(51) |
自1995年Miyaura报道钯催化的卤代芳烃硼基化反应合成芳基硼酸以来, 大批化学工作者致力于开发更高活性的钯催化剂, 拓展底物的适应性.与此同时, 镍、铜、锌、铁、钴等一系列非贵金属催化剂也被应用于芳基硼酸类化合物合成.上述金属催化硼基化反应成功关键在于配体及碱的选择, 因此新配体的开发将会促进现有反应的优化及新型硼基化反应的产生.
此外, 近几年来, 非金属催化及光诱导的硼基化反应也取得了一定的进展, 与传统的有机金属试剂或过渡金属相比, 其官能团耐受性相对较好, 而且具有条件温和, 操作简单, 环境友好等优点, 是对金属催化有益的补充.但是非金属催化及光诱导的硼基化反应仍存在挑战: (1)开发新的反应底物, 拓展反应底物的范围, 不仅局限于芳胺、芳基重氮盐、芳香偶氮化合物、芳香卤代物和芳香NHPI酯等反应底物, 提高反应的普适性. (2)芳杂环底物通常反应活性较差, 硼基化产率低下或难以得到相应产物. (3) UV诱导的反应需要石英反应管等特殊实验装置, 发展新型的可见光诱导的硼基化反应及反应装置. (4)将该类反应应用于工业化生产, 不再局限于实验室研究. (5)发展自由基类型的硼基化反应, 将自由基化学与硼化学进一步关联, 这不仅具有理论上的意义, 而且具有潜在的合成应用价值.相信在不久的将来, 芳基硼酸类化合物的合成方法将取得更大的突破, 并在有机合成、药物化学及材料科学中有更广泛的应用.
(a) Lacina, K. ; Skladal, P. ; James, T. D. Chem. Cent. J. 2014, 8, 60.
(b) Li, D. -J. ; Chen, Y. ; Liu, Z. Chem. Soc. Rev. 2015, 44, 8097.
(c) Sun, X. ; Zhai, W. ; Fossey, J. S. ; James, T. D. Chem. Commun. 2016, 52, 3456.
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457. doi: 10.1021/cr00039a007
Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508. doi: 10.1021/jo00128a024
Murata, M. Heterocycles 2012, 85, 1795. doi: 10.3987/REV-12-736
Chow, W. K.; Yuen, O. Y.; Choy, P. Y.; So, C. M.; Lau, C. P.; Wong, W. T.; Kwong, F. Y. RSC Adv. 2013, 3, 12518. doi: 10.1039/c3ra22905j
Kubota, K.; Iwamoto, H.; Ito, H. Org. Biomol. Chem. 2017, 15, 285. doi: 10.1039/C6OB02369J
Li, P.; Meng, G.; Chen, K.; Wang, L. Synthesis 2017, 49, 4719. doi: 10.1055/s-0036-1590913
Yan, G.-B.; Huang, D.-Y.; Wu, X.-M. Adv. Synth. Catal. 2018, 360, 1040. doi: 10.1002/adsc.v360.6
(a) Ros, A. ; Fernandez, R. ; Lassaletta, J. M. Chem. Soc. Rev. 2014, 43, 3229.
(b) Xu, L. ; Wang, G. -H. ; Zhang, S. ; Wang, H. ; Wang, L. -H. ; Liu, L. ; Jiao, J. ; Li, P. -F. Tetrahedron 2017, 73, 7123.
(a) Ishiyama, T. ; Itoh, Y. ; Kitano, T. ; Miyaura, N. Tetrahedron Lett. 1997, 38, 3447.
(b) Murata, M. ; Watanabe, S. ; Masuda, Y. J. Org. Chem. 1997, 62, 6458.
(c) Willis, D. M. ; Strongin, R. M. Tetrahedron Lett. 2000, 41, 8683.
Ishiyama, T.; Ishida, K.; Miyaura, N. Tetrahedron 2001, 57, 9813. doi: 10.1016/S0040-4020(01)00998-X
Billingsley, K. L.; Barder, T. E.; Buchwald, S. L. Angew. Chem., Int. Ed. 2007, 46, 5359. doi: 10.1002/(ISSN)1521-3773
Billingsley, K. L.; Buchwald, S. L. J. Org. Chem. 2008, 73, 5589. https://www.google.de/patents/WO2004056994A3?cl=en
(a) Molander, G. A. ; Trice, S. L. ; Dreher, S. D. J. Am. Chem. Soc. 2010, 132, 17701.
(b) Molander, G. A. ; Trice, S. L. J. ; Kennedy, S. M. ; Dreher, S. D. ; Tudge, M. T. J. Am. Chem. Soc. 2012, 134, 11667.
Chow, W. K.; Yuen, O. Y.; So, C. M.; Wong, W. T.; Kwong, F. Y. J. Org. Chem. 2012, 77, 3543. doi: 10.1021/jo202472k
Yamamoto, Y.; Matsubara, H.; Yorimitsu, H.; Osuka, A. ChemCat-Chem 2016, 8, 2317. doi: 10.1002/cctc.201600456
Baudoin, O.; Guénard, D.; Guéritte, F. J. Org. Chem. 2000, 65, 9268. doi: 10.1021/jo005663d
Fang, H.; Kaur, G.; Yan, J.; Wang, B. Tetrahedron Lett. 2005, 46, 1671. doi: 10.1016/j.tetlet.2005.01.064
Rodriguez, S.; Qu, B.; Haddad, N.; Reeves, D. C.; Tang, W.-J.; Lee, H.; Krishnamurthy, D.; Senanayake, C. H. Adv. Synth. Catal. 2011, 353, 533. doi: 10.1002/adsc.201000878
Kawamorita, S.; Ohmiya, H.; Iwai, T.; Sawamura, M. Angew. Chem., Int. Ed. 2011, 50, 8363. doi: 10.1002/anie.v50.36
Morgan, A. B.; Jurs, J. L.; Tour, J. M. J. Appl. Polym. Sci. 2000, 76, 1257. doi: 10.1002/(ISSN)1097-4628
Rosen, B. M.; Huang, C.; Percec, V. Org. Lett. 2008, 10, 2597. doi: 10.1021/ol800832n
Moldoveanu, C.; Wilson, D. A.; Wilson, C. J.; Corcoran, P.; Rosen, B. M.; Percec, V. Org. Lett. 2009, 11, 4974. doi: 10.1021/ol902155e
Yamamoto, T.; Morita, T.; Takagi, J.; Yamakawa, T. Org. Lett. 2011, 13, 5766. doi: 10.1021/ol202267t
Molander, G. A.; Cavalcanti, L. N.; Garcia-Garcia, C. J. Org. Chem. 2013, 78, 6427. doi: 10.1021/jo401104y
Zhang, H.; Hagihara, S.; Itami, K. Chem.-Eur. J. 2015, 21, 16796. doi: 10.1002/chem.v21.47
Hu, J.-F.; Sun, H.-Q.; Cai, W.-S.; Pu, X.-H.; Zhang, Y.-M.; Shi, Z.-Z. J. Org. Chem. 2016, 81, 14. doi: 10.1021/acs.joc.5b02557
Liu, X.-W.; Echavarren, J.; Zarate, C.; Martin, R. J. Am. Chem. Soc. 2015, 137, 12470. doi: 10.1021/jacs.5b08103
Tobisu, M.; Zhao, J.-N.; Kinuta, H.; Furukawa, T.; Igarashi, T.; Chatani, N. Adv. Synth. Catal. 2016, 358, 2417. doi: 10.1002/adsc.201600336
Hu, J.; Zhao, Y.; Liu, J.; Zhang, Y.; Shi, Z. Angew. Chem., Int. Ed. Engl. 2016, 55, 8718. doi: 10.1002/anie.201603068
Pu, X.-H.; Hu, J.-F.; Zhao, Y.; Shi, Z.-Z. ACS Catal. 2016, 6, 6692. doi: 10.1021/acscatal.6b01956
Guo, L.; Rueping, M. Chem.-Eur. J. 2016, 22, 16787. doi: 10.1002/chem.v22.47
Zhu, W.; Ma, D.-W. Org. Lett. 2006, 8, 261. doi: 10.1021/ol052633u
Kleeberg, C.; Dang, L.; Lin, Z.; Marder, T. B. Angew. Chem., Int. Ed. 2009, 48, 5350. doi: 10.1002/anie.200901879
Yan, G.-B.; Yang, M.-H.; Yu, J. Lett. Org. Chem. 2012, 9, 71. doi: 10.2174/157017812799304015
Yu, H.; Zhang, J.; Wang, X.; Ye, J. Synlett 2012, 23, 1394. doi: 10.1055/s-0031-1290960
Nagashima, Y.; Takita, R.; Yoshida, K.; Hirano, K.; Uchiyama, M. J. Am. Chem. Soc. 2013, 135, 18730. doi: 10.1021/ja409748m
Bose, S. K.; Marder, T. B. Org. Lett. 2014, 16, 4562. doi: 10.1021/ol502120q
Bose, S. K.; Deissenberger, A.; Eichhorn, A.; Steel, P. G.; Lin, Z.; Marder, T. B. Angew. Chem., Int. Ed. 2015, 54, 11843. doi: 10.1002/anie.201505603
Qi, X.-X.; Jiang, L.-B.; Zhou, C.; Peng, J.-B.; Wu, X.-F. ChemistryOpen 2017, 6, 345. doi: 10.1002/open.v6.3
Tobisu, M.; Kinuta, H.; Kita, Y.; Remond, E.; Chatani, N. J. Am. Chem. Soc. 2012, 134, 115. doi: 10.1021/ja2095975
Kinuta, H.; Tobisu, M.; Chatani, N. J. Am. Chem. Soc. 2015, 137, 1593. doi: 10.1021/ja511622e
Marciasini, L. D.; Richy, N.; Vaultier, M.; Pucheault, M. Adv. Synth. Catal. 2013, 355, 1083. doi: 10.1002/adsc.v355.6
Bedford, R. B.; Brenner, P. B.; Carter, E.; Gallagher, T.; Murphy, D. M.; Pye, D. R. Organometallics 2014, 33, 5940. doi: 10.1021/om500847j
Yoshida, T.; Ilies, L.; Nakamura, E. ACS Catal. 2017, 7, 3199. doi: 10.1021/acscatal.7b00310
Yao, W.-B.; Fang, H.-Q.; Peng, S.-H.; Wen, H.-N.; Zhang, L.; Hu, A.-G.; Huang, Z. Organometallics 2016, 35, 1559. doi: 10.1021/acs.organomet.6b00161
Komeyama, K.; Kiguchi, S.; Takaki, K. Chem. Commun. 2016, 52, 7009. doi: 10.1039/C6CC03086F
Mo, F.-Y.; Jiang, Y.-B.; Qiu, D.; Zhang, Y.; Wang, J.-B. Angew. Chem., Int. Ed. 2010, 49, 1846. doi: 10.1002/anie.v49:10
Qiu, D.; Jin, L.; Zheng, Z.-T.; Meng, H.; Mo, F.-Y.; Wang, X.; Zhang, Y.; Wang, J.-B. J. Org. Chem. 2013, 78, 1923. doi: 10.1021/jo3018878
Zhu, C.; Yamane, M. Org. Lett. 2012, 14, 4560. doi: 10.1021/ol302024m
Erb, W.; Hellal, A.; Albini, M.; Rouden, J.; Blanchet, J. Chem.-Eur. J. 2014, 20, 6608. doi: 10.1002/chem.v20.22
Zhao, C.-J.; Xue, D.; Jia, Z.-H.; Wang, C.; Xiao, J. Synlett 2014, 25, 1577. doi: 10.1055/s-00000083
Yamamoto, E.; Izumi, K.; Horita, Y.; Ito, H. J. Am. Chem. Soc. 2012, 134, 19997. doi: 10.1021/ja309578k
Yamamoto, E.; Ukigai, S.; Ito, H. Chem. Sci. 2015, 6, 2943. doi: 10.1039/C5SC00384A
Uematsu, R.; Yamamoto, E.; Maeda, S.; Ito, H.; Taketsugu, T. J. Am. Chem. Soc. 2015, 137, 4090. doi: 10.1021/ja507675f
Zhang, J.-M.; Wu, H.-H.; Zhang, J.-L. Eur. J. Org. Chem. 2013, 6263. https://doaj.org/article/8ddcc4ba9b974e78875955bcac337aca
Lee, Y.; Baek, S. Y.; Park, J.; Kim, S. T.; Tussupbayev, S.; Kim, J.; Baik, M. H.; Cho, S. H. J. Am. Chem. Soc. 2017, 139, 976. doi: 10.1021/jacs.6b11757
Zhang, L.; Jiao, L. J. Am. Chem. Soc. 2017, 139, 607. doi: 10.1021/jacs.6b11813
Pucheault, M.; Pinet, S.; Liautard, V.; Debiais, M. Synthesis 2017, 49, 4759. doi: 10.1055/s-0036-1588431
Miralles, N.; Romero, R. M.; Fernández, E.; Mu iz, K. Chem. Commun. 2015, 51, 14068. doi: 10.1039/C5CC04944J
Cheng, W.-M.; Shang, R.; Zhao, B.; Xing, W.-L.; Fu, Y. Org. Lett. 2017, 19, 4291. doi: 10.1021/acs.orglett.7b01950
Chen, K.; Zhang, S.; He, P.; Li, P.-F. Chem. Sci. 2016, 7, 3676. doi: 10.1039/C5SC04521E
Chen, K.; Cheung, M. S.; Lin, Z.-Y.; Li, P.-F. Org. Chem. Front. 2016, 3, 875. doi: 10.1039/C6QO00109B
Mfuh, A. M.; Doyle, J. D.; Chhetri, B.; Arman, H. D.; Larionov, O. V. J. Am. Chem. Soc. 2016, 138, 2985. doi: 10.1021/jacs.6b01376
Liu, W.-B.; Yang, X.-B.; Gao, Y.; Li, C.-J. J. Am. Chem. Soc. 2017, 139, 8621. doi: 10.1021/jacs.7b03538
Yu, J.; Zhang, L.; Yan, G.-B. Adv. Synth. Catal. 2012, 354, 2625. doi: 10.1002/adsc.v354.14/15
Ahammed, S.; Nandi, S.; Kundu, D.; Ranu, B. C. Tetrahedron Lett. 2016, 57, 1551. doi: 10.1016/j.tetlet.2016.02.097
Jiang, M.; Yang, H.-J.; Fu, H. Org. Lett. 2016, 18, 5248. doi: 10.1021/acs.orglett.6b02553
Teders, M.; Pitzer, L.; Hopkinson, M. N.; Glorius, F. Angew. Chem., Int. Ed. 2017, 56, 902. doi: 10.1002/anie.201609393
Hu, D.-W.; Wang, L.-H.; Li, P.-F. Org. Lett. 2017, 19, 2770. doi: 10.1021/acs.orglett.7b01181
Candish, L.; Teders, M.; Glorius, F. J. Am. Chem. Soc. 2017, 139, 7440. doi: 10.1021/jacs.7b03127
Fawcett, A.; Pradeilles, J.; Wang, Y.; Mutsuga, T.; Myers, E. L.; Aggarwal, V. K. Science 2017, 357, 283. doi: 10.1126/science.aan3679

Scheme 3 氯代芳烃与四羟基二硼的钯催化硼基化反应
Scheme 3 Pd-catalyzed borylation of aryl chlorides with bisboronic acids

Scheme 4 镍催化脱羧硼化反应及机理
Scheme 4 Ni-catalyzed decarbonylative borylation and proposed mechanism

Scheme 5 锌催化卤代芳烃硼基化可能机理
Scheme 5 Proposed mechanism of zinc-catalyzed borylation of aryl halides

Scheme 6 锌催化的芳香重氮盐硼基化可能机理
Scheme 6 Proposed mechanism of zinc-catalyzed borylation of aryl diazonium salts

Scheme 8 非金属催化的芳香胺硼基化可能机理
Scheme 8 Proposed mechanism of metal-free borylation of arylamines

Scheme 9 BF3•OEt2催化的芳香三氮烯硼基化可能机理
Scheme 9 Plausible mechanism of BF3•OEt2-mediated borylation of aryltriazenes

Scheme 10 烷氧基碱介导的有机卤代物硼基化及可能机理
Scheme 10 Alkoxy base mediated borylation of organic halides and plausible mechanism

Scheme 11 1, 1-二[(频哪醇)硼基]烷烃为硼源的非金属催化硼基化反应
Scheme 11 Metal-free borylation reaction using 1, 1-bis[(pina-colato)boryl]alkanes as the boron source

Scheme 12 吡啶催化的芳香卤代物硼基化可能机理
Scheme 12 Proposed mechanism of pyridine-catalyzed borylation of aryl halides

Scheme 13 氟离子介导的芳香碘代物硼基化可能机理
Scheme 13 Plausible mechanism of fluoride mediated borylation of aryl iodides

Scheme 14 可见光催化的芳香重氮盐硼基化可能机理
Scheme 14 Proposed mechanism of photocatalytic borylation of aryldiazonium salts

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



















































 下载:
下载:



 下载:
下载: