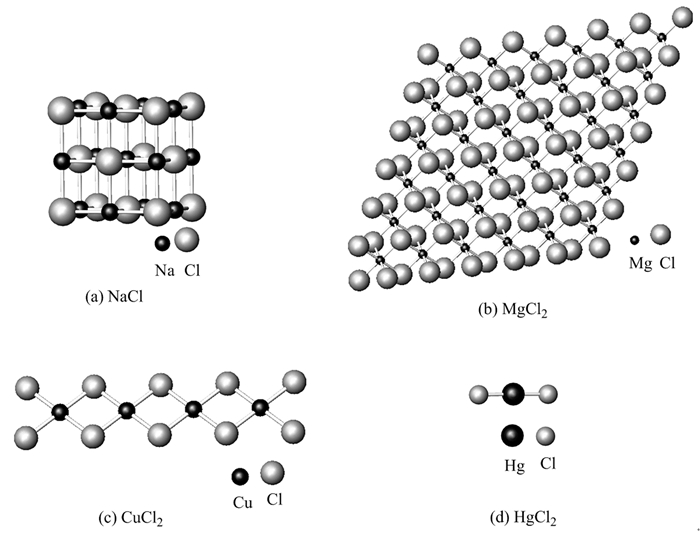
图 1.
金属卤化物的典型结构
(a)三维;(b)二维;(c)一维;(d)零维
Figure 1.
Typical structures of metal halides

在中学化学教学中,盐类在水中的溶解度规律都是通过口诀传诵。在大一无机化学教学中,教师们尝试着概括出更多盐类在水溶液中溶解度规律,将此规律传授给学生[1, 2]。虽然可以采用热力学数据来解释盐类在水中溶解度的规律,但深层次的原因并未详细给出。例如,在无机化学教学中,虽然向学生讲授了金属卤化物的水溶性规律,但该规律所基于的更深刻原因就是学生经常困惑的问题。以NaCl、AlCl3和AgCl的水溶性为例,为什么前二者易溶于水,而后者却难溶?有些教科书用离子极化的概念来解释AgCl的难溶现象,但显然,用相同的概念却无法解释同样受极化影响很大的AlCl3的易溶现象。本文从多个角度探讨了金属卤化物的水溶性原因,并推出了卤化物的水溶性依据及其规律。
通过大量晶体结构数据[3] (所有晶体结构数据均取自文献[3])分析得出,金属卤化物共具有四种典型结构,包括三维空间结构、二维层状结构、一维链状结构和零维小分子结构,见图 1。其中三维空间结构又包括以离子键为结合力的离子晶体和以共价键为结合力的原子晶体,它们均属于巨型分子。图 1(a)是NaCl晶体结构,它是典型的三维巨型分子,是金属卤化物中最常见的结构;图 1(b)是MgCl2的晶体结构,典型的二维层状巨型分子,也是金属卤化物比较常见的结构;一维链状结构(如CuCl2)和零维小分子结构(如HgCl2)相对较少。一维、二维和三维结构都构成巨型分子,均可以视为零维最小结构基元(化学式)的聚合物。
用MXn来表示金属卤化物的组成,其中M代表金属,X代表卤素,再用分子轨道理论处理M-X间的化学键时可以得到金属卤化物中存在如图 2中的六种典型的化学键。
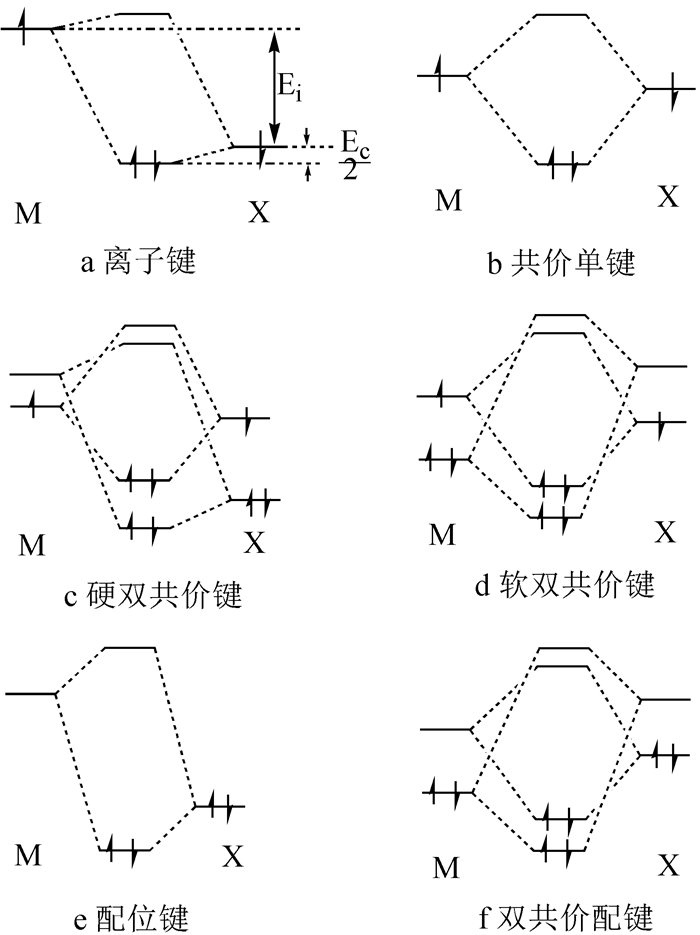
(a)离子键;(b)共价单键;(c)硬共价双键;(d)软共价双键;(e)配位单键;(f)配位双键
一般教科书中将离子晶体用静电模型来处理,离子键的强弱用晶格能U来衡量,有波恩-朗德方程:
|
$ U = \frac{{A{N_{\rm{A}}}{Z_ + }{Z_-}{e^2}}}{{4\pi {\varepsilon _0}d}}\left( {1-\frac{1}{m}} \right) $ |
其中, A为马德隆常数,NA为阿伏加德罗常数,Z+、Z-分别为正负离子的氧化数,e为电子电荷,ε0为真空介电常数,d为阴阳离子的平衡核间距,m为波恩指数。该模型非常简单,在离子性成分高时能得到较好的结果。
离子键也可以用分子轨道理论处理,能同时突出离子性成分和共价键成分的共存,见图 2(a)。先考虑M和X双原子间的成键,由于M和X的价层电子所在轨道能量差异大,经过原子轨道线性组合成键后形成成键轨道和反键轨道,两个电子优先占据能量低的成键轨道,其能量贡献可以分为纯离子键贡献Ei和纯共价键贡献Ec。其中, Ei对应M价轨道上的电子向X的价轨道上转移,Ec则指在发生转移后再通过电子对的共用,进一步降低的能量。总能量贡献为Ei + Ec,能量大小比较为Ei > Ec,故表现为离子键。离子晶体中的能量贡献还应包括结构基元MXn聚合成晶体环节的能量变化。离子键最容易出现在活泼的卤素(主要指F、Cl)与活泼金属(主要是s区和f区元素)之间形成的金属卤化物中。
如图 2(b),M和X的价层电子能量差异小,原子轨道线性组合后电子填充入成键轨道,此时离子键贡献Ei与共价键贡献Ec的关系为Ei < Ec,即以共价键为主。共价单键最容易出现在高氧化态金属中,尤其是高氧化态d区和p区金属氯化物、溴化物和碘化物(如AlCl3、SnI4等化合物)中均存在共价单键。
硬共价双键M原子除了有电子转移成键外,还有能量相对较低的空轨道(p轨道或d轨道),而X原子则除了接受电子转移成键外,还有孤电子对(见图 2(c))。经原子轨道线性组合后,除了有部分离子键成份的贡献外,最主要的能量贡献来自两条共价键形成,而其共价键的总贡献一般也远大于离子键贡献,即Ei
M原子除了有电子转移成键外,还有能量也相对较高的内层的成对价电子,一般是指d电子;而X原子则除了有接受电子转移成键的轨道外,还有能量也相对较低的空轨道,也是指d空轨道。这样,卤素中F价层无d空轨道而不符合这一要求,Cl、Br、I均有nd价层空轨道而符合要求(见图 2(d))。与硬共价双键相似,软共价双键中的总共价键能量贡献也明显大于离子键的贡献,即Ei
配位键(见图 2(e))是纯共价贡献,由于无离子键成份,且一方单独供电子,因此一般都较弱,明显弱于前面所描述的四种键,因此是化学反应中最易断裂的化学键。它是金属卤化物分子单体聚合的两种驱动力之一。由于金属都是缺电子原子,金属原子M最外层达不到8电子结构,所以必须通过配位键来稳定化,这就自然导致聚合。例如,Al2Cl6的形成是由于由共价单键构成的AlCl3属于缺电化合物,通过配位单键产生二聚而达到稳定结构。配位单键存在于形成离子键和共价单键之后的结构基元的聚合产物中。
当M具有高能量的d电子,且X具有孤电子对和低能量的空轨道时,便同时形成配位σ键和配位π键(见图 2(f));F由于无低能量的空轨道,故不能形成配位双键。金属d电子多而氧化态低时,除了利用空轨道接受孤电子对形成σ键外,内层的(n-1)d轨道能量相当高,与Cl、Br和I的价层nd轨道能量相当接近,再形成π键,且配位π键与配位σ键相互加强,导致配位双键的强度规律为:M-Cl < M-Br < M-I。由于具有双键性质,因此其作用力比配位单键强,却仍比另外四种键弱。这种化学键也是化学反应中相对易断裂的化学键,同时也是金属卤化物分子单体聚合的两种驱动力之一。与软共价双键相似,配位双键也一般存在于d电子多而又氧化态低的金属,尤其是指重后过渡金属和重p区金属与Cl、Br、I形成的化合物中,是其结构基元聚合的驱动力。
以上六种化学键中,前四种键的形成都会改变元素的氧化态,为描述方便,这类化学键统称为价键;后两种键的形成不会改变元素的氧化态,这类化学键统称为配位键,它们只存在于卤化物聚合物中。键的极性主要来自价键中的离子键能量贡献比例,而配位键的形成会增大共价键的比例,因此也会影响键的极性,即使键的极性降低。
价键可以单独形成各种结构的金属卤化物,而配位键不能。配位键只起将卤化物的结构基元聚合形成多聚体,如形成一维长链、二维层状和三维网状结构,其中配位双键只存在于共价化合物中。
离子键是形成离子型金属卤化物的核心作用力,但形成离子晶体必须还要有配位单键的参与,这是由于结构基元中金属的缺电子性所致。离子晶体中,离子键稳定性最主要的贡献来自电子的转移,而单体MXn的聚合过程则通过配位键来实现,通过离子晶体的升华焓,即式(1),可以计算出聚合过程中配位键的总贡献。
|
$ {\rm{M}}{{\rm{X}}_n}\left( {\rm{s}} \right) \to {\rm{M}}{{\rm{X}}_n}\left( {\rm{g}} \right)\Delta {H^\Theta } $ |
(1) |
其贡献比例则由每个单元平均的配位键数量来决定。离子晶体中聚合形成的配位键数量往往较高,因此配位键对稳定性的总能量贡献仍然相当高。如化学键K—Cl的键能[4]为433kJ·mol-1,而其单体聚合成晶体的总配位键的键能,即升华焓,也达到222kJ·mol-1。KCl的晶体结构与NaCl的相似,K+周围紧靠有6个Cl-,形成八面体结构,即由原来的每个KCl单元的K-Cl间一条价键再增加5条配位键(非经典配位键,考虑1个s轨道和3个p轨道在轴向与八面体中的6个外围原子配位,再平移形成晶体中的离域配位键),聚合成KCl晶体,聚合后6条化学键平均化。尽管配位键数量多,但其总能量贡献仍然比不过1条价键,这说明价键的稳定性远大于配位键。
金属卤化物原子晶体也不能由纯共价单键来单独构建。共价单键或硬共价双键与配位单键以及共价双键与配位双键,这两种组合形式都可以构建原子晶体,当然也可以构成低维聚合物。由纯共价单键构建的金属卤化物只能是零维小分子,最终化合物是形成三维原子晶体还是其他低维聚合物还取决于配位键形成的状况。如PdF4形成原子晶体,其价键可以看成硬共价双键,晶体结构中Pd的配位数均为6,一半的F通过配位单键将[PdF6]单元连接成三维结构。AgI也形成原子晶体,晶体中每个原子的配位数均为4,其中每个原子形成1条共价双键,再通过Ag和I各自的空轨道和孤电子对形成3条配位双键。
有关化学键强弱比较,我们已有文章进行详细分析[5]。上述金属卤化物的六种化学键的强弱规律为:(1)价键远强于配位键;(2)形成离子键、共价单键和硬共价双键时,原子半径越小,其化学键越强;(3)形成软共价双键和配位双键时,金属原子和卤素原子越软(通常原子半径越大)化学键越强,这是由于σ键与其同时形成的π键相互加强;当卤素原子的半径大或电负性小,以及金属的d电子多或电负性大时,化学键的反馈效果更好;(4)同为配位键,原子形成的键的数量越多则配位键越弱,即聚合度越高,单条配位键越弱,但配位键数量的增多对体系总能量的下降贡献增大;配位双键强于配位单键;(5)同为价键,原子形成的键的数量越多,则价键越弱,即氧化态绝对值越高,则单条价键越弱;且氧化态越高,其成键对氟化物来说越容易表现为硬共价双键,对氯化物、溴化物和碘化物来说越容易表现为共价单键;但氧化态增高引起成键数增多,价键引起的总能量的下降幅度增大;(6)硬共价双键明显强于共价单键;(7)对同一金属原子,软共价双键明显强于共价单键,即低氧化态时键更强。
结合金属卤化物的结构和化学键可以得出,金属卤化物中至少存在离子键、共价单键和硬软共价双键中的一种,而配位键则可能存在,也有可能不存在。配位键存在于聚合的巨型分子中,图 1中的三维、二维和一维结构的卤化物中大都是通过配位键而聚合起来的。小分子,如Al2Cl6中,仍然可以存在配位键;结构基元小分子,如GeCl4中,则只有共价单键而没有配位键;再如图 1(d)中的HgCl2也是结构基元小分子,Hg和Cl之间也只存在共价双键。根据小分子都以共价键构建这一事实,可以推出晶体结构中聚合度低的化合物,化学键的共价成份高。但化学键的共价成份高的却并不一定聚合度低,如原子晶体虽然聚合度高,但化学键的共价成份也高。如AgI也是三维巨型分子,而构建其结构的化学键则由配位双键和共价双键构成,它们均属于共价键。
水是小分子,对于巨型分子的金属卤化物来说在水中的溶解必然要形成小分子或离子碎片,以便分散溶解,此时发生的是卤化物与水间的化学反应,即溶解过程是化学变化。由于金属的缺电子性和水的配位特性,金属卤化物,即使是小分子,在水中发生纯物理溶解的可能性也很小。金属卤化物化学变化后在水中的微观存在形式因卤化物的不同甚至浓度不同会有明显差别。水能作为金属卤化物的良好溶剂,溶解过程涉及到溶剂分子向缺电子的金属中心的亲核进攻[6]。在此重点考虑盐类与水作用后形成的微粒。
由于金属都是缺电子原子,在形成金属卤化物时往往通过配位键而聚合成巨型分子,因此当溶剂的供电子能力更强或是浓度更高时,必然发生配位取代反应,即溶剂化。配位实现电荷分散能稳定金属阳离子,而非金属阴离子也需要稳定化,质子溶剂能通过氢键稳定阴离子。水是典型的配位质子溶剂,即O上孤电子对配位稳定金属阳离子,而OH基团通过氢键稳定阴离子,所以金属卤化物在水中能实现离子化。如果形成稀溶液,则MXn在水中可以水合离子化,按式(2)进行。
|
$ \begin{gathered} {\rm{M}}{{\rm{X}}_{\rm{n}}} + \left( {p + nq} \right){{\rm{H}}_2}{\rm{O}} \to {\left[{{\rm{M}}{{\left( {{\rm{O}}{{\rm{H}}_{\rm{2}}}} \right)}_p}} \right]^{n + }} + \hfill \\ n{\left[{{\rm{X}}{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_q}} \right]^ -} \hfill \\ \end{gathered} $ |
(2) |
首先,金属中心通过接受自来H2O中O上的孤电子对形成配位键,构成水合离子的紧密层,此部分水被束缚较牢;其次,由于离子电场的存在,诱导紧密层H2O上的电荷重新分配,即紧密层水中O上的电子被金属阳离子拉走一部分,导致与该O成键的H上更加缺少电子,更易与外围H2O形成氢键,这样在配位层之外还会通过氢键有序地结合水,阳离子的电荷密度越高,则电场越强,结合外围水的层数越多,其结果是使金属阳离子电荷得到充分分散而稳定化。不同的是卤素阴离子X-,其与水分子间无法形成配位键,只能通过与紧密层的H2O发生质子快速共振传递而稳定化。然而由于其电场依然存在,因此外围水分子也会与紧密层水分子间通过氢键而有序化,同时也会参与与内层水分子交换质子而共振传递质子,以进一步稳定阴离子。相对而言,阴离子由于半径明显比阳离子大,形成的电场也弱,氢键也不如配位键强,所以阴离子的水合热一般小于阳离子。
当形成浓溶液时,水合离子外围层减少,离子之间也靠得更近,水合离子之间会相互影响,争抢水分子,甚至争抢紧密层的水分子,引起一系列作用。如果按式(3)和式(4)进行:
|
$ {\left[{{\rm{X}}{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_q}} \right]^ - } \to {\left[{{\rm{X}}{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_{q-1}}} \right]^ -} + {{\rm{H}}_2}{\rm{O}} $ |
(3) |
|
$ \begin{gathered} {\left[{{\rm{M}}{{\left( {{\rm{O}}{{\rm{H}}_{\rm{2}}}} \right)}_p}} \right]^{n + }} + {\left[{{\rm{X}}{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_{q-1}}} \right]^ - } \to \hfill \\ {\left[{{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_{p-1}}{\rm{MHO}}-{\rm{H}} \cdots {\rm{X}}{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_{q-1}}} \right]^{\left( {n -1} \right) + }} \hfill \\ \end{gathered} $ |
(4) |
则产生溶剂共享型离子对。离子对的形成过程是阴离子X-易脱去一个相对弱的以氢键结合的H2O,再与水合阳离子上的水作用时形成相对更强的氢键,即阳离子的强电场使配位水上的H更缺电子,该H更易以H+形式传递到阴离子上;同时阴阳离子靠得更近,因此库仑作用力也更强,所以在能量上形成离子对是有利的。不过在稀溶液时,阴阳水合离子被溶剂分隔较远,能量优势被熵增大抵消,导致稀溶液时不易形成离子对。如果金属M的氧化态高于1,则在高浓度时可以结合不同数量的阴离子,产生多种形式的离子对,如{(H2O)p-2M[HO—H…X(H2O)q-1]2}(n-2)+、{(H2O)p-3M[HO—H…X(H2O)q-1]3}(n-3)+等。
溶液的浓度越高,越易形成离子对。离子对的形成会降低溶液的表观离子浓度,从而降低溶液的导电性。
如果金属的电负性大,电荷高,则形成的水合阳离子不稳定,可以通过降低电荷密度的方法来稳定,其中最有效的稳定方式就是将配位键价键化:
|
$ {\left[{{\rm{M}}{{\left( {{\rm{O}}{{\rm{H}}_2}} \right)}_p}} \right]^{n + }} + {{\rm{H}}_2}{\rm{O}} \to {\left[{{\rm{M}}{{\left( {{\rm{O}}{{\rm{H}}_2}} \right)}_{p-1}}\left( {{\rm{OH}}} \right)} \right]^{\left( {n -1} \right) + }} + {{\rm{H}}_3}{\rm{O}} $ |
(5) |
此过程虽然能使金属配位阳离子稳定化,但却生成了稳定性更差的H3O+,因此会被后者抑制,可以通过调整溶液的酸碱性来控制产物的生成或抑制。对于高电荷的水配位阳离子,能生成多种去质子后的产物,如[M(OH2)p-2(OH)2](n-2)+、[M(OH2)p-3(OH)3](n-3)+等。由于OH-带负电荷,且能形成价键,其配位能力明显强于H2O,它还能与多个金属离子配位形成多核聚合离子:
|
$ \begin{gathered} 2{\left[{{\rm{M}}{{\left( {{\rm{O}}{{\rm{H}}_2}} \right)}_{p-1}}\left( {{\rm{OH}}} \right)} \right]^{\left( {n - 1} \right) + }} \to \hfill \\ {\left[{{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_{p-2}}{\rm{M}}{{\left( {{\rm{OH}}} \right)}_2}{\rm{M}}{{\left( {{\rm{O}}{{\rm{H}}_2}} \right)}_{p-2}}} \right]^{\left( {2n -2} \right) + }} + \hfill \\ 2{{\rm{H}}_2}{\rm{O}} \hfill \\ \end{gathered} $ |
(6) |
显然,此过程发生的前提是水合金属离子的正电荷已被OH-中和到相当低,否则高电荷的阳离子之间同性互斥很难靠近而共用配体OH-。由于聚合的阳离子体积显著增大,容易与大体积的阴离子结合而沉淀出来,工业上常用此原理从溶液中除铁。水合阳离子的聚合度随溶液的碱性提高而迅速增大,聚合的终点通常形成层状结构的氢氧化物。s区金属离子和f区低氧化态金属离子,除Be2+外,一般不容易发生此类水解。d区和p区金属离子容易发生此类水解,且氧化态越高,水解趋势越大,甚至能形成阴离子,如[Sb(OH)6]-。
在卤化物中,只有F-在水溶液中是弱酸根,也会发生酸碱水解,即将氢键转化为共价键:
|
$ \begin{gathered} {\left[{{\rm{OH}}-{\rm{H}} \cdots {\rm{F}}{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_{q-1}}} \right]^ - } \to \hfill \\ {\left[{{\rm{HO}} \cdots {\rm{H}}-{\rm{F}}{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_{q-1}}} \right]^ -} \to {\rm{O}}{{\rm{H}}^ -}\left( {{\rm{aq}}} \right) + {\rm{H}} -{\rm{F}}{\left( {{{\rm{H}}_2}{\rm{O}}} \right)_{q - 1}} \hfill \\ \end{gathered} $ |
(7) |
由于此过程生成了稳定性更差的水合OH-,因此加质子过程会被生成的水合OH-所抑制。如果溶液中存在高浓度的HF,则向F-提供H形成氢键的分子将是HF,形成超强氢键,稳定程度相当于化学键:
|
$ \begin{gathered} {\left[{{\rm{F}}{{\left( {{{\rm{H}}_2}{\rm{O}}} \right)}_q}} \right]^ - } + q{\rm{HF}} \to {\left[{{{\left( {{\rm{FH}}} \right)}_{q-1}}{\rm{F}} \cdots {\rm{H}}-{\rm{F}}} \right]^ -} \hfill \\ + q{{\rm{H}}_2}{\rm{O}} \hfill \\ \end{gathered} $ |
(8) |
该氢键的形成会导致难溶于水的氟化物都易溶于高浓度的HF酸中,HF是金属卤化物的理想溶剂。
如果盐MXn中的M-X化学键强于M-O,则在水溶时保留M-X化学键,即难形成水合阴离子[X(H2O)q]-。此时可能发生的溶解反应有:
|
$ {\rm{M}}{{\rm{X}}_n} + {\rm{p}}{{\rm{H}}_2}{\rm{O}} \to {\rm{M}}{\left( {{\rm{O}}{{\rm{H}}_2}} \right)_p}{{\rm{X}}_n} $ |
(9) |
|
$ \begin{gathered} {\rm{M}}{{\rm{X}}_n} + {\rm{p}}{{\rm{H}}_2}{\rm{O}} \to {\left[{{\rm{M}}{{\left( {{\rm{O}}{{\rm{H}}_2}} \right)}_p}{{\rm{X}}_{n-x}}} \right]^{x + }} + \hfill \\ {\left[{{\rm{M}}{{\rm{X}}_{n + y}}} \right]^{y -}} \hfill \\ \left( {x, y = 0, 1, 2} \right) \hfill \\ \end{gathered} $ |
(10) |
例如,HgCl2在水中的溶解主要采取式(9)的配位方式进行,形成的溶液导电性差;而PbCl2在冷水中难溶,但在热水中可以溶解,主要采用式(10)的方式溶解,即形成的阳离子可以为[Pb(OH2)p]2+和[Pb(OH2)p-1Cl]+,形成的阴离子可以为[Pb(OH2)p-3Cl3]-、[Pb(OH2)p-4Cl4]2-。高氧化态过渡金属或p区金属氟化物在水中溶解易采取式(10)方式,这是由于此时F参与形成的化学键均含有p-dπ键,即形成硬共价双键,稳定性很高,难以破坏;而同时金属元素的氧化态高且有低能量的空的d轨道,因此形成配合物的能力较强。低氧化态的重过渡元素或p区重金属元素的溴或碘化物也可采用式(10)方式溶解,否则难溶解。这是因为这些重金属的d电子多,与Br或I之间易形成软共价双键和配位双键,亲O能力差,而与软阴离子形成配合物的能力强,如[HgBr4]2-、[PbI4]2-等。
一些配位能力弱的阴离子,在溶液较稀时无法参与金属离子的配位,但当水溶液浓度高时,配位取代反应也会发生,即配位水部分甚至全部被阴离子取代:
|
$ {{\rm{[M(O}}{{\rm{H}}_2}{{\rm{)}}_p}{\rm{]}}^{n + }} \to {{\rm{[M(O}}{{\rm{H}}_2}{{\rm{)}}_{p-1}}{\rm{]}}^{n + }}{\rm{ + }}{{\rm{H}}_2}{\rm{O}} $ |
(11) |
|
$ \begin{gathered} {{\rm{[M(O}}{{\rm{H}}_2}{{\rm{)}}_{p-1}}{\rm{]}}^{n + }}{\rm{ + [X(}}{{\rm{H}}_2}{\rm{O}}{{\rm{)}}_{q-1}}{{\rm{]}}^ - } \to \hfill \\ {\left[{{{{\rm{(}}{{\rm{H}}_2}{\rm{O)}}}_{p-1}}{\rm{M}}-{\rm{X}}} \right]^{\left( {n -1} \right) + }} + \left( {q -1} \right){{\rm{H}}_2}{\rm{O}} \hfill \\ \end{gathered} $ |
(12) |
其中式(11)反应有时不需要发生,而是按式(13)一步完成:
|
$ \begin{gathered} {{\rm{[M(O}}{{\rm{H}}_2}{{\rm{)}}_p}{\rm{]}}^{n + }}{\rm{ + [X(}}{{\rm{H}}_2}{\rm{O}}{{\rm{)}}_{q-1}}{{\rm{]}}^ - } \to \hfill \\ {\left[{{{{\rm{(}}{{\rm{H}}_2}{\rm{O)}}}_{p-1}}{\rm{M}}-{\rm{X}}} \right]^{\left( {n -1} \right) + }} + \left( q \right){{\rm{H}}_2}{\rm{O}} \hfill \\ \end{gathered} $ |
(13) |
如果配位取代反应只是在浓度较高时才进行,说明X-的半径比O明显大,配位能力与水相近或甚至比水还弱。因为X-为阴离子,在半径相近时总是阴离子电子更富有,配位更容易。阴离子的配位取代常常分多步逐级进行,形成一系列配位取代产物,如[(H2O)p-2MX2](n-2)+、[(H2O)p-3MX3](n-3)+等,甚至形成配位阴离子,如[MXp](p-n)-,因此溶液中常出现水合离子与各级配位组分并存的情况。例如,当浓度较低时,CuCl2在水溶液中主要表现为水合离子,即[Cu(OH2)4]2+和[Cl(OH2)q]-,而在浓溶液中,尤其是富Cl-的水溶液中[7],则除了水合离子外,还存在配阳离子和配阴离子,如[Cu(OH2)3Cl]+、[Cu(OH2)2Cl2]、[Cu(OH2)Cl3]-和[CuCl4]2-,导致溶液的颜色从稀溶液的浅蓝色过渡到绿色、黄色。
阴离子配位也被称为接触型离子对,与溶剂共享型离子对相比,其作用方式明显不同,配位型的金属离子与阴离子之间通过配位键作用,而溶剂共享型离子对中金属离子与阴离子之间则是通过静电场作用和氢键结合在一起的。
如果阴离子配位能力弱于水,则只有在高浓度水溶液,尤其是接近饱和溶液时,才能出现阴离子配位,相当于结晶的前驱体。
讨论金属卤化物在水中化学溶解前后的能量变化,以探寻溶解度的规律性。金属卤化物在水中的存在形式多样而复杂,首先需要确定溶解后的存在形式才能处理溶解前后的能量变化,以便推导出溶解规律。金属卤化物酸碱性水解虽然存在,但由于受酸碱水解产物的抑制,因此其水解产物在大多数情况下并不是主流,所以讨论的重点放在形成水合离子和形成卤素配合物两种溶解方式上。
在金属卤化物水溶性方面,氟化物与其他卤化物有显著不同,而氯化物、溴化物和碘化物相似,因此将后三种卤化物归并为一类讨论。F与O同周期,价轨道相同,半径也相近,因此成键方式相同,成键能力相近;而Cl、Br和I之间的价轨道类似,且半径都明显大于O,因此成键方式和成键能力均与O有明显差异。
在金属氟化物中,活泼金属或低氧化态金属形成离子键,并以配位单键聚合成离子化合物;而高氧化态的金属氟化物则形成硬共价双键,如果聚合则以配位单键形成大分子。s区元素和f区元素氟化物,除了高氧化态氟化物外,都形成离子晶体,过渡金属和p区金属的低氧化态氟化物也形成离子晶体,M-F配位键键能只有在M呈+1氧化态时与M-O配位键相近且略大,但随着金属氧化态的提高,即M-F键的数量增多,累积的键能差异被放大,即氧化态越高的氟化物配位键转化为M-O配位键将更难;并且随着金属半径的减小键能差异也被放大,即金属半径越小,氟化物配位键也更难转化为M-O配位键。在高氧化态共价氟化物中,同样存在配位键强弱关系M-F>M-O,由于键能差异的累积,前者转化为后者难度更大,因此在水溶时趋向于保留M-F配位键,这导致与离子晶体氟化物的溶解方式不同,即离子晶体类氟化物水溶时形成的阴离子是水合氟离子,而共价型金属氟化物水溶时则趋向于形成含氟配离子,甚至配阴离子。
在金属氯化物、溴化物和碘化物中,活泼的金属形成离子键,并以配位单键聚合成离子化合物。高氧化态的金属则与卤素间形成共价单键,如果聚合则以配位单键形成大分子。而d电子多的低氧化态的d区和p区金属则形成共价双键,并通过配位双键聚合成大分子。活泼的s区元素和f区元素易形成离子化合物,由于Cl、Br、I的半径都明显比O大,因此形成的M-X配位键键强不如M-O配位键,容易转化为水配位,且随金属的氧化态的提高而放大累积的键能差异,也会随金属阳离子半径的减小而放大键能差异,还会随阴离子半径的增大而放大键能的差异,即溶解过程的放热量更大、更有利于溶解。高氧化态的金属氯化物、溴化物和碘化物一般都是共价化合物,包括少数活泼性较差的s区金属,如Be,但更多地出现在过渡金属和p区金属,如Fe、Cr、Al等。其M-X配位键依然不如M-O配位键,且规律相同,因此也易溶于水。只是该类金属阳离子容易发生酸碱水解,根据酸碱水解程度不同而需要加酸来抑制水解。对于d电子多的低氧化态金属卤化物,其情况很不相同,由于M-X配位键是配位双键,而形成的水合离子中的M-O配位键则为配位单键,配位双键强于配位单键,即键能M-X>M-O,所以,水合过程能量上不利,因此较难溶于水。这些低氧化态金属包括过渡金属Cu(Ⅰ)、Ag(Ⅰ)、Au(Ⅰ)、Pd(Ⅱ)、Pt(Ⅱ)、Hg(Ⅱ)等,还有p区金属Tl(Ⅰ)、Pb(Ⅱ)、Bi(Ⅰ)等。
用热力学数据处理溶解是可靠而常用的方法[2, 8],但在教学中对每一种盐的溶解过程都这么处理并不合理,通过典型计算得出规律才更有价值。
根据式(2)形成水合阴阳离子溶解方式,溶解前后的吉布斯自由能变有:
|
$ \Delta G = \Delta H-T\Delta S $ |
(14) |
从熵变上看,金属卤化物一般情况下是晶体,形成离子分散到水中形成溶液,则熵增大;而形成水合离子会将水分子束缚在离子周围导致高度有序,熵减少。由于阳离子通常以配位键结合水,形成的水合离子稳定性高,电场也更强,即会使外围水分子更有序,所以阳离子对熵减影响幅度大,尤其是高氧化金属阳离子,为水溶前后熵变的核心决定因素。对于金属卤化物,通过增熵环节和减熵环节综合考虑得出如下一般规律[9](所有热力学原始数据均来自文献[9],经处理后使用)。
(1) 单核1价阴、阳离子水合,尤其是大体积阴、阳离子,绝大多数情况熵增大,但Li+和F-例外,其半径太小,导致熵减小;2价及以上单核金属离子水合,熵减少。
(2) 金属离子氧化态越高,其水合电场越强,有序度越高,熵减少幅度越大。
(3) 金属离子半径越小,其电场越强,熵减少幅度越大。
从热效应上看,溶解过程主要破坏卤化物中原有的配位键。由于溶解产物是水合离子,金属原子和卤素原子之间在形成化合物时电子转移已经完成,所以水合前后忽略离子键成分Ei影响;而价键中共价成分Ec则相当于阴离子与阳离子间的配位键,属于溶解过程中要破坏的作用力。即,卤化物溶解过程中要破坏的作用力包括包含于价键中的共价键和单体聚合所需要的配位键,这是消耗能量的部分。新产生的作用力则主要是阳离子与水间的新配位键以及阴离子与水间的氢键,这是释放能量的部分。阳离子由于半径往往更小且电荷可以更高,不管是溶解前的化学键还是溶解后形成水合离子,都是溶解热效应的决定因素。
根据前面氟化物和氯化物、溴化物、碘化物中的配位键分析与比较,同时参考水溶解的大量热力学数据,卤化物形成水合离子的溶解热效应有如下规律。
(1) 金属氧化态+1的卤化物的溶解热效应都较小,一般小于±20kJ·mol-1,熵效应对溶解影响较大。
(2) 金属氧化态+2及以上的氟化物,溶解形成水合离子吸热,且氧化态越高,吸热越多,金属半径越小吸热也越多,在吸热多的同时熵减少也越明显。
(3) 不含配位双键的氯化物、溴化物和碘化物溶解形成水合离子一般放热,且金属氧化态越高,放热量越大,金属半径越小放热量越大;同一金属形成的卤化物,水合时放热量由小到大顺序为:氯化物 < 溴化物 < 碘化物,即非金属原子与金属原子结合越不牢,溶解形成水合离子时放热量越大。
(4) 含配位双键的氯化物、溴化物和碘化物溶解形成水合离子的过程是吸热的,同一金属卤化物,溶解焓由小到大顺序为:氯化物 < 溴化物 < 碘化物,即配位双键越强,溶解形成水合离子时吸热量越大。
对于溶解热效应小的卤化物,借助熵增大会有较大溶解度;对于溶解显著吸热的卤化物,则熵效应也很难显著改变溶解度,即一般难溶;对于溶解过程显著放热的盐类,其熵也往往显著减小,尤其在高浓度时熵减幅度更大,因此溶解度虽然很大,但也不会无限可溶。
在金属氟化物中,只有离子晶体类氟化物才以氟水合阴离子为溶解产物,对应s区、f区和低氧化态过渡金属氟化物。根据前面的规律,金属离子半径越大,氟化物的溶解度越大,金属氧化态越低,氟化物的溶解度越大,所以低氧化态而大体积的金属阳离子形成的氟化物均易溶于水。在元素周期表中,从上到下随着原子半径增大,氟化物在水中的溶解度增大,如LiF < NaF < KF;而从左到右离子型氟化物随金属氧化态的提高而溶解度减小,如RbF>SrF2>YF3。
不含配位双键的氯化物、溴化物和碘化物,不管是离子化合物还是共价化合物,都以水合离子方式溶解。由于金属氧化态越高,溶解反应放热越多,因此溶解度均相当大。只是溶解熵随氧化态升高熵减幅度越大,因此溶解度随氧化态并不是单调上升。如NaCl、MgCl2和AlCl3,它们的水溶性都很高,但按溶液中离子总浓度mol·kg-1来计量,393K下溶解度最高的是MgCl2(见表 1)。究其原因,NaCl溶于水的热效应几乎为零,主要靠熵效应促进溶解;而MgCl2和AlCl3的阳离子电荷均很高且半径小,尤其是AlCl3,溶于水时显著放热,但其熵减也随阳离子电荷升高而更显著,导致AlCl3按离子计的溶解度并不是最高。对于同一金属的高氧化态卤化物,由于键的强弱关系为:M-Cl>M-Br>M-I,所以溶解焓满足关系:碘化物 < 溴化物 < 氯化物。
 下载:
导出CSV
下载:
导出CSV
| 卤化物 | 溶解度*/(mol·kg-1) | ΔHΘ(aq)/(kJ·mol-1) | ΔSΘ(aq)/(J·K-1·mol-1) |
| NaCl | 12.3 | 3.9 | 157.6 |
| MgCl2 | 17.2 | -159.6 | -114.7 |
| AlCl3 | 13.7 | -328.0 | -261.7 |
| AgF | -22.5 | -24.7 | |
| AgCl | 65.5 | 33.1 | |
| AgBr | 84.4 | 48.1 | |
| AgI | 112.3 | 68.7 | |
| *以离子总浓度计量 | |||
含配位双键的氯化物、溴化物和碘化物都为共价化合物。由于形成的配位σ键和配位π键相互加强,而转化为水合离子时形成的M-O配位键则只是单键,因此转化在能量上不利,主要体现在溶解热效应上。以低氧化态的银的卤化物为例,见表 1,AgF溶解焓为负,熵变很小,导致其水溶性很高;而其他三种银卤化物溶解焓均为不小的正值,而溶解过程的熵变都不大,因此其溶解度主要由溶解焓决定。由于溶解焓依AgCl < AgBr < AgI的顺序递增,所以溶解度依次下降。
在水溶液中,有三种情况导致配位溶解:高氧化态金属(硬共价双键型)氟化物、共价单键型氯化物的浓溶液和d电子多且氧化态低的金属(软共价双键型)氯化物、溴化物和碘化物。
高氧化态氟化物有多重因素导致M-F化学键的稳定性很高,难以转化为水合离子,这些因素包括F的半径小成键能力强、氧化态高时的稳定性累积效应以及高氧化态时还会存在硬共价双键。这些因素均会导致F-难以形成水合离子,而是直接配位在金属原子上。由于F-的配位,导致金属阳离子配位后的产物在电荷密度上显著下降,提高溶解产物的稳定性,而水分子则可以部分配位,其结果表示如下:
|
$ \begin{gathered} {\rm{M}}{{\rm{F}}_n} + {{\rm{H}}_2}{\rm{O}} \to {\left[{{\rm{MF}}{{\left( {{\rm{O}}{{\rm{H}}_2}} \right)}_{q-1}}} \right]^{\left( {n - 1} \right) + }} + \hfill \\ {\left[{{\rm{M}}{{\rm{F}}_2}{{\left( {{\rm{O}}{{\rm{H}}_2}} \right)}_{q-2}}} \right]^{\left( {n - 2} \right) + }} + \cdots + \hfill \\ {\left[{{\rm{M}}{{\rm{F}}_{q-1}}\left( {{\rm{O}}{{\rm{H}}_2}} \right)} \right]^{\left( {q - n + 1} \right) - }} + {\left[{{\rm{M}}{{\rm{F}}_q}} \right]^{\left( {q -n} \right) -}} \hfill \\ \end{gathered} $ |
(15) |
水分子参与配位的数量越少,要破坏的M-F配位键数量越少,其稳定性越高;配离子所带电荷越少,即外围水分子有序程度越低,溶解时的熵增越明显;形成的配阳离子和配阴离子并存在于溶液中。可以理解,当接近饱和溶液时,还会出现F-作为桥配体配位的情况,相当于结晶前的结晶单元。对于s区和f区元素的典型的离子型氟化物,其金属空的价轨道能量高,难以形成稳定的配位键,因此不会以配位方式溶解,M-F键强只能表现为难溶于水;而d区过渡元素尤其是前过渡元素,均具有低能量的内层d空轨道,因此形成配合物能力很强,容易通过配位方式溶解;p区高氧化态金属也会将外层d轨道能量拉低,因此也具有较强的形成配位键能力,可以配位方式溶解。高氧化态氟化物虽然可以通过配位方式溶解阻止水合氟离子的形成,并降低溶解产物的电荷引起溶解熵增大而有利于溶解;但氟化物结构基元聚合时形成的M-F配位键在水溶时必须要破坏,并用M-O配位键取代,这是不利于溶解的因素;但由于M-O键虽弱于M-F键,在键能上差异却并不太大,金属的高氧化态必然导致要破坏的M-F配位键比例相对较低,配位溶解中吸热量并不太大,熵增因素导致氟化物具有一定的溶解度,总体说来溶解度一般不会很高。如果通过含氟的可溶性盐类(如KF)或氢氟酸,引入外来F-,则溶解过程中所有M-F键都保持不被M-O键替代,配位溶解将更加容易,溶解度会显著提高。
以离子键或共价单键结合为主的氯化物、溴化物和碘化物都易溶于水,其中Cl-的半径虽然大于O,但它为阴离子,比中性的H2O更易供电子,因此两方面综合结果是Cl-的配位能力接近而稍弱于H2O。Br-和I-的半径显著大于O,尽管也带负电荷,其配位能力仍然与H2O差距较大。所以对于那些金属氧化态高、半径小的溴化物和碘化物,即使接近饱和溶液,Br-和I-也一般难以形成配位溶解,其从水溶液中结晶析出时也是形成结晶水配合物,Br-、I-通常在外界。相对而言,氯化物的浓溶液中更易表现Cl-的配位。同理可以推出:(1) Cl-配位时形成与水共配位的配阳离子和配阴离子;(2) s区和f区的氯化物不易表现配位溶解,只有配位能力强的d过渡金属和p区金属才能形成配位溶解;(3)高浓度的Cl-离子溶液(如浓盐溶液和浓盐酸均可)促进配位方式溶解。共价单键型氯化物、溴化物和碘化物,不管是否以配位方式溶解,溶解度均很大。
此类化合物的结构基元通过共价双键结合,而结构基元的聚合则通过配位双键实现,由于金属的氧化态低,聚合时配位双键比例较高。在纯水中,这类化合物既难以以水合离子方式溶解,也不会以配位方式溶解。这是因为不管是形成水合离子溶解还是卤素阴离子配位溶解,都需要将M-X配位双键转化为M-O配位单键,能量上不利;也不能象高氧化态氟化物那样通过阴离子配位来显著降低电荷密度以增大熵效应,因为金属的氧化态已经足够低。但如果创造条件保持原配位双键不被M-O键替代,则溶解仍然可以进行。如使用卤化物的易溶性盐类或氢卤酸提供卤素阴离子,再通过配位而溶解:
|
$ {\rm{M}}{{\rm{X}}_n} + m{{\rm{X}}^- } \to {\left[{{\rm{M}}{{\rm{X}}_{m + n}}} \right]^{m -}}\left( {{\rm{aq}}} \right) $ |
(16) |
因为引入的X-与金属中心之间仍然以配位双键结合,能量上并无不利,所以溶解度会大幅度提高。对于一些金属氧化态趋高的氯化物,M-Cl键与M-O键键能差异还不是很大,通过受热后的熵增效应也能促进配位溶解(降低阴阳离子电荷及电荷密度,溶解熵增大);如:
|
$ \begin{gathered} {\rm{PbC}}{{\rm{l}}_2} + \left( {2p- 4} \right){{\rm{H}}_2}{\rm{O}} \to \hfill \\ {\left[{{\rm{PbCl}}{{\left( {{\rm{O}}{{\rm{H}}_2}} \right)}_{p-1}}} \right]^ + } + {\left[{{\rm{PbC}}{{\rm{l}}_3}{{\left( {{\rm{O}}{{\rm{H}}_2}} \right)}_{p-3}}} \right]^ -} \hfill \\ \end{gathered} $ |
(17) |
而溴化物和碘化物则由于M-X键与M-O键的键能差异太大,不易以这种方式溶解。此类配位溶解的规律为:(1)配位溶解的能力除与卤素阴离子浓度有关外,还与卤素阴离子种类有关;卤素阴离子浓度越高,溶解度越大;卤素阴离子半径越大,形成的配位双键越强,因此阴离子促进溶解由小到大的顺序为Cl- < Br- < I-;(2)d区过渡金属,尤其是后过渡金属,以及p区重金属,都富有d电子,氧化态低时都易形成配位双键,容易以此配位方式溶解。
在水溶性方面,金属氟化物与金属氯化物、溴化物和碘化物有明显差异。对于金属氟化物,当金属呈+1氧化态时,除LiF微溶外,其他均易溶于水,且随金属离子半径的增大而更易溶,主要是熵效应导致其溶解;碱土金属和f区金属氟化物难溶于水,溶解焓变和熵变均导致其难溶;过渡金属和p区金属的高氧化态氟化物主要以阴离子配位方式溶解,这是由于金属的配位能力强所致,在该过程中形成的配离子电荷密度低,以及相应的熵效应也都有利于这类氟化物的溶解,其溶解度一般不会很大。
对于金属氯化物、溴化物和碘化物,当金属为s区、f区以及d区和p区高氧化态时,其卤化物均易溶于水,其中碱金属卤化物主要是熵效应导致溶解;当金属氧化态为+2及以上时,主要是溶解焓效应导致其溶解。d区和p区低氧化态金属卤化物,尤其是碘化物,水溶性小,主要是因为溶解焓效应导致其难以溶解,但可以通过引入重卤素离子配位溶解。
如果将热力学数据与卤化物结构和化学键关联,则更易理解卤化物的水溶性规律,解决学生的困惑。
M Monroe, K Abrams. J. Chem. Edu., 1984, 61:885. doi: 10.1021/ed061p885
W G Van Der Sluys. J. Chem. Edu. 2001, 78:111~115. doi: 10.1021/ed078p111
Findit Database, 2009.
CRC Handbook of Chemistry and Physics, 84th Ed. 2003~2004.
王稼国, 荆西平.化学通报, 2016, 79(9):864~875. http://www.hxtb.org/ch/reader/view_abstract.aspx?flag=1&file_no=20160113005&journal_id=hxtb
王稼国, 荆西平.化学通报, 2017, 80(4):400~407. http://www.hxtb.org/ch/reader/view_abstract.aspx?flag=1&file_no=20160819001&journal_id=hxtb
M D Collings, D M Sherman, K V Ragnarsdottir. Chem. Geol., 2000, 167(1/2):65~73.
L Eisen, N Marano, S Glazier. J. Chem. Edu., 2014, 91:484~491. doi: 10.1021/ed4005563
Langes Chemistry Handbook, 15th Ed. 2003.

图 2 金属卤化物中的化学键
Figure 2 Chemical bonds of metal halides
(a)离子键;(b)共价单键;(c)硬共价双键;(d)软共价双键;(e)配位单键;(f)配位双键
表 1 典型卤化物的溶解度和水溶热力学数据
Table 1. Solubilities and hydrolytic thermodynamic data of typical halides
| 卤化物 | 溶解度*/(mol·kg-1) | ΔHΘ(aq)/(kJ·mol-1) | ΔSΘ(aq)/(J·K-1·mol-1) |
| NaCl | 12.3 | 3.9 | 157.6 |
| MgCl2 | 17.2 | -159.6 | -114.7 |
| AlCl3 | 13.7 | -328.0 | -261.7 |
| AgF | -22.5 | -24.7 | |
| AgCl | 65.5 | 33.1 | |
| AgBr | 84.4 | 48.1 | |
| AgI | 112.3 | 68.7 | |
| *以离子总浓度计量 | |||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: