 图1
基于不同换能器的内毒素检测生物传感器分类
Figure1.
Classification of biosensor for endotoxin based on transduction methods
图1
基于不同换能器的内毒素检测生物传感器分类
Figure1.
Classification of biosensor for endotoxin based on transduction methods
引用本文:
温路新, 徐溢, 项松涛, 陈李. 基于生物传感器的内毒素检测研究进展[J]. 化学通报,
2018, 81(1): 29-36.

Citation: Wen Luxin, Xu Yi, Xiang Songtao, Chen Li. Progress in Endotoxin Detection Based on Biosensor[J]. Chemistry, 2018, 81(1): 29-36.
Citation: Wen Luxin, Xu Yi, Xiang Songtao, Chen Li. Progress in Endotoxin Detection Based on Biosensor[J]. Chemistry, 2018, 81(1): 29-36.
基于生物传感器的内毒素检测研究进展
English
Progress in Endotoxin Detection Based on Biosensor
Abstract:
Endotoxin is well established as a main reason for endoxemia and organs failure, which are severely harmful to human health. So far, much more attention has been paid to develop a efficient and sensitive detection method with high specificity. Because of the superior efficiency and sensitivity, easy to automation and miniaturization, biosensors which is confronted with substantial development space is playing an increasingly important role in the field of endotoxin detection. In this paper, the detection methods of endotoxin in recent years were introduced briefly, and then paid special emphasis on summarizing the optical and electrochemical biosensors applied in endotoxin detection. The challenges and development prospects of endotoxin detection by biosensors are also reviewed.
-
Key words:
- Endotoxin
- / LPS
- / Biosensor
- / Ultrasensitive detection
-
细菌内毒素是革兰氏阴性菌细胞壁的成分[1],于细菌死亡解体后释放出来,由脂多糖(LPS)、蛋白质和磷脂组成,其活性成分是LPS。LPS是一种具有结构多样性的大分子复合物,同时其又具有抗原性和毒素性质,它由O-特异性链、核心多糖和类脂A(Lipid A)三部分组成。其中,类脂A是LPS活性不可缺少的部分,可视作LPS的“生物活性中心”,是抗原活性部位,保存着LPS的各种生物学活性[2]。细菌内毒素在体内会作用于单核巨噬细胞,产生多种炎症细胞因子,如肿瘤坏死因子、白细胞介素(IL)-1、前列腺素、凝血素、干扰素、血小板激活因子等。这些因子适量时可激活免疫系统,对机体产生有益作用,过量时则通过粘膜进入血液,造成内毒素血症[3],从而引起机体严重的病理生理反应,表现为发热[4]、多器官功能衰竭(MOF)[5]、低血压、心动过速、休克[6]甚至死亡。因而,内毒素对食品加工、药物治疗和药物生产等过程的安全存在着巨大的威胁,内毒素的高效检测成为医疗器械、食品、药物以及生物制品等使用前必不可少的环节。
细菌内毒素检测最早采用的方法是家兔热源法(The rabbit pyrogen test,RPT)[7],由于其反应存在个体差异、敏感度不高、费时等缺点导致其有被逐渐淘汰的趋势。鲎试验法(Limulus amoebocyte lysate assays,LAL)[8~10]是目前最主要的内毒素检测方法,具有快速、简便、重复性好、灵敏度高(0.01~1 ng/mL)、易推广和标准化的优点,成为各国药典细菌内毒素检测的首选方法。但其检测结果容易受到酶和杂质的影响,反应条件苛刻,据中国药典(2015版)记载,鲎试验法需要使用无热源的水,并在pH范围6.0至8.0、温度37C±1℃的恒温条件下进行。此方法不仅容易对碳水化合物类似的分子如β-葡聚糖产生响应而造成假阳性结果[11],还存在非热源底物同时参与凝胶形成的问题,难以排除增强或抑制凝胶形成的其他因素[12]。酶联免疫测定法(Enzyme linked immunosorbent assay,ELISA)[13]是一种将抗原和抗体的特异性免疫反应和酶的高效催化作用有机结合起来的检测技术,具有简便、快速、灵敏等特点,近年来逐步得到了广泛的发展和应用,但该类方法建立在抗原与抗体相互作用基础之上,在高温、强酸、强碱等检测环境中容易变性,复杂样品的基质干扰大,使得检测结果易出现假阳性。
生物传感器是利用生物识别探针与被测分析物相互作用,由此产生光学、电学或磁性信号等响应信号,以此实现目标物检测的分析器件或系统[14, 15],其通常由生物敏感元件和转换器两部分构成。生物传感器的生物敏感元件主要来源于生物活性物质,如酶、抗原、抗体和各种功能蛋白质等,而信号转换器主要有微电极(如电位、电流的测量)、光学检测元件、热敏电阻、压电石英晶体及表面等离子共振器件等。当待测物与生物敏感层发生相互作用后,产生的物理化学信号变化可通过信号转换器转换而输出电化学、光学、热学、压电等响应信号,解析这些信号与待测物之间的关联性,从而达到分析检测待测物的目的。大量研究显示,生物传感器具有选择性好、灵敏度高、分析速度快、成本低、能在复杂的体系中进行在线连续监测等优势[16],尤其是它高度自动化、微型化与集成化的特点,近年来在研究与技术开发领域获得蓬勃发展。生物传感器的巨大优势使其在内毒素研究中也显示出良好的应用前景,目前已有大量生物传感器用于内毒素的检测。光学生物传感器[17]、电化学生物传感器[18~20]和压电晶体生物传感器[21, 22]是内毒素检测中备受关注的热点(图 1)。
图1
基于不同换能器的内毒素检测生物传感器分类
Figure1.
Classification of biosensor for endotoxin based on transduction methods
1 光学生物传感器
光学生物传感器[23]是将具有分子识别和换能作用的指示剂、染料、酶、受体、抗原抗体等固定在光导纤维、平面波导等换能器上,通过其对样品中的待测物进行选择性识别作用,然后转换获得各种光信号(如荧光、化学发光或生物发光、光吸收和折射率变化、拉曼散射光等)输出的一类传感器。根据传感界面光信号的产生方式,将其分为荧光生物传感器(光信号作为激发源)、电化学发光生物传感器(电信号作为激发源)、表面等离子体共振生物传感器(无激发源)。
1.1 荧光生物传感器
荧光生物传感器通过将生物识别元件上的生物信号转换成荧光信号来实现对内毒素的检测。传感器中采用的生物敏感层是内毒素的受体和识别探针,包括适配体、结合蛋白CD14、多粘菌素B(Polymyxin B,PMB)等,其结构设计和优化是这类生物传感器实现高选择性、高灵敏度和快速响应的核心和研究热点。
Jamaes等[24]]提出将PMB共价固定在光纤探头表面,构建光导纤维生物荧光传感器,其能选择性结合荧光标记的LPS,而未标记的LPS可以通过与荧光标记的LPS竞争,使之减少。用于血浆和污染的水中内毒素的测量,可在30s产生响应,进而根据荧光的减弱程度对内毒素浓度进行检测,检出限可达到10ng/mL。该传感器不仅能探测LPS,同时其还能对LPS的血清类型以及其他能俘获LPS分子的物质,如抗体、外源凝集素或抗生素等进行测试。Voss等[25]将羧基四甲基若丹明和羧基荧光素两种荧光染料分别接在CD14的氨基端和羧基端,制备了荧光标记的分化抗原14(Cluster of differentiation 14,CD14),构建了CD14衍生多肽-LPS生物荧光传感器。两种荧光基团结合到多肽上形成二聚体,测试时LPS可改变CD14衍生多肽构象,引起荧光猝灭,对LPS的检出限可达到10μmol/L。
为了进一步提高传感器的选择性和灵敏度,Wu等[26]针对LPS的结构特征,以萘二甲酸为荧光探针,丁二炔衍生的脂质a和丁二炔衍生的脂质b在紫外光的照射下发生聚合作用,形成聚乙二炔脂质体,以其作为生物荧光传感器的敏感层。两个不同的丁二炔脂质物a、b以1:9的量混合时,彼此空间结构改变,能量转移,使脂质体a的荧光完全被猝灭。当溶液中存在LPS时,LPS与脂质a结合,使得荧光恢复,该方法对LPS的检测限达到0.1μmol/L。Lan等[27]合成了基于水溶性复合噻吩的3-苯基噻吩(CPT1)荧光探针,CPT1的季铵结构和1, 4-二氮杂二环辛烷(DABCO)不仅提高了水溶性而且提供两个正电荷中心,通过静电作用与LPS的负电荷相结合;同时,DABCO还对磷酸基具有很强的亲和力。噻吩环3位引入苯基形成一个空缺结构,促使噻吩形成弯曲的结构,当存在LPS时,CPT1与LPS结合,构型会发生改变,构型变化从而引起吸收波长和荧光波长的红移,荧光强度也会发生明显的变化,可用于内毒素的超灵敏检测,检测限可达到270pmol/L。
由于传统的羟基荧光素、若丹明等有机荧光染料存在光稳定性差、容易发生光漂白且激发谱窄等局限和不足,促成了量子点的提出和快速发展,其大大提升了荧光标记的效率。Ebrahim等[28]采用巯基乙酸(TGA)包覆CdTe量子点,再将碳量子点连接上具有许多糖类结合位点的伴刀豆球蛋白素A(Con A),构建了一种新的CdTe QDS-ConA生物敏感层,用于LPS选择性和特异性的测定。另外,CdTe QDS-ConA还可作为荧光指示剂来捕获粘质沙雷氏菌,通过与粘质沙雷氏菌产生的LPS结合,显示出极高的检测灵敏度,检出限可达到10fg/mL。
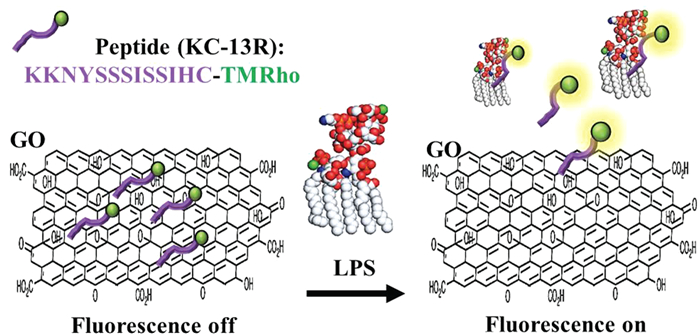
近年来,基于石墨烯材料的荧光共振能量转移(FRET)传感器的研究逐渐增多。石墨烯和碳纳米管等纳米材料具有高度离域化的大π键,作为天然的荧光猝灭剂,ssDNA可通过π-π堆积作用吸附在纳米材料表面,将其用于构建分子信标模式检测时,可以省去标记的复杂步骤。Lim等[29]在氧化石墨烯(GO)的表面吸附上以四甲基若丹明乙酯为标记试剂的多肽,构建了一种灵敏且具有高选择性的LPS生物荧光传感器。多肽的半胱氨酸中的羧基通过硫醇与四甲基若丹明乙酯(TMRho)反应形成带有荧光的多肽(KC-13R)。由于GO的静电作用和π-π堆积作用,使得KC-13R能量转移而荧光猝灭;存在LPS时,其与石墨烯表面的多肽结合,结合物从石墨烯表面脱落,体系的荧光恢复(图 2)。其对LPS的检出限可达到130pmol/L,用于临床注射液的检测,能从不同的菌株中选择性地检测LPS。
荧光生物传感器具有极高的检测灵敏度和光学稳定性以及快速的信号产生和读取速度,加上近年来光纤技术的发展与应用,使得基于荧光检测的生物传感器在内毒素的检测中展现了巨大的前景。但该类生物传感器的传感层设计与构建耗时长,容易受到背景光的影响,抗干扰能力差,存在非特异性结合,限制了其应用范围。随着新材料制备技术的发展,各种新型荧光标记材料大量涌现,荧光生物传感器在光学生物传感器领域依旧占据最主要的地位。
1.2 电化学发光生物传感器
电化学发光生物传感器是将生物分子固定在电极上,并将生物识别分子和目标分析物结合产生的生物信号转换为可检测的电化学发光信号的一种分析器件,兼具有电化学和电化学发光的优点。光信号的产生由电压控制,使其具有能方便控制发光反应的时间和位置[28]的独特优势。Chaney等[30]利用钌标记的核苷酸探针与细菌的rRNA杂化,链霉亲和素包覆的磁珠捕获标记的rRNA,富集后的rRNA沉积在电极表面,通电后即获得电化学发光信号。将其用于革兰氏阴性菌和革兰氏阳性菌的检测,检测范围在105~108 CFU/mL之间。Shelton等[31]设计了一个“三明治”抗体型检测系统,采用磁珠捕获富集E.coliO157,利用电化学发光法实现对E.coliO157的高效检测,检测限达到25CFU/100mL。Xie等[32]基于自组装四面体DNA树枝状聚合物构建了用于LPS的超灵敏检测的电化学发光适体传感器,将阿霉素(Dox)与电化学发光体N-氨基丁基N-乙基异鲁米洛(ABEI)偶联,形成新类型的指示剂(Dox-ABEI),用于非共价吸附dsDNA。自组装的四面体DNA树枝状大分子,作为有效的纳米载体,为Dox-ABEI提供了高效的结合位点,从而有效地放大电化学发光信号,方法检测限可达0.18fg/mL。
电化学发光传感器具有反应速度可控制和发光物质稳定性高两个突出的优点。但它也存在传感层材料可选择性不大、测试过程较为复杂、许多发光试剂的发光效率不高且毒性大等缺点,从而极大限制了其应用。因此寻求高效低毒价廉的新型发光试剂己成为研究者的探索的热点之一。
1.3 表面等离子体共振检测
表面等离子体共振(Surface plasmon resonance,SPR)生物传感器正成为测量分子相互作用的有效方法。SPR生物传感器法目前多用于检测在薄膜金属表面附近络合物形成导致结构变化引起的折射率变化值。其核心是先固定一种反应物在薄膜金属表面上,然后测定另一种反应物与之相互作用而带来的表面结构和光学性能的变化。Barlen等[33]通过调整金属膜材料和棱镜材料以及形状等参数,对实验光路进行优化。利用该SPR生物传感器对沙门氏菌和沙门氏菌血清同时进行检测,所需样本量少(只需要10μL),操作简单,且无需样品稀释,检测限达到102CFU/mL。SPR生物传感器具有能实时检测生物分子的相互作用和无需标记物等独特优点[34, 35],因而被广泛应用。
2 电化学生物传感器
电化学生物传感器是将生物敏感元件与电化学换能器结合的生物传感器[36],其需要在电极表面固定识别敏感层,并通过其与待测物之间特异性的识别作用可选择性识别待测物并将待测物捕获固定到电极表面。待测物在电化学体系的固/液接触面发生物理或化学变化引起电极表面的电化学特性改变,通过电极作为信号传导器对电极表面的敏感层的固定情况、生化反应动力学过程等信息进行测试,最终达到分析检测的目的。这方面检测技术主要包括电流分析法、伏安法、电位分析法和阻抗分析法等[37, 38]。电化学生物传感器具有分析速度快、选择性好、灵敏度高等特点,其已成功用于革兰氏阴性菌及其内毒素的鉴别和定量分析[39]。
2.1 电化学阻抗传感器
电化学阻抗生物传感器是通过在电极表面固定受体(如适配体、核酸、噬菌体或凝集素),细菌内毒素与生物受体结合后,电极表面的电流减小而阻抗值增加,通过阻抗值的变化或电容的变化来检测内毒素。由于电化学阻抗法具有良好的界面表征作用,微小振幅正弦电压或电流不会对内毒素分子造成干扰[40~42],敏感性高,使其在内毒素检测中具有广阔的应用前景。
Su等[43]以能特异性识别LPS的适配体(单链DNA,ssDNA)作为电化学生物传感器的探针,设计了一种基于核酸适配体的电化学阻抗传感器。首先将适配体固定在具有自组装单层巯基丙酸(MPA)膜的金电极表面,MPA的羧基与适配体5位的氨基结合产生肽键,使适配体牢固地固定在金电极表面,适配体对LPS具有高度的结合力,且与LPS浓度在1pg/mL~1ng/mL范围内有良好的线性关系,检测限达到1pg/mL。Cho等[44]利用Cu2+和氮三乙酸(NTA)对脂多糖O-侧链的吸附性,设计了基于NTA-Cu的电化学生物传感器,检测限达到0.1pg/mL,进一步提高了检测灵敏度。
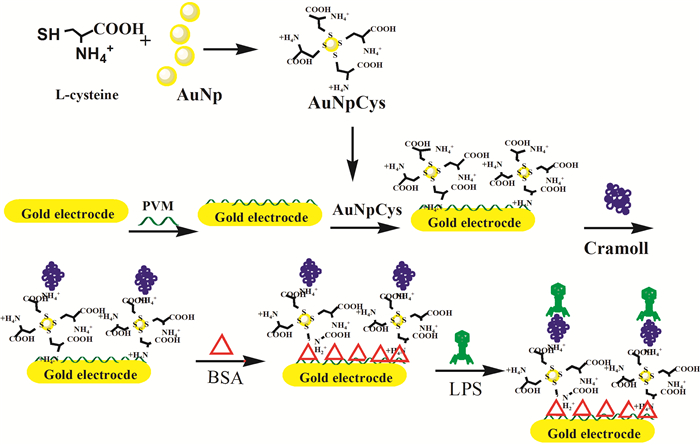
金纳米粒子(AuNPs)具有较大的比表面积、较好的生物相容性以及较高的表面能量,允许电活性粒子和电极材料之间的电子快速和直接转移,能为生物分子提供良好的固定界面,使其在生物传感器中得到广泛应用。Oliveira等[45]在金电极表面设计制作了PVM-AuNpCys-CramoLL-BSA模拟仿生界面,即在金电极表面先结合上带负电荷的PVM (Poly vinyl chloride-co-vinyl acetate-co-maleic acid),PVM与表面结合了L-半胱氨酸的AuNPs进一步结合得到基底。PVM能与CramoLL凝集素通过静电作用结合,最后加入牛血清蛋白BSA,形成PVM-AuNpCys-CramoLL-BSA界面(图 3),由于CramoLL凝集素能选择性地与不同细菌内毒素结合,采用电阻抗分析法可实现对内毒素的灵敏检测。
金纳米粒子(AuNPs)具有较大的比表面积、较好的生物相容性以及较高的表面能量,允许电活性粒子和电极材料之间的电子快速和直接转移,能为生物分子提供良好的固定界面,使其在生物传感器中得到广泛应用。Oliveira等[45]在金电极表面设计制作了PVM-AuNpCys-CramoLL-BSA模拟仿生界面,即在金电极表面先结合上带负电荷的PVM (Poly vinyl chloride-co-vinyl acetate-co-maleic acid),PVM与表面结合了L-半胱氨酸的AuNPs进一步结合得到基底。PVM能与CramoLL凝集素通过静电作用结合,最后加入牛血清蛋白BSA,形成PVM-AuNpCys-CramoLL-BSA界面(图 3),由于CramoLL凝集素能选择性地与不同细菌内毒素结合,采用电阻抗分析法可实现对内毒素的灵敏检测。
Su等[46]利用AuNPs的信号放大作用在金电极表面沉积上AuNPs,设计了一个基于适配体的阻抗传感器。在AuNPs表面沉积上硫醇化的核酸适配体(ssDNA),使适配体覆盖在AuNPs表面,用来测量浓度范围在0.01~10.24 ng/mL的内毒素,检出限低至0.005ng/mL,可实现内毒素的超灵敏检测。
2.2 电位型传感器
电位型传感器是基于待测物质与识别元件相结合后诱导电位发生变化进行检测的一类生物传感器[47, 48],目前广泛应用于水体中内毒素检测。Gustavo等[49]以适配体作为受体,构建的以网状单璧碳纳米管(SWCNTs)为换能器的电位型传感器的响应时间不超过30s,重现性能与稳定性能良好。用于金黄色酿脓葡萄球菌的实时监测,检测限可达到8×102CFU/mL。
电位型传感器与DNA杂交技术相结合,制备具有单一识别性能的“三维探针”电位型传感器是近年来传感器研究的热点课题之一。Bai等[50]设计了一个结合DNA杂交技术和纳米放大技术的电化学生物传感器,对内毒素实现了快速超灵敏检测。在电极表面固定上捕获探针,捕获探针可以与DNA1杂交,当加入辅助探针后,辅助探针打开发卡结构,捕获探针、DNA1和辅助探针形成三向的“Y”结构。当存在内切酶时,DNA1能够从杂交“Y”结构中释放出来并与LPS结合。释放出DNA1以后,大量的捕获探针能在这循环过程中结合上用纳米金和石墨烯标记DNA2,而纳米金和石墨烯材料有利于信号的放大。此方法用于内毒素检测时灵敏度极高,检测限达到8.7fg/mL。在实际样本中也能获得良好的效果。
2.3 电流型传感器
电流型传感器是通过给电极和电解质溶液组成的界面施加一个恒定电位,将目标物氧化或还原,由于氧化还原反应中电流大小与浓度成正比,因此通过将电流作为信号输出,得到电流与目标物质浓度的线性关系,从而实现对目标物质的检测。电流型传感器以其低消耗、高信噪比以及应用电压可控等优点[51~54]在内毒素检测中发挥着巨大的作用。
Miao等[55]在三电极体系下完成了鲎试剂凝胶形成过程,采用电流分析法实现了内毒素的快速检测,检测限达到0.06ng/mL。Lin等[56]在丝网印刷碳电极(Screen-printed carbon electrodes,SPCEs)上设计了一个基于电流测定的生物传感器,用于大肠杆菌E.coli O157:H7的特异性测定。SPCEs由银和碳墨混合组成,再采用双抗体“三明治”的酶联免疫法测定。在电极上固定上第一种大肠杆菌E.coli O157:H7的抗体,在第二种大肠杆菌E.coli O157:H7抗体上连接上过氧化氢酶(Horseradis peroxidase,HRP)。在恒定+300mV电压下,过氧化氢与HRP、二茂铁甲酸(FeDC)以及AuNPs结合。在外加电压作用下,AuNPs被吸附到工作电极上,响应电流被放大达到13倍,显示出AuNPs和FeDC的联合效应,最后检测限可达到6CFU·strip-1。
总之,电化学生物传感器作为一种将生物组织或细胞中的生物大分子和活性小分子(如酶、蛋白、抗体、微生物、核酸等)与电化学信号检测手段相结合构建起来的装置,在临床医学[57, 58]、药物检验[59, 60]、食品分析[61]和环境监测[62, 63]等领域得到广泛应用,是目前发展最成熟的一种传感器。针对内毒素检测,电化学生物传感器相对于光学生物传感器而言具有稳定性更好、灵敏度更高的特点。但其也同样存在传感界面的构建复杂、成本高、生物识别原件的寿命不长以及非特异性结合等问题和难点。因此,开发低成本与集成化的高通量电化学生物传感器是当前及今后的重点研究方向。
3 压电晶体生物传感器
压电晶体生物传感器广泛应用于细菌、病毒等的检测,其基本原理是利用固定在压电传感器表面的生物功能物质选择性或特异性地结合待测物,引起电极上质量的微小变化(质量型)或电极所处的被测溶液的物理性质(密度、粘度、电导率、介电常数)的微小变化(非质量型),这个微小的变化使谐振电路的谐振频率发生变化[64]。通过测量频率的变化值,从而推算出被测溶液的浓度。
Keat等[65]发现了一种压磁应力传感器(Stress-respinsivemagnetoelastic sensors),将带状的传感器浸入鲎试验的溶液中,其能感应鲎试验法中的凝胶形成过程,内毒素浓度和由振幅时间曲线一阶导数决定的最大凝结速度有着极好的相关性。在0.12ng/mL鲎凝胶试验中,该传感器能在20min内探测到0.03ng/mL的内毒素。Muramatsu等[66]在内毒素样品溶液中加入鲎试剂,根据溶液粘度的变化和镀钯的石英晶体的共振频移与电阻变化关系,可检测内毒素浓度,方法的最低检测限为1pg/mL,用类似方法在纤维蛋白质浓度测量中也获得满意结果。
Sheikh等[67]设计了一个电磁压电传感器(Elect-romagnetic piezoelectric acoustic sensor,EMPAS),利用多粘菌素B与内毒素的结合能力,将PMB固定在压电晶体上用于测定内毒素,测试的频率较大、特异性好;同时,在石英谐振器表面的硅氧烷对内毒素不仅具有吸附作用,且对其他生物基质具有防阻塞的作用,可消除由于被测液粘度引起的非质量效应。在血液样本中检测限达到10ng/mL以下。
 表 1
不同类型生物传感器的比较
Table 1.
Comparison in terms of general advantages and disadvantages associated with individual biosensor
表 1
不同类型生物传感器的比较
Table 1.
Comparison in terms of general advantages and disadvantages associated with individual biosensor
传感器 分类 优点 缺点 检测限 光学生物传感器 荧光生物传感器 检测灵敏度高,信号读取速度快,传感界面设计简单 容易受到背景光的影响,抗干扰能力差,存在非特异性结合 10fg/mL 电化学发光生物传感器 具有电化学和电化学发光的优点,能方便控制发光反应的时间和位置的独特优势 传感层材料可选择性不大,测试过程较为复杂,发光试剂的发光效率不高且毒性大 0.18fg/mL SPR生物传感器 能检测生物分子的相互作用,且无需标记物 检测过程复杂,对敏感层材料要求高 0.7ng/mL 电化学生物传感器 电化学阻抗传感器 具有良好的界面表征作用,微小振幅正弦电压或电流不会对内毒素分子造成干扰 传感界面的构建复杂,成本高,生物识别元件的寿命短 0.1pg/mL 电位型传感器 响应时间快,对样本不会造成破坏 需要参比电极,电极容易被污染 8.7fg/mL 电流型传感器 低消耗,高信噪比,应用电压可控 分析物消耗多,需要氧化还原介质 0.06ng/mL 压电晶体生物传感器 - 检测灵敏度高,对微小质量变化都具有快速响应,抗干扰能力强 质量变化和频率转换之间缺乏精确的相关性 0.03ng/mL 压电传感技术广泛用于生物传感领域,测量技术对晶体表面的质量变化和石英晶体处的环境如溶液的粘度、密度、导电率等的变化都非常敏锐。通常具有纳克级的质量检测灵敏度,能够敏感地测量微观反应过程中的微小变化并转化为可以定量检测的频率信号,为内毒素检测提供了一种敏锐、快捷且可靠的检测手段。
4 结语
内毒素是革兰阴性菌感染的致病因子,是威胁人畜健康的主要因素。传统标准内毒素检测方法(鲎试剂法)检测速率快、灵敏度高,但抗干扰能力差,不能完全满足诊断的要求。生物传感器的问世为内毒素的检测带来了新希望,不仅提高了灵敏度,使测定过程变得更为简单,而且便于实现自动化,使内毒素的快速高效检测成为可能。光学生物传感器和电化学生物传感器是内毒素检测中应用最为广泛的两类传感器,其均具有检测速度快、灵敏度高的优势。但同时其也存在识别元件和传感界面设计与构建复杂及使用寿命短的局限。
传感界面上生物分子探针的构建和效能,很大程度上决定了生物传感检测的性能,因此发展具有更好选择性、特异性和稳定性的识别元件和传感界面是目前生物传感器的重要研发方向,而新颖的生物偶联技术是未来内毒素生物传感器设计和研制的基石。如何调控生物探针在界面上的分布,减少生物探针与待测物以外其他分子在传感界面的非特异性吸附已成为该领域的挑战性问题之一。通过引入纳米技术发展出刚性的三维结构探针,可实现探针之间的有序的布控,为生物分子界面提供新的途径,更提高了界面生物识别能力,进而显著提升了生物传感器的检测能力。同时,未来生物传感器应开发适合不同领域的新型换能器,如在临床注射剂的内毒素检测中,为了保证卫生和避免交叉感染,应设计廉价的一次性传感器;在药物生产或环境的内毒素检测中,应设计寿命长、可以实现在线检测的传感器。总之,伴随材料技术、生物技术、计算机技术等的快速发展,生物传感器面临的诸多挑战将有望得到完善,在微生物毒素检测中的应用会越来越广泛。
-
-
[1]
A H Ding, F Porteu, E Sanchez et al. Science, 1990, 248(4953):370~372. doi: 10.1126/science.1970196
-
[2]
APreston, R E Mandrell, B W Gibson et al. Crit. Rev. Microbiol., 1996, 22(3):139~180. doi: 10.3109/10408419609106458
-
[3]
J Bhattacharyya, S Biswas, A G Datta. Curr. Med. Chem., 2004, 11(3):359~368. doi: 10.2174/0929867043456098
-
[4]
N Hirata, Y Yanagawa, M Satoh et al. Cell. Immunol., 2010, 261(1):37~41. doi: 10.1016/j.cellimm.2009.10.009
-
[5]
K Lawrence. Crit. Care Nurs. Clin. North Am., 2011, 23(2):323~327. doi: 10.1016/j.ccell.2011.02.005
-
[6]
K Brandenburg, A B Schromm, T Gutsmann. Curr. Top. Med. Chem., 2004, 4(11):1127~1146. doi: 10.2174/1568026043388213
-
[7]
C Y Park, S H Jung, J P Bak et al. Biologicals, 2005, 33(3):145~151. doi: 10.1016/j.biologicals.2005.04.002
-
[8]
J F Cooper, J Levin, W H Jr. J. Lab. Clin. Med., 1971, 78(1):138~143.
-
[9]
J S Bolden, R E Warburton, R Phelan et al. Biologicals, 2016, 44(5):434~440. doi: 10.1016/j.biologicals.2016.04.009
-
[10]
T Liu, W Zhang, L Zhou et al. Anal. Chim. Acta, 2017, 961(1):106~111.
-
[11]
J F Cooper, M E Weary, F T Jordan. PDA J. Pharm. Sci. Tech., 1997, 51(1):2~6.
-
[12]
P F Roslansky, T J Novitsky. J. Clin. Microbiol., 1991, 29(11):2477~2483.
-
[13]
A H Mohammed, D E Mccallus, N L Norcross. Vet. Microbiol., 1988, 18(1):27~39. doi: 10.1016/0378-1135(88)90113-7
-
[14]
M I Prodromidis. Electrochim. Acta, 2010, 55(14):4227~4233. doi: 10.1016/j.electacta.2009.01.081
-
[15]
A Turner. Trends Biotechnol., 2013, 31(3):119~120. doi: 10.1016/j.tibtech.2012.10.002
-
[16]
G S Wilson, R Gifford. Biosens. Bioelectron., 2005, 20(12):2388~2403. doi: 10.1016/j.bios.2004.12.003
-
[17]
X Fan, I M White, S I Shopova et al. Anal. Chim. Acta, 2008, 620(1/2):8~26.
-
[18]
A Zuzuarregui, D Souto, E Pérez-Lorenzo et al. Analyst, 2015, 140(2):654~660. doi: 10.1039/C4AN01324G
-
[19]
A Zuzuarregui, M C Morant-Minana, E Perez-Lorenzo et al. IEEE Sens. J., 2013, 14(1):270~277.
-
[20]
C Luo, Y Yan, T Yu et al. Electroanalysis, 2012, 24(5):1186~1191. doi: 10.1002/elan.201100700
-
[21]
江奇峰, 陈龙聪, 高斌等.重庆医学, 2013, 42(19):2185~2187. doi: 10.3969/j.issn.1671-8348.2013.19.001
-
[22]
X L Xiong, S M Wang, Y Zhang et al. Appl. Mech. Mater., 2012, 195~196(1):874~878. http://www.scientific.net/AMM.195-196.874
-
[23]
S M Borisov, O S Wolfbeis. Chem. Rev., 2008, 108(2):423~427. doi: 10.1021/cr068105t
-
[24]
E A James, K Schmeltzer, F S Ligler. Appl. Biochem. Biotechnol., 1996, 60(3):189~202. doi: 10.1007/BF02783583
-
[25]
S Voss, R Fischer, G Jung et al. J. Am. Chem. Soc., 2012, 129(3):554~561.
-
[26]
J Wu, A Zawistowski, M Ehrmann et al. J. Am. Chem. Soc., 2011, 133(25):9720~9723. doi: 10.1021/ja204013u
-
[27]
M Lan, J Wu, W Liu et al. J. Am. Chem. Soc., 2012, 134(15):6685~6694. doi: 10.1021/ja211570a
-
[28]
S Ebrahim, M Reda, A Hussien et al. Spectrochim. Acta A, 2015, 150(1):212~219.
-
[29]
K L Seng, P Chen, F L Lee et al. Anal. Chem., 2015, 87(18):9408~9412. doi: 10.1021/acs.analchem.5b02270
-
[30]
R Chaney, J Rider, D Pamphilon. Transfusion Med., 1999, 9(3):177~188. doi: 10.1046/j.1365-3148.1999.00196.x
-
[31]
D R Shelton, J S Karns. Appl. Environ. Microbiol., 2001, 67(7):2908~2915. doi: 10.1128/AEM.67.7.2908-2915.2001
-
[32]
S Xie, Y Dong, Y Yuan et al. Anal. Chem., 2016, 88(10):5218~5224. doi: 10.1021/acs.analchem.6b00276
-
[33]
B Barlen, S D Mazumdar, O Lezrich et al. Sensors, 2007, 7(8):1427~1446.
-
[34]
P D Keathley, J T Hastings. Plasmonics, 2012, 7(1):59~69. doi: 10.1007/s11468-011-9276-6
-
[35]
X Dou, B M Phillips, P Y Chung et al. Optics Lett., 2012, 37(17):3681~3683. doi: 10.1364/OL.37.003681
-
[36]
U Yogeswaran, S M Chen. Sensors, 2008, 8(1):290~313. http://www.emeraldinsight.com/servlet/linkout?suffix=b39&dbid=16&doi=10.1108%2F13565361211219167&key=10.3390%2Fs8010290
-
[37]
X Luo, J J Davis. Chem. Soc. Rev., 2013, 42(13):5944~5947. doi: 10.1039/c3cs60077g
-
[38]
M Mehrvar, M Abdi. Anal. Sci., 2004, 20(8):1113~1126. doi: 10.2116/analsci.20.1113
-
[39]
L Lu, G Chee. Biosens. Bioelectron., 2013, 42(1):492~495.
-
[40]
E Katz, I Willner. Electroanalysis, 2010, 15(11):913~947.
-
[41]
J T Cao, X Y Hao, Y D Zhu et al. Anal. Chem., 2012, 84(15):6775~6782. doi: 10.1021/ac3013048
-
[42]
J A Ho, H C Chang, W T Su. Anal. Chem., 2012, 84(7):3246~3253. doi: 10.1021/ac203362g
-
[43]
W Su, M Lin, H Lee et al. Biosens. Bioelectron., 2012, 32(1):32~36. doi: 10.1016/j.bios.2011.11.009
-
[44]
M Cho, L Chun, M Lin et al. Sens. Actuat. B, 2012, 174(3):490~494. http://www.researchgate.net/publication/257354646_Sensitive_electrochemical_sensor_for_detection_of_lipopolysaccharide_on_metal_complex_immobilized_gold_electrode?ev=auth_pub
-
[45]
M D Oliveira, C A Andrade, M T Correia et al. J. Colloid Interf. Sci., 2011, 362(1):194~201. doi: 10.1016/j.jcis.2011.06.042
-
[46]
W Su, S E Kim, M Cho et al. Innate Immun., 2013, 19(4):388~397. doi: 10.1177/1753425912465099
-
[47]
P Miao. RSC Adv., 2013, 3(25):9606~9617. doi: 10.1039/c3ra00047h
-
[48]
N Y Wu, W Gao, X L He et al. Biosens. Bioelectron., 2013, 39(1):210~214. doi: 10.1016/j.bios.2012.07.038
-
[49]
G A Zelada-Guillén, J L Sebastián-Avila, P Blondeau et al. Biosens. Bioelectron., 2012, 31(1):226~232. doi: 10.1016/j.bios.2011.10.021
-
[50]
L Bai, Y Chai, X Pu et al. Nanoscale, 2014, 6(5):2902~2908. doi: 10.1039/c3nr05930h
-
[51]
M R Guascito, D Chirizzi, C Malitesta et al. Electrochem. Commun., 2012, 22(8):45~48.
-
[52]
Z D Gao, Y Qu, T Li et al. Sci. Reports, 2014, 137(4):5113~5122.
-
[53]
A D Ryabov, R Cerón-Camacho, O Saavedra-Díaz et al. Anal. Chem., 2012, 84(21):9096~9100. doi: 10.1021/ac301714r
-
[54]
K Y Inoue, M Matsudaira, R Kubo et al. Lab Chip, 2012, 12(18):3481~3490. doi: 10.1039/c2lc40323d
-
[55]
P Miao, K Han, J Qi et al. Electrochem. Commun., 2013, 26(1):29~32.
-
[56]
Y H Lin, S H Chen, Y C Chuang et al. Biosens. Bioelectron., 2008, 23(12):1832~1837. doi: 10.1016/j.bios.2008.02.030
-
[57]
J Wang. Biosens. Bioelectron., 2006, 21(10):1887~1892. doi: 10.1016/j.bios.2005.10.027
-
[58]
R Monošík, M Stred' ansky', E Šturdík. J. Clin. Lab. Anal., 2012, 26(1):22~34. doi: 10.1002/jcla.2012.26.issue-1
-
[59]
G Yang, F Zhao, B Zeng. Biosens. Bioelectron., 2014, 53(9):447~452. http://europepmc.org/abstract/med/24211456
-
[60]
D S Hélder, J O G Pacheco, M M E Júlia et al. Biosens. Bioelectron., 2014, 52(2):56~61.
-
[61]
T Gan, J Sun, W Meng et al. Food Chem., 2013, 141(4):3731~3737. doi: 10.1016/j.foodchem.2013.06.084
-
[62]
L Rassaei, F Marken, M Sillanpää et al. Trends Anal. Chem., 2011, 30(11):1704~1715. doi: 10.1016/j.trac.2011.05.009
-
[63]
M I Mead, O A M Popoola, G B Stewart et al. Atmos. Environ., 2013, 70(2):186~203.
-
[64]
R L Bunde, E J Jarvi, J J Rosentreter. Talanta, 1998, 46(6):1223~1236. doi: 10.1016/S0039-9140(97)00392-5
-
[65]
K G Ong, J M Leland, K Zeng et al. Biosens. Bioelectron., 2006, 21(12):2270~2274. doi: 10.1016/j.bios.2005.11.007
-
[66]
H Muramatsu, M Suda, T Ataka et al. Sens. Actuat. A, 1990, 21(1):362~368.
-
[67]
S Sheikh, C Blaszykowski, A Romaschin et al. RSC Adv., 2016, 6(44):38037~38041. doi: 10.1039/C6RA02745H
-
[1]
-

图 1 基于不同换能器的内毒素检测生物传感器分类
Figure 1 Classification of biosensor for endotoxin based on transduction methods


表 1 不同类型生物传感器的比较
Table 1. Comparison in terms of general advantages and disadvantages associated with individual biosensor
传感器 分类 优点 缺点 检测限 光学生物传感器 荧光生物传感器 检测灵敏度高,信号读取速度快,传感界面设计简单 容易受到背景光的影响,抗干扰能力差,存在非特异性结合 10fg/mL 电化学发光生物传感器 具有电化学和电化学发光的优点,能方便控制发光反应的时间和位置的独特优势 传感层材料可选择性不大,测试过程较为复杂,发光试剂的发光效率不高且毒性大 0.18fg/mL SPR生物传感器 能检测生物分子的相互作用,且无需标记物 检测过程复杂,对敏感层材料要求高 0.7ng/mL 电化学生物传感器 电化学阻抗传感器 具有良好的界面表征作用,微小振幅正弦电压或电流不会对内毒素分子造成干扰 传感界面的构建复杂,成本高,生物识别元件的寿命短 0.1pg/mL 电位型传感器 响应时间快,对样本不会造成破坏 需要参比电极,电极容易被污染 8.7fg/mL 电流型传感器 低消耗,高信噪比,应用电压可控 分析物消耗多,需要氧化还原介质 0.06ng/mL 压电晶体生物传感器 - 检测灵敏度高,对微小质量变化都具有快速响应,抗干扰能力强 质量变化和频率转换之间缺乏精确的相关性 0.03ng/mL  下载: 导出CSV
下载: 导出CSV
-




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 33
- 文章访问数: 7090
- HTML全文浏览量: 2030
 下载:
下载:
