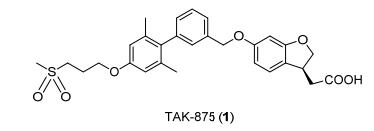
 图 1
TAK-875 (1) 的结构
Figure 1.
Structure of TAK-875 (1)
图 1
TAK-875 (1) 的结构
Figure 1.
Structure of TAK-875 (1)
引用本文:
闫玉刚, 陈雪英, 杨新颖, 徐文方, 张颖杰. G蛋白偶联受体40激动剂TAK-875亚砜类似物的合成、绝对构型确证及生物活性研究[J]. 有机化学,
2017, 37(4): 858-865.
doi:
10.6023/cjoc201612041

Citation: Yan Yugang, Chen Xueying, Yang Xinying, Xu Wenfang, Zhang Yingjie. Sulfoxide Analogs of TAK-875 as G Protein Coupled Receptor 40 Agonists: Synthesis, Determination of Absolute Configuration and Biological Activity[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 858-865. doi: 10.6023/cjoc201612041
Citation: Yan Yugang, Chen Xueying, Yang Xinying, Xu Wenfang, Zhang Yingjie. Sulfoxide Analogs of TAK-875 as G Protein Coupled Receptor 40 Agonists: Synthesis, Determination of Absolute Configuration and Biological Activity[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 858-865. doi: 10.6023/cjoc201612041
G蛋白偶联受体40激动剂TAK-875亚砜类似物的合成、绝对构型确证及生物活性研究
摘要:
G蛋白偶联受体40(GPR40)是治疗二型糖尿病的潜在靶点.本研究首先合成TAK-875(1)作为检测GPR40激动活性的对照药物.进一步合成化合物1的亚砜类似物11.手性制备HPLC分离化合物11得到光学纯化合物12(S,S,100.0% de)和13(R,S,100.0% de).其绝对构型通过圆二色谱确证.评价化合物1(EC50=84.3 nmol·L-1),11(EC50=77.5 nmol·L-1),12(EC50=76.1 nmol·L-1),13(EC50=114.0 nmol·L-1)对GPR40的激动活性,发现所合成的化合物体外保持了对照药物的活性.并通过计算机模拟对接的方法对活性保持的原因进行了解释.鉴于化合物12和13的良好活性与绝对构型的差别,下一步可分别对它们的成药性进行评价.
English
Sulfoxide Analogs of TAK-875 as G Protein Coupled Receptor 40 Agonists: Synthesis, Determination of Absolute Configuration and Biological Activity
Abstract:
G protein coupled receptor 40 (GPR40) is a potential target for treatment of type 2 diabetes. Herein, the well-known GPR40 agonist TAK-875 (compound 1) was synthesized as a positive control. Besides, an epimeric mixture 11, which was the sulfoxide analog of compound 1 was also synthesized. The following chiral HPLC separation of 11 led to optically pure compounds 12 (S, S, 100.0% de) and 13 (R, S, 100.0% de), of which the absolute configurations were determined by circular dichroism spectra analysis. In vitro biological activity evaluation results showed that the GPR40 agonistic potency of epimeric mixture 11 (EC50=77.5 nmol·L-1) and its two optically pure epimers (12, EC50=76.1 nmol·L-1; 13, EC50=114.0 nmol·L-1) were comparable to that of compound 1 (EC50=84.3 nmol·L-1), which was rationalized by docking analysis. Compounds 12 and 13 warrant further drug-like property evaluation due to their promising potency and novel structures.
-
Key words:
- chirality
- / GPR40
- / sulfoxide
- / Circular dichroism
-
糖尿病已经成为威胁人类健康的重大代谢性疾病, 根据国际糖尿病联盟 (IDF) 2015年的报道, 全世界有4.15亿人患有糖尿病, 预计, 2040年糖尿病患者将增加2.27亿, 世界各地的糖尿病患者中有90%为二型糖尿病病人.二型糖尿病通常在成人中发病, 但是目前在儿童和青少年中发病率也在增加.二型糖尿病发病是由人体无法分泌足够的胰岛素或无法有效利用胰岛素 (胰岛素抵抗) 造成的, 从而导致高血糖.高血糖是二型糖尿病致病的根本原因[1].
随着人们对糖尿病药物不断的需求, 众多治疗糖尿病的靶点被发现.其中, G蛋白偶联受体40 (GPR40) 是一种新型的治疗二型糖尿病的靶标[2], 主要表达在胰岛β细胞和小肠的内分泌细胞, 在体内由中 (C6~C12) 长 (C14~C24) 链饱和与不饱和自由脂肪酸激活[3~7].由于通过GPR40介导的胰岛素释放是葡糖糖依赖性的[3, 8, 9], 所以可以有效降低药物引起的低血糖风险[10].
当前, 有很多的GPR40激动剂被报道[11], 例如TAK-875[12~14], JTT-851[14], P11187[14], LY2881835[14], AMG837[14, 15], GW9508[16], DS-1558[16], Yhhu4488[17], AM-3189[18], AS2575959[19], CNX-011-67[20]等.其中TAK-875, JTT-851, P11187, LY2881835和AMG837进入到了临床研究阶段[14]. TAK-875有潜在的肝毒性, 日本武田制药公司主动终止了其临床试验[21], 但是它仍然是到现在为止唯一一个以GPR40为治疗靶标的进入到三期临床试验的, 活性良好且口服有效地治疗二型糖尿病的药物[12~14].随着研究的深入, 2014年, 《自然》杂志报道了TAK-875与人源GPR40的结晶复合物[22].本研究将TAK-875的砜基团换成亚砜基团, 增加化合物的极性, 减小化合物的脂溶性, 降低化合物的脂毒性[23], 并考察亚砜基团的立体化学对化合物活性的影响.
1 结果与讨论
1.1 化合物的合成
化合物TAK-875 (1) 作为对照药物 (图 1), 按照文献报道的方法合成[13, 24].
图 1
TAK-875 (1) 的结构
Figure 1.
Structure of TAK-875 (1)
2经过磺化反应得3. 4与3-甲酰基苯硼酸发生Suzuki反应得5.在碳酸钾存在的条件下, 3与5发生亲核取代反应得6, 继而经过还原反应得7. 7与关键中间体8经Mitsunobu反应得9, 再经氧化反应, 水解反应得到11 (Scheme 1).
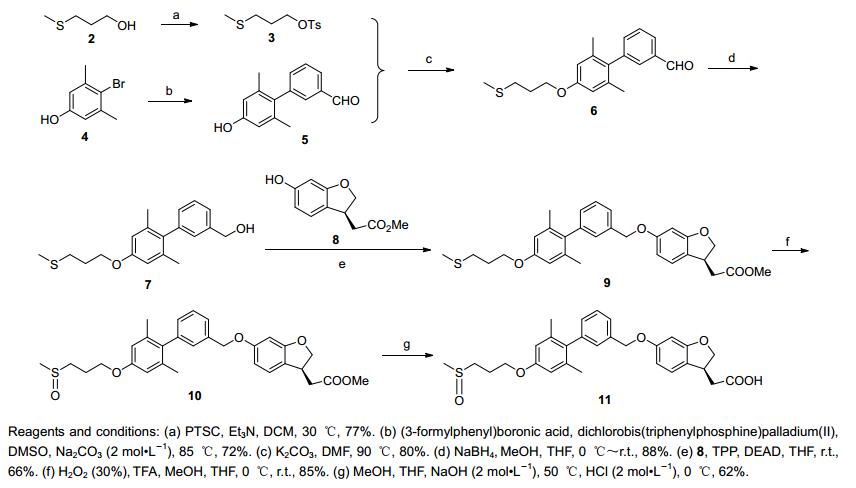 图 图式1
目标化合物11的合成路线
Figure 图式1.
Synthetic route of target compound 11
图 图式1
目标化合物11的合成路线
Figure 图式1.
Synthetic route of target compound 11
1.2 化合物光学纯度及绝对构型的确证
合成化合物TAK-875 (1) 作为对照药物, 其ee值为100%.紧接着合成了化合物11, 采用分析手性CHIRALPAK IA柱做高效液相色谱 (HPLC) 得知化合物11为两个差向异构体的混合物 (图 2A).采用制备手性CHIRALPAK ADH柱, 通过制备高效液相分离化合物11得到了一对差向异构体12和13 (Eq. 1).采用分析化合物11相同的条件做分析高效液相, 确证化合物12和13的de值都是100% (图 2B, 图 2C).
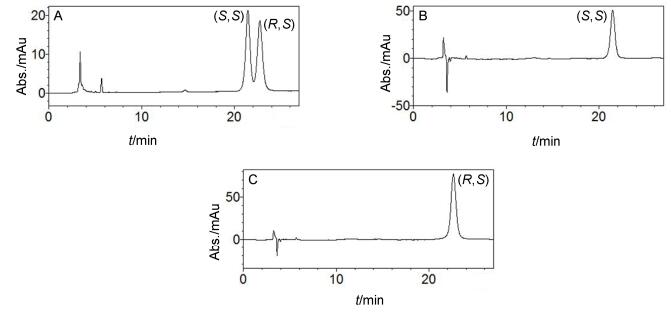 图 2
化合物11 (A), 12 (B) 和13 (C) 的高效液相图谱
Figure 2.
Analytical chiral HPLC spectra of 11 (A), 12 (B) and 13 (C)
图 2
化合物11 (A), 12 (B) 和13 (C) 的高效液相图谱
Figure 2.
Analytical chiral HPLC spectra of 11 (A), 12 (B) and 13 (C)
根据文献报道[24], 中间体8的绝对构型是 (S).因为1, 11, 12和13衍生自关键中间体8, 所以这些化合物手性碳的绝对构型都应该为 (S).为了确定12和13亚砜部分的绝对构型, 分别做了它们的圆二色谱. 12和13的Cotton效应是化合物在乙腈中以相同的浓度 (50 μg/mL), 25 ℃下, 由Chirascan CD光谱仪检测得到.从图 3A可知, 由于手性碳Cotton效应的影响, 12和13亚砜部分的Cotton效应不能清晰得到.为了消除手性碳Cotton效应的影响, 用12的Cotton效应值减去13的Cotton效应值, 消除手性碳Cotton效应值的影响, 得到信号放大两倍的12亚砜部分的Cotton效应值, 如图 3B所示, 其最大吸收 (λmax) 在197 nm处为正Cotton效应, 而根据文献报道己基甲基亚砜 (S) 异构体的最大吸收 (λmax) 在208 nm处为正Cotton效应[25](图 3C), 于是得出, 两者CD谱类似, 所以, 12亚砜基团上的手性硫的立体化学绝对构型为 (S).相似的推导方法可知化合物13亚砜基团上的手性硫的立体化学绝对构型为 (R).因此, 12和13的绝对构型分别为 (S, S) 和 (R, S).
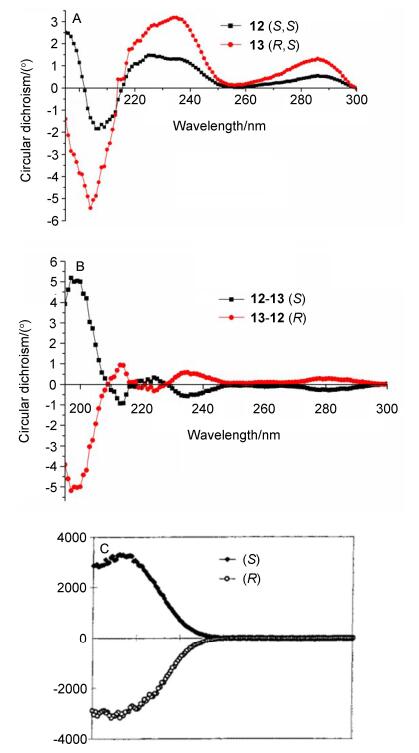 图 3
(A) 12和13、(B) (12-13) 和 (13-12)、(C) 己基甲基亚砜的圆二色谱
Figure 3.
Shows Cotton effects in CD spectra of 12 and 13 (A),
12-13 and 13-12 (B), and hexyl methyl sulfoxide (C)
图 3
(A) 12和13、(B) (12-13) 和 (13-12)、(C) 己基甲基亚砜的圆二色谱
Figure 3.
Shows Cotton effects in CD spectra of 12 and 13 (A),
12-13 and 13-12 (B), and hexyl methyl sulfoxide (C)
1.3 亚砜类似物与TAK-875水溶性比较
利用标准曲线法, 测试了化合物1, 11, 12和13室温下水中溶解度.由表 1可知, 消旋体亚砜类似物11的水溶性要大于砜类化合物1, 光学纯亚砜化合物12和13的水溶性表现出了较大差异, 在这四种化合物中, 12的溶解性最好, 而13的溶解性最差.
 表 1
室温下化合物1, 11, 12和13的水中溶解度
Table 1.
Solubility of 1, 11, 12 and 13 in water at room temperature
表 1
室温下化合物1, 11, 12和13的水中溶解度
Table 1.
Solubility of 1, 11, 12 and 13 in water at room temperature
Compd. 1 11 12 13 Solubilitya/
(µg•mL-1)0.79±0.05 1.18±0.04 2.04±0.03 0.32±0.04 aResults expressed as the mean±standard deviation of three separate determinations. 表 1 室温下化合物1, 11, 12和13的水中溶解度
Table 1. Solubility of 1, 11, 12 and 13 in water at room temperature1.4 生物活性测试
1, 11, 12和13体外测试GPR40的激动活性, 结果列在表 2中.从表中数据可以得出所合成的化合物体外保持了对照药物的活性.由此可见对照药物的砜基团, 合成化合物的消旋的亚砜基团, 以及不同立体构型的亚砜基团, 对化合物的体外活性没有明显影响.这样鉴于12和13良好的体外活性与其立体构型的不同, 下一步分别考察其成药性将是十分有意义的工作.
表 2
化合物1, 11, 12, 13和γ-亚麻酸的立体构型及体外对GPR40的激动活性
Table 2.
Configurations and in vitro agonist activities for GPR40 of 1, 11, 12, 13 and γ-linolenic acid
Compd. Stereo EC50a/(nmol•L-1) 1 S 84.3 11 (±, S) 77.5 12 (S, S) 76.1 13 (R, S) 114.0 γ-Linolenic acid 5.2b aValues are means of three experiments, the standard derivations are<20% of the mean. bThe EC50 of γ-linolenic acid, unit is µmol•L-1, values are means of three experiments, and the standard derivations are<20% of the mean. 表 2 化合物1, 11, 12, 13和γ-亚麻酸的立体构型及体外对GPR40的激动活性
Table 2. Configurations and in vitro agonist activities for GPR40 of 1, 11, 12, 13 and γ-linolenic acid1.5 计算机模拟对接
为了理解TAK-875亚砜类似物活性保持的原因, 我们对化合物TAK-875, 12和13与GPR40做了对接研究.从图 4可以看出12和13的亚砜基团与TAK-875的砜基团一样都没有跟蛋白结合, 不影响化合物的活性.
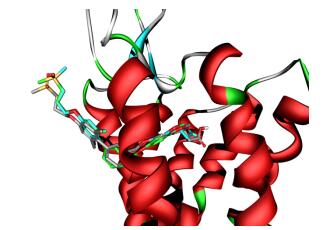 图 4
TAK-875及其衍生物与GPR40活性位点的对接模型
Figure 4.
TAK-875, 12 and 13 docked into the active site of GPR40
图 4
TAK-875及其衍生物与GPR40活性位点的对接模型
Figure 4.
TAK-875, 12 and 13 docked into the active site of GPR40
2 结论
将TAK-875 (1) 的磺酰基结构改造为亚磺酰基而得到了差向异构体混合物11, 通过手性制备液相拆分11得到了光学纯差向异构体12 (S, S, 100.0% de) 和13 (R, S, 100.0% de), 利用圆二色谱的方法确定了其绝对构型.体外测定目标化合物激动GPR40的EC50, 发现本实验的砜类化合物与亚砜的类似物具有相当的体外活性, 这也可以由目标化合物与hGPR40的结合模式得到解释. 12和13体外保持了良好的活性, 同时这两个化合物的亚砜部分的立体构型相反, 下一步的工作可以分别考察它们的成药性.
3 实验部分
3.1 仪器与试剂
除特殊说明, 所有原料、试剂均购自试剂公司, 未进一步纯化使用.反应进度和化合物初步纯度由薄层层析板 (TLC, 0.25 mm硅胶GF254) 结合紫外灯监测.化合物纯化是通过柱层析的方法来实现的.柱层析用的硅胶为正相300~400目C18硅胶, 仪器为Combiflash Rf 200 (Teledyne Isco). 1H NMR和13C NMR由山东大学化学院Brucker DRX (300 MHz与400 MHz) 型核磁共振仪和山东省科学院Brucker DRX (600 MHz) 型核磁共振仪测定, 所使用的溶剂为DMSO-d6), 内标为四甲基硅烷 (TMS); HRMS由山东省科学院和山东大学化学院1200RRLC-6520 Accurate-Mass Q-TOF LC/MS型质谱仪测定, ESIMS由山东大学药学院分析测试中心API 4000型质谱仪测定; IR由山东省科学院和山东大学药学院分析测试中心Nicolet 6700 FT-IR Spectrometer型红外光谱仪测定, 样品处理采用溴化钾压片法; 熔点由RY-1熔点测定仪测定 (温度计未经校正); 手性制备由大赛璐药物手性技术 (上海) 有限公司提供; 手性HPLC分析采用Shimadzu 20A工作站进行, 色谱柱为CHIRALPAK IA柱; 旋光度由MCP 200旋光仪测定; 圆二色谱 (CD) 由山东大学药学院分析测试中心Chirascan CD光谱仪测定.
3.2 实验方法
3.3 HEK 293/GPR40/NFAT/βla细胞系钙流检测实验
温度为37 ℃同时含有5% CO2的条件下, 将HEK 293/GPR40/NFAT/βla细胞系 (HD Biosciences) 培养在含有10%胎牛血清的dulbecco's modified eagle medium (DMEM) 培养基中. 24 h后, 清除培养基.之后加入Fluo8 (不含钙离子的染料).在37 ℃下培养1 h.孵育完成后, 将细胞板、化合物板和枪头放到FLIPR (Molecular Devices) 的相应板位上进行读板.将实验结果用Screen Works软件导出.利用作图软件Prism 5 (GraphPad) 制作化合物浓度曲线, 计算EC50值.
辅助材料 (Supporting Information) 化合物1~13的1H NMR和13C NMR谱图; 化合物1, 6, 8~13的HRMS谱图; 化合物1, 8的高效液相谱图.这些材料可以免费从本刊网站 (http://sioc-journal.cn/) 上下载.
3.2.9 2-((3S)-6-((2', 6'-二甲基-4'-(3-(甲亚磺酰基) 丙氧基) 联苯基-3-基) 甲氧基)-2, 3-苯并二氢呋喃-3-基) 乙酸 (11) 的合成
称量10 (2.62 g) 溶于甲醇 (13 mL) 和四氢呋喃 (26 mL) 混合液中, 加入2 mol•L-1氢氧化钠 (8.0 mL), 50 ℃反应2 h.移至冰浴中2 mol•L-1盐酸酸化, 有大量白色固体析出, 用乙酸乙酯萃取三遍, 再用饱和氯化钠溶液洗两遍, 无水硫酸镁干燥后, 用甲醇/二氯甲烷体系过flash柱, 蒸干, 真空干燥得1.57 g淡黄色固体, 产率62%. [α]D25+5.89 (c 1.0, acetonitrile); 1H NMR (DMSO-d6, 300 MHz) δ: 1.92 (s, 6H), 2.05~2.14 (m, 2H), 2.44~ 2.51 (m, 1H), 2.58 (s, 3H), 2.66~2.98 (m, 3H), 3.62~ 3.72 (m, 1H), 4.09 (t, J=6.0 Hz, 2H), 4.16~4.21 (m, 1H), 4.68 (t, J=9.0 Hz, 1H), 5.09 (s, 2H), 6.46~6.49 (m, 2H), 6.71 (s, 2H), 7.04~7.14 (m, 3H), 7.37~7.47 (m, 2H), 12.35 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ: 20.70, 22.15, 37.04, 38.03, 39.10, 49.86, 66.01, 69.29, 77.07, 96.89, 106.92, 113.24, 121.90, 124.51, 125.78, 128.52, 128.72, 133.81, 136.55, 137.31, 140.23, 157.01, 159.07, 160.64, 173.07; IR (KBr) ν: 3418, 2918, 2578, 1718, 1619, 1599, 1496, 1470, 1316, 1275, 1182, 1154, 1004, 829, 797, 712, 633 cm-1; ESI-MS m/z: 507.3 [M-H]-. HRMS (AP-ESI) calcd for C29H31O6S [M-H]- 507.1920; found 507.1857.
3.2.2 3-(甲硫基) 丙基-4-苯磺酸甲酯 (3) 的合成
称取对甲苯磺酰氯 (11.2 g) 于250 mL反应瓶中, 加入100 mL无水二氯甲烷, 再加入12 mL三乙胺, 室温下滴加3-甲硫基丙醇 (2)(4.0 mL), 30 ℃反应过夜.加入大量二氯甲烷, 用2 mol•L-1盐酸洗至酸性, 饱和氯化钠溶液洗两遍, 无水硫酸镁干燥后, 用乙酸乙酯/石油醚体系过flash柱, 蒸干得8.0 g无色油状液体3[12], 产率77%. 1H NMR (DMSO-d6, 400 MHz) δ: 1.80~1.86 (m, 2H), 1.96 (s, 3H), 2.41~2.50 (m, 5H), 4.11 (t, J=6.2 Hz, 2H), 7.47 (d, J=8.1 Hz, 2H), 7.79 (d, J=8.3 Hz, 2H); 13C NMR (DMSO-d6, 100 MHz) δ: 14.84, 21.51, 28.20, 29.37, 69.82, 128.02, 130.59, 132.89, 145.34; IR (KBr) ν: 2960, 2919, 1598, 1363, 1189, 1097, 958, 915, 815, 665, 576, 555 cm-1; ESI-MS m/z: 261.2 [M+H]+.
3.2.4 2', 6'-二甲基-4'-[3-(甲硫基) 丙氧基]联苯基-3-甲醛 (6) 的合成
称量3 (1.76 g)、5 (1.84 g) 和碳酸钾 (0.93 g), 加入15 mL二甲基甲酰胺, 90 ℃反应24 h.冷却至室温, 加入大量水, 用乙酸乙酯萃取三遍, 1 mol•L-1盐酸洗乙酸乙酯层至酸性, 再用饱和氯化钠溶液洗两遍, 无水硫酸镁干燥后, 用乙酸乙酯/石油醚体系过flash柱, 蒸干得1.7 g无色油状液体, 产率80%. 1H NMR (DMSO-d6, 400 MHz) δ: 1.93 (s, 6H), 1.95~2.01 (m, 2H), 2.04 (s, 3H), 2.61 (t, J=7.2 Hz, 2H), 4.05 (t, J=6.2 Hz, 2H), 6.72 (s, 2H), 7.41 (d, J=7.6 Hz, 1H), 7.61 (t, J=7.6 Hz, 1H), 7.66 (s, 1H), 7.86 (d, J=7.7 Hz, 1H), 10.03 (s, 1H); 13C NMR (DMSO-d6, 100 MHz) δ: 15.14, 21.11, 28.86, 30.37, 66.20, 113.78, 127.98, 129.66, 131.18, 133.08, 136.09, 136.98, 137.02, 141.72, 158.04, 193.21; IR (KBr) ν: 3042, 2916, 2865, 2724, 1693, 1602, 1578, 1466, 1320, 1281, 1177, 1162, 1150, 1065, 855, 800, 704, 651 cm-1; ESI-MS m/z: 315.3 [M+H]+. HRMS (AP-ESI) calcd for C19H22O2SNa [M+Na]+ 337.1341; found 337.1296.
3.2.6 (S)-2-(6-羟基-2, 3-苯并二氢呋喃-3-基) 乙酸甲酯 (8) 的合成
化合物8按照文献报道的方法合成[24]. m.p. 64~ 66 ℃; 100.0% ee [柱子: CHIRALPAK IA (4.6 mm I.D.× 250 mm, 5 μm); 流动相: V(正己烷)/V(异丙醇)=80/20;流速: 0.8 mL/min; 检测: UV 286 nm; 温度:室温]; [α]D25+10.5 (c 1.2, acetonitrile); 1H NMR (DMSO-d6, 300 MHz) δ: 2.56 (dd, J=7.5, 9.0 Hz, 1H), 2.74 (dd, J=10.5, 6.0 Hz, 1H), 3.63~3.71 (m, 4H), 4.16 (dd, J=2.4, 6.6 Hz, 1H), 4.64 (t, J=9.0 Hz, 1H), 6.17 (d, J=2.1 Hz, 1H), 6.24 (dd, J=6.0, 2.1 Hz, 1H), 6.94~6.97 (m, 1H), 9.30 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ: 37.03, 51.37, 76.68, 97.03, 107.17, 119.50, 124.42, 158.10, 160.59, 172.01; IR (KBr) ν: 3446, 3425, 1711, 1621, 1607, 1507, 1457, 1366, 1344, 1303, 1184, 1171, 1102, 984, 864, 804, 764, 638, 631, 578 cm-1; ESI-MS m/z: 209.3 [M+H]+. HRMS (AP-ESI) calcd for C11H12O4 [M-H]- 207.0736; found 207.0657.
3.2.8 2-((3S)-6-((2', 6'-二甲基-4'-(3-(甲亚磺酰基) 丙氧基) 联苯基-3-基) 甲氧基)-2, 3-苯并二氢呋喃-3-基) 乙酸甲酯 (10) 的合成
称量9 (3.0 g) 于100 mL反应瓶中, 加入甲醇 (36 mL) 和四氢呋喃 (18 mL), 冰浴下, 滴加30%双氧水 (9.0 mL), 滴加完双氧水后再滴加1.0 mL三氟乙酸, 反应0.5 h后, 撤冰浴, 室温反应2 d.用甲醇/二氯甲烷体系过flash柱, 蒸干得2.6 g无色油状液体, 产率85%. [α]D25+ 6.39 (c 0.3, methanol); 1H NMR (DMSO-d6, 300 MHz) δ: 1.91 (s, 6H), 2.04~2.14 (m, 2H), 2.54~2.63 (m, 4H), 2.73~2.97 (m, 3H), 3.63 (s, 3H), 3.65~3.75 (m, 1H), 4.09 (t, J=6.0 Hz, 2H), 4.20 (dd, J=2.4, 6.6 Hz, 1H), 4.67 (t, J=9.0 Hz, 1H), 5.09 (s, 2H), 6.46~6.49 (m, 2H), 6.71 (s, 2H), 7.04~7.09 (m, 2H), 7.14 (s, 1H), 7.36~7.47 (m, 2H); 13C NMR (DMSO-d6, 75 MHz) δ: 20.68, 22.15, 37.03, 38.06, 38.59, 49.89, 51.41, 66.04, 69.32, 76.86, 96.94, 107.02, 113.27, 121.62, 124.50, 125.77, 128.51, 128.73, 133.82, 136.55, 137.30, 140.25, 157.02, 159.16, 160.65, 171.94; IR (KBr) ν: 3416, 2970, 2918, 1735, 1620, 1598, 1497, 1470, 1438, 1316, 1277, 1156, 1112, 1049, 881, 792, 713, 630 cm-1; ESI-MS m/z: 523.5 [M+H]+. HRMS (AP-ESI) calcd for C30H34O6SNa [M + Na] + 545.2076; found 545.1969.
3.2.3 4'-羟基-2', 6'-二甲基联苯基-3-甲醛 (5) 的合成
称量4 (2.0 g)、3-甲酰基苯硼酸 (1.6 g) 和双三苯基膦二氯化钯 (0.07 g) 于100 mL双颈瓶中, 加入20 mL二甲基亚砜.抽真空, 冲氩气保护.注入2 mol•L-1碳酸钠溶液 (6 mL), 抽真空, 冲氩气保护. 85 ℃反应过夜.冷却至室温, 加入大量水, 用乙酸乙酯萃取三遍, 1 mol•L-1盐酸洗乙酸乙酯层至酸性, 再用饱和氯化钠溶液洗两遍, 无水硫酸镁干燥后, 用乙酸乙酯/石油醚体系过flash柱, 蒸干得1.62 g白色固体5[12], 产率72%. m.p. 110~112 ℃; 1H NMR (DMSO-d6, 300 MHz) δ: 1.90 (s, 6H), 6.57 (s, 2H), 7.45~7.48 (m, 1H), 7.63~7.68 (m, 2H), 7.86~7.89 (m, 1H), 9.27 (s, 1H), 10.05 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ: 20.59, 114.18, 127.24, 129.25, 130.95, 131.03, 135.93, 136.29, 136.39, 141.47, 156.29, 193.23; IR (KBr) ν: 3343, 1683, 1679, 1611, 1577, 1456, 1382, 1309, 1245, 1184, 1152, 1029, 853, 726, 705, 651 cm-1; ESI-MS m/z: 225.4 [M-H]-.
3.2.7 (S)-2-(6-((2', 6'-二甲基-4'-(3-(甲硫基) 丙氧基) 联苯基-3-基) 甲氧基)-2, 3-苯并二氢呋喃-3-基) 乙酸甲酯 (9) 的合成
称量7 (2.85 g)、8 (2.25 g) 和三苯基膦 (3.6 g), 溶于50 mL无水四氢呋喃中, 室温下, 将偶氮二甲酸二乙酯 (2.2 mL) 溶于10 mL无水四氢呋喃, 滴加到反应液中, 反应过夜.用乙酸乙酯/石油醚体系过flash柱, 蒸干得3.0 g无色油状液体, 产率66%. [α]D25+9.35 (c 1.7, acetonitrile); 1H NMR (DMSO-d6, 300 MHz) δ: 1.92 (s, 6H), 1.96~2.02 (m, 2H), 2.07 (s, 3H), 2.54~2.65 (m, 3H), 2.77 (dd, J=10.8, 5.7 Hz, 1H), 3.63 (s, 3H), 3.68~3.82 (m, 1H), 4.04 (t, J=6.0 Hz, 2H), 4.20 (dd, J=2.4, 6.6 Hz, 1H), 4.67 (t, J=9.0 Hz, 1H), 5.09 (s, 2H), 6.46~6.49 (m, 2H), 6.69 (s, 2H), 7.03~7.09 (m, 2H), 7.15 (s, 1H), 7.36~ 7.46 (m, 2H); 13C NMR (DMSO-d6, 75 MHz) δ: 20.66, 28.33, 29.78, 37.05, 38.59, 51.37, 65.69, 69.33, 76.88, 96.92, 106.97, 113.17, 121.60, 124.47, 125.71, 128.49, 128.73, 133.68, 136.52, 137.27, 140.31, 157.19, 159.18, 160.67, 171.91; IR (KBr) ν: 2951, 2915, 1735, 1619, 1598, 1499, 1458, 1437, 1355, 1316, 1279, 1199, 1155, 1146, 1111, 1074, 1034, 989, 893, 841, 792, 713, 645 cm-1; ESI-MS m/z: 507.5 [M+H]+. HRMS (AP-ESI) calcd for C30H34O5SNa [M+Na]+ 529.2127; found 529.2029.
3.2.10 手性柱制备光学纯化合物2-((S)-6-((2', 6'-二甲基-4'-(3-((S)-甲亚磺酰基) 丙氧基) 联苯基-3-基) 甲氧基)-2, 3-苯并二氢呋喃-3-基) 乙酸 (12) 和2-((S)-6-((2', 6'-二甲基-4'-(3-((R)-甲亚磺酰基) 丙氧基) 联苯基-3-基) 甲氧基)-2, 3-苯并二氢呋喃-3-基) 乙酸 (13)
化合物11的手性制备是在大赛璐药物手性技术有限公司进行的.手性制备条件:柱子: CHIRALPAK ADH, 0.46 cm I.D.×25 cm•L; 流动相: V(二氧化碳)/ V(甲醇)=60/40;流速: 2.0 mL/min; 检测: UV 208 nm; 温度: 35 ℃.化合物12的保留时间为14.2 min, 化合物13的保留时间17.6 min.
(S, S)-异构体12: 100.0% de [柱子: CHIRALPAK IA (4.6 mm I.D.×250 mm, 5 μm); 流动相: V(正己烷): V(乙醇):V(乙酸乙酯):V(三氟乙酸)=75:10:15: 0.1;流速: 1 mL/min; 检测: UV 284 nm; 温度:室温]; [α]25D+35.93 (c 0.8, acetonitrile); 1H NMR (DMSO-d6, 600 MHz) δ: 1.92 (s, 6H), 2.04~2.12 (m, 2H), 2.46~2.51 (m, 1H), 2.58 (s, 3H), 2.69 (dd, J=5.4, 10.8 Hz, 1H), 2.77~2.82 (m, 1H), 2.90~2.95 (m, 1H), 3.66~3.68 (m, 1H), 4.09 (t, J=6.0 Hz, 2H), 4.17~4.20 (m, 1H), 4.68 (t, J=9.0 Hz, 1H), 5.09 (s, 2H), 6.46~6.48 (m, 2H), 6.71 (s, 2H), 7.05~7.14 (m, 3H), 7.36~7.45 (m, 2H); 13C NMR (DMSO-d6, 150 MHz) δ: 21.19, 22.62, 37.53, 38.49, 39.50, 50.31, 66.47, 69.74, 77.57, 97.34, 107.38, 113.70, 122.40, 124.91, 126.26, 129.00, 129.01, 129.20, 134.28, 137.03, 137.78, 140.71, 157.47, 159.53, 161.11, 173.60; IR (KBr) ν: 3423, 2918, 2579, 1717, 1618, 1600, 1496, 1470, 1316, 1275, 1182, 1154, 1005, 826, 797, 712, 632 cm-1. HRMS (AP-ESI) calcd for C29H31O6S [M-H]- 507.1920; found 507.1724.
(R, S)-异构体13: 100.0% de [柱子: CHIRALPAK IA (4.6 mm I.D.×250 mm, 5 μm); 流动相: V(正己烷)/V(乙醇)/V(乙酸乙酯)/V(三氟乙酸)=75/10/15/0.1;流速: 1 mL/min; 检测: UV 284 nm; 温度:室温]; [α]D25 -20.32 (c 0.7, acetonitrile); 1H NMR (DMSO-d6, 600 MHz) δ: 1.92 (s, 6H), 2.07~2.11 (m, 2H), 2.47 (dd, J=7.8, 9.0 Hz, 1H), 2.58 (s, 3H), 2.69 (dd, J=5.4, 11.4 Hz, 1H), 2.77~ 2.82 (m, 1H), 2.90~2.95 (m, 1H), 3.66~3.68 (m, 1H), 4.09 (t, J=6.0 Hz, 2H), 4.17~4.20 (m, 1H), 4.68 (t, J= 9.0 Hz, 1H), 5.09 (s, 2H), 6.4~6.48 (m, 2H), 6.71 (s, 2H), 7.05~ 7.14 (m, 3H), 7.37~ 7.45 (m, 2H); 13C NMR (DMSO-d6, 150 MHz) δ: 21.18, 22.61, 37.55, 38.48, 39.50, 39.64, 50.31, 66.49, 69.73, 77.59, 97.33, 107.37, 113.69, 122.43, 124.98, 126.26, 128.99, 129.01, 129.19, 134.27, 137.03, 137.78, 140.70, 157.47, 159.51, 161.11, 173.63; IR (KBr) ν: 3421, 2918, 1713, 1619, 1598, 1496, 1470, 1317, 1278, 1182, 1156, 1001, 824, 791, 712, 632 cm-1. HRMS (AP-ESI) calcd for C29H31O6S [M-H]- 507.1920; found 507.1723.
3.2.5 (2', 6'-二甲基-4'-(3-(甲硫基) 丙氧基) 联苯基-3-基) 甲醇 (7) 的合成
称量6 (1.7 g) 于50 mL反应瓶中, 加入无水甲醇 (8 mL) 和无水四氢呋喃 (16 mL).冰浴搅拌下缓缓加入0.2 g硼氢化钠, 反应0.5 h, 撤冰浴, 室温反应过夜.加入大量水, 用乙酸乙酯萃取三遍, 1 mol•L-1盐酸洗乙酸乙酯层至酸性, 再用饱和氯化钠溶液洗两遍, 无水硫酸镁干燥后, 用乙酸乙酯/石油醚体系过flash柱, 蒸干得1.5 g无色油状液体[26], 产率88%. 1H NMR (DMSO-d6, 400 MHz) δ: 1.95 (s, 6H), 1.97~2.02 (m, 2H), 2.06 (s, 3H), 2.63 (t, J=7.2 Hz, 2H), 4.05 (t, J=6.1 Hz, 2H), 4.56 (s, 2H), 5.20 (s, 1H), 6.70 (s, 2H), 6.96 (d, J=7.4 Hz, 1H), 7.06 (s, 1H), 7.29 (d, J=7.6 Hz, 1H), 7.38 (t, J=7.6 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz) δ: 15.16, 21.24, 28.86, 30.31, 63.35, 66.18, 113.61, 125.12, 127.80, 128.11, 128.58, 134.61, 136.99, 140.55, 143.19, 157.60; IR (KBr) ν: 3416, 2917, 2869, 1606, 1468, 1437, 1316, 1181, 1155, 1066, 1039, 855, 796, 713, 640 cm-1; ESI-MS m/z: 317.5 [M+H]+.
3.2.1 (S)-2-(6-((2', 6'-二甲基-4'-(3-(甲磺酰基) 丙氧基) 联苯-3-基) 甲氧基)-2, 3-苯并二氢呋喃-3-基) 乙酸 (1) 的合成
TAK-875 (1) 作为对照药物, 按照文献报道的方法合成[13, 24]. m.p. 120~122 ℃(文献值[13] 127~129 ℃); 100.0% ee [柱子: CHIRALPAK IA (4.6 mm I.D.×250 mm, 5 μm); 流动相: V(正己烷)/V(异丙醇)=80/20;流速: 0.8 mL/min; 检测: UV 286 nm; 温度:室温]; [α]D25 +5.2 (c 0.24, methanol); 1H NMR (DMSO-d6, 300 MHz) δ: 1.92 (s, 6H), 2.11~2.20 (m, 2H), 2.50~2.53 (m, 1H), 2.70 (dd, J=11.1, 5.4 Hz, 1H), 3.04 (s, 3H), 3.26~3.31 (m, 2H), 3.63~3.73 (m, 1H), 4.09 (t, J=6.3 Hz, 2H), 4.19 (dd, J=2.1, 6.9 Hz, 1H), 4.68 (t, J=9.0 Hz, 1H), 5.09 (s, 2H), 6.46~6.50 (m, 2H), 6.71 (s, 2H), 7.04~7.12 (m, 2H), 7.15 (s, 1H), 7.37~7.47 (m, 2H), 12.37 (s, 1H); 13C NMR (DMSO-d6, 75 MHz) δ: 20.69, 22.04, 37.04, 39.10, 40.19, 50.55, 65.36, 69.29, 77.08, 96.89, 106.92, 113.26, 121.90, 124.51, 125.79, 128.53, 128.72, 133.90, 136.58, 137.31, 140.22, 156.92, 159.07, 160.65, 173.08; IR (KBr) ν: 3020, 2924, 2885, 1720, 1704, 1614, 1597, 1498, 1315, 1279, 1156, 1127, 1071, 1035, 993, 974, 825, 788, 711, 634, 533 cm-1; ESI-MS m/z: 523.6 [M-H]-. HRMS (AP-ESI) calcd for C29H31O7S [M-H]- 523.1869; found 523.1808.
-
-
[1]
The Diabetes Education Consultative Section (DECS); Jeannete, A.; Nizar, A. B.; Maria, H. H.; Sir, M. H.; Ute, L.; Dianna, M.; Farheen, O.; Chris, P.; Nasheeta, P.; Andrey, P.; Mohammad, M. A.S.; Elena, S.; Teresa, T.; Juliet, U. S.; Zhang, X.; Samrawit, Y.; George, A.; Peter, B.; Juliana, C.; Adel, A. E. S.; Beatriz, Y. J.; Ji, L.; Kerry, L.; Viswanathan, M.; Lyudmil, N.; Graham, O.; Lorenzo, P.; Marie, A. T.; Sarah, H. W.; Paul, Z.; Bernard, Z. IDF Diabetes Atlas, 7th ed.; International Diabetes Federation, 2015; http://www.diabetesatlas.org. http://www.diabetesatlas.org
-
[2]
Choi, Y. J.; Shin, D.; Lee, J. Y. Arch. Pharm. Res. 2014, 37, 435. doi: 10.1007/s12272-013-0283-3
-
[3]
Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; Tanaka, H.; Maruyama, M.; Satoh, R.; Okubo, S.; Kizawa, H.; Komatsu, H.; Matsumura, F.; Noguchi, Y.; Shinohara, T.; Hinuma, S.; Fujisawa, Y.; Fujino, M. Nature 2003, 422, 173. doi: 10.1038/nature01478
-
[4]
Latour, M. G.; Alquier, T.; Oseid, E.; Tremblay, C.; Jetton, T. L.; Luo, J.; Lin, D. C.; Poitout, V. Diabetes 2007, 56, 1087. doi: 10.2337/db06-1532
-
[5]
Lu, H.; Fei, H.; Yang, F.; Zheng, S.; Hu, Q.; Zhang, L.; Yuan, J.; Feng, J.; Sun, P.; Dong, Q. Bioorg. Med. Chem. Lett. 2013, 23, 2920. doi: 10.1016/j.bmcl.2013.03.060
-
[6]
Tikhonova, I. G.; Sum, C. S.; Neumann, S.; Thomas, C. J.; Raaka, B. M.; Costanzi, S.; Gershengorn, M. C. J. Med. Chem. 2007, 50, 2981. doi: 10.1021/jm0614782
-
[7]
Briscoe, C. P.; Tadayyon, M.; Andrews, J. L.; Benson, W. G.; Chambers, J. K.; Eilert, M. M.; Ellis, C.; Elshourbagy, N. A.; Goetz, A. S.; Minnick, D. T.; Murdock, P. R.; Sauls, H. R., Jr.; Shabon, U.; Spinage, L. D.; Strum, J. C.; Szekeres, P. G.; Tan, K. B.; Way, J. M.; Ignar, D. M.; Wilson, S.; Muir, A. I. J. Biol. Chem. 2003, 278, 11303. doi: 10.1074/jbc.M211495200
-
[8]
Shapiro, H.; Shachar, S.; Sekler, I.; Hershfinkel, M.; Walker, M. D. Biochem. Biophys. Res. Commun. 2005, 335, 97. doi: 10.1016/j.bbrc.2005.07.042
-
[9]
Fujiwara, K.; Maekawa, F.; Yada, T. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E670. doi: 10.1152/ajpendo.00035.2005
-
[10]
Tan, C. P.; Feng, Y.; Zhou, Y. P.; Eiermann, G. J.; Petrov, A.; Zhou, C.; Lin, S.; Salituro, G.; Meinke, P.; Mosley, R.; Akiyama, T. E.; Einstein, M.; Kumar, S.; Berger, J. P.; Mills, S. G.; Thornberry, N. A.; Yang, L.; Howard, A. D. Diabetes 2008, 57, 2211. doi: 10.2337/db08-0130
-
[11]
李鹤, 龙亚秋, 有机化学, 2016, 36, 736.Li, H.; Long Y. Q. Chin. J. Org. Chem. 2016, 36, 736 (in Chinese).
-
[12]
Negoro, N.; Sasaki, S.; Mikami, S.; Ito, M.; Suzuki, M.; Tsujihata, Y.; Ito, R.; Harada, A.; Takeuchi, K.; Suzuki, N.; Miyazaki, J.; Santou, T.; Odani, T.; Kanzaki, N.; Funami, M.; Tanaka, T.; Kogame, A.; Matsunaga, S.; Yasuma, T.; Momose, Y. ACS Med. Chem. Lett. 2010, 1, 290. doi: 10.1021/ml1000855
-
[13]
Negoro, N.; Sasaki, S.; Mikami, S.; Ito, M.; Tsujihata, Y.; Ito, R.; Suzuki, M.; Takeuchi, K.; Suzuki, N.; Miyazaki, J.; Santou, T.; Odani, T.; Kanzaki, N.; Funami, M.; Morohashi, A.; Nonaka, M.; Matsunaga, S.; Yasuma, T.; Momose, Y. J. Med. Chem. 2012, 55, 3960. doi: 10.1021/jm300170m
-
[14]
Defossa, E.; Wagner, M. Bioorg. Med. Chem. Lett. 2014, 24, 2991. doi: 10.1016/j.bmcl.2014.05.019
-
[15]
Liu, J. J.; Wang, Y.; Ma, Z.; Schmitt, M.; Zhu, L.; Brown, S. P.; Dransfield, P. J.; Sun, Y.; Sharma, R.; Guo, Q.; Zhuang, R.; Zhang, J.; Luo, J.; Tonn, G. R.; Wong, S.; Swaminath, G.; Medina, J. C.; Lin, D. C.; Houze, J. B. ACS Med. Chem. Lett. 2014, 5, 517. doi: 10.1021/ml400501x
-
[16]
Takano, R.; Yoshida, M.; Inoue, M.; Honda, T.; Nakashima, R.; Matsumoto, K.; Yano, T.; Ogata, T.; Watanabe, N.; Hirouchi, M.; Yoneyama, T.; Ito, S.; Toda, N. ACS Med. Chem. Lett. 2015, 6, 266. doi: 10.1021/ml500391n
-
[17]
Guo, D. Y.; Li, D. W.; Ning, M. M.; Dang, X. Y.; Zhang, L. N.; Zeng, L. M.; Hu, Y. H.; Leng, Y. Biochem. Biophys. Res. Commun. 2015, 466, 740. doi: 10.1016/j.bbrc.2015.09.130
-
[18]
Ma, Z.; Lin, D. C.; Sharma, R.; Liu, J.; Zhu, L.; Li, A. R.; Kohn, T.; Wang, Y.; Liu, J. J.; Bartberger, M. D.; Medina, J. C.; Zhuang, R.; Li, F.; Zhang, J.; Luo, J.; Wong, S.; Tonn, G. R.; Houze, J. B. Bioorg. Med. Chem. Lett. 2016, 26, 15. doi: 10.1016/j.bmcl.2015.11.050
-
[19]
Tanaka, H.; Yoshida, S.; Minoura, H.; Negoro, K.; Shimaya, A.; Shimokawa, T.; Shibasaki, M. Life Sci. 2014, 94, 115. doi: 10.1016/j.lfs.2013.11.010
-
[20]
Sunil, V.; Verma, M. K.; Oommen, A. M.; Sadasivuni, M.; Singh, J.; Vijayraghav, D. N.; Chandravanshi, B.; Shetty, J.; Biswas, S.; Dandu, A.; Moolemath, Y.; Venkataranganna, M. V.; Somesh, B. P.; Jagannath, M. R. BMC Pharmacol. Toxicol. 2014, 15, 19. doi: 10.1186/2050-6511-15-19
-
[21]
Lead GPR40 agonist bites the dust Nat. Rev. Drug Discovery 2014, 13, 91.
-
[22]
Srivastava, A.; Yano, J.; Hirozane, Y.; Kefala, G.; Gruswitz, F.; Snell, G.; Lane, W.; Ivetac, A.; Aertgeerts, K.; Nguyen, J.; Jennings, A.; Okada, K. Nature 2014, 513, 124. doi: 10.1038/nature13494
-
[23]
McGarry, J. D.; Dobbins R. L. Diabetologia 1999, 42, 128. doi: 10.1007/s001250051130
-
[24]
Yamano, M.; Goto, M.; Kajiwara, T.; Maeda, H.; Konishi, T.; Sera, M.; Kondp, Y.; Yamasaki, S. WO 2012/111849, 2012[Chem. Abstr. 2012, 157, 410099].
-
[25]
Cho, H.; Plapp, B. V. Biochemistry 1998, 37, 4482. doi: 10.1021/bi9727040
-
[26]
Kang, X. S.; Chen, Z. H. WO 2015/024526, 2015[Chem. Abstr. 2015, 162, 353242].
-
[1]
-

图 2 化合物11 (A), 12 (B) 和13 (C) 的高效液相图谱
Figure 2 Analytical chiral HPLC spectra of 11 (A), 12 (B) and 13 (C)

图 3 (A) 12和13、(B) (12-13) 和 (13-12)、(C) 己基甲基亚砜的圆二色谱
Figure 3 Shows Cotton effects in CD spectra of 12 and 13 (A), 12-13 and 13-12 (B), and hexyl methyl sulfoxide (C)

图 4 TAK-875及其衍生物与GPR40活性位点的对接模型
Figure 4 TAK-875, 12 and 13 docked into the active site of GPR40
Carbon atom colored gray is TAK-875, blue is 12, green is 13. The docking processes were performed using Glide and GOLG 5.0
表 1 室温下化合物1, 11, 12和13的水中溶解度
Table 1. Solubility of 1, 11, 12 and 13 in water at room temperature
Compd. 1 11 12 13 Solubilitya/
(µg•mL-1)0.79±0.05 1.18±0.04 2.04±0.03 0.32±0.04 aResults expressed as the mean±standard deviation of three separate determinations.  下载: 导出CSV
下载: 导出CSV
表 2 化合物1, 11, 12, 13和γ-亚麻酸的立体构型及体外对GPR40的激动活性
Table 2. Configurations and in vitro agonist activities for GPR40 of 1, 11, 12, 13 and γ-linolenic acid
Compd. Stereo EC50a/(nmol•L-1) 1 S 84.3 11 (±, S) 77.5 12 (S, S) 76.1 13 (R, S) 114.0 γ-Linolenic acid 5.2b aValues are means of three experiments, the standard derivations are<20% of the mean. bThe EC50 of γ-linolenic acid, unit is µmol•L-1, values are means of three experiments, and the standard derivations are<20% of the mean.
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 7
- 文章访问数: 1589
- HTML全文浏览量: 197
 下载:
下载:
