
图式 1.
目标化合物STL127705的逆合成路线分析
Scheme 1.
Retrosynthesis of target compound STL127705

DNA依赖的蛋白激酶(DNA-PK)是由DNA依赖的蛋白激酶催化亚单位(DNA-PKcs)与Ku蛋白(由Ku70和Ku80组成的蛋白异构体)组成的一种蛋白复合物,其功能是启动DNA双链断裂的非同源末端连接修复(NHEJ)[1~4]。DNA损伤诱导剂和放疗已经广泛应用于临床肿瘤治疗,其抗肿瘤机制为诱导肿瘤细胞发生致死性的DNA双链断裂(DSB),DSB主要是通过非同源末端连接来修复的,因此在此修复过程中起关键作用的DNA-PK的抑制剂就具有增强放化疗敏感性的作用[5, 6]。奥拉帕尼是一种多聚二磷酸腺苷核糖聚合酶(PARP)抑制剂,其可阻断参与修复受损DNA的酶[7~9]。2014年,美国FDA批准奥拉帕尼应用于BRCA基因缺陷相关的卵巢癌的治疗。随着奥拉帕尼的临床获批,DNA修复抑制剂引起了广大研究者的注意,越来越多的新型DNA修复抑制剂被研发并投入到临床试验当中。研究发现,STL127705是一种具有嘧啶并嘧啶二酮骨架结构的DNA-PK的抑制剂,其对肿瘤细胞的杀伤效应不大,但其能够通过阻断DNA修复通路,极大地增强DNA损伤诱导剂或放疗的抗肿瘤功效[10]。
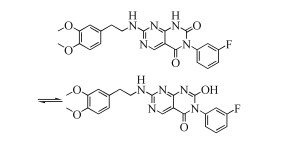
有关STL127705的合成方法之前未见报道,从其化学结构分析,可由3, 4-二甲氧基苯乙胺与含有嘧啶并嘧啶二酮结构的衍生物缩合而成;嘧啶并嘧啶二酮结构化合物可由4-氨基嘧啶-5-酰胺衍生结构合环反应制得;4-氨基嘧啶-5-酰胺衍生物可由相应的4-氨基-5-嘧啶甲酸衍生物与3-氟苯胺酯化缩合来得到(图式 1)。
通过对目标化合物的逆合成分析,本文设计的合成路线是以2-甲基硫-4-氯嘧啶-5-基甲酸乙酯为起始原料,先对其4位进行氨化生成化合物1,再水解化合物1中的甲酸乙酯,游离的羧酸基团与3-氟苯胺进行缩合酰化反应,生成化合物3。化合物3与二(三氯甲基)碳酸酯发生合环反应,生成化合物4。然后将化合物4中的甲硫醚基团氧化为甲磺酰基,化合物5与3, 4-二甲氧基苯乙胺反应生成最终的目标化合物STL127705(图式 2)。

实验中相关化学试剂均为国产分析纯级,无水溶剂用常规方法干燥处理。使用E. Merck (GF254) 0.25mm TLC硅胶板监测反应;柱层析分离纯化采用快速制备仪器Biotage Isolera I,所用的硅胶为300~400目;反应收率由过柱纯化后计算而得;熔点采用X4型显微熔点仪测定;质谱通过LCQ-Deca XP/Ad型(Thermo Electron)离子阱质谱仪测定,离子源采用电喷雾离子化(ESI);1H NMR和13C NMR谱由Bruker Avance Ⅲ 400MHz核磁共振波谱仪测定。
将5.00g(21.5mmol)2-甲基硫-4-氯嘧啶-5-基甲酸乙酯溶于20mL四氢呋喃,向反应液中加入5mL 25%氨水溶液,室温下反应2h,TLC监测至反应结束后加入30mL饱和氯化铵溶液淬灭反应,用乙酸乙酯(20mL×3)萃取,合并有机相,经无水硫酸钠干燥,过滤,滤液减压浓缩,残留物经柱层析纯化(乙酸乙酯/石油醚,1:1(体积比,下同))得到白色固体4.12g,收率89.3%,熔点130~131℃。ESI-MS m/z:214.0[M+H]+;1H NMR (400MHz,DMSO-d6)δ:8.61(s,1H,H-4),7.84(s,2H,-NH2),3.47~3.41(q,J=6.8Hz,2H,-OCH2CH3),2.45(s,3H,-SCH3),1.04~1.00(t,J=6.8Hz,3H,-OCH2CH3)。
将4.12g(19.3mmol)化合物1溶于15mL甲醇,滴加3mL 0.50g/mL氢氧化锂水溶液,在50℃条件下反应5h,TLC监测至反应结束,加入30mL饱和氯化铵溶液,用乙酸乙酯(20mL×3)萃取,合并有机相,经无水硫酸钠干燥,过滤,滤液减压浓缩,残留物经柱层析纯化(乙酸乙酯/石油醚,1:3)得到白色固体3.25g,收率91.0%,熔点122~124 ℃。ESI-MS m/z:183.9 [M-H]-;1H NMR (400MHz,DMSO-d6)δ:13.17(s,1H,-COOH),8.54(s,1H,H-4),7.89(s,1H,-NH2),7.82(s,1H,-NH2),2.47(s,3H,-SCH3)。
将3.25g(17.6mmol)化合物2和8.01g(21.1mmol)2-(7-氧化苯并三氮唑)-N, N, N′, N′-四甲基脲六氟磷酸酯(HATU)溶于20mL干燥的N, N-二甲基甲酰胺(DMF)中,在氮气保护和室温条件下,向反应液中加入4.6mL(26.4mmol)N, N-二异丙基乙胺(DIPEA),滴加完毕后反应10min,再向反应液中加入2.40g(21.1mmol)3-氟苯胺,室温下反应12h,待反应结束后向反应液中加入30mL饱和氯化铵溶液,用乙酸乙酯(20mL×4)萃取,合并有机相,经无水硫酸钠干燥,过滤,滤液减压浓缩,残留物经柱层析纯化(乙酸乙酯/石油醚,2:3)得到淡黄色固体4.55g,收率93.0%,熔点145~147 ℃。ESI-MS m/z:279.4 [M+H]+;1H NMR (400MHz,DMSO-d6)δ:10.33(s,1H,-CONH-),8.65(s,1H,H-4),7.82(s,2H,-NH2),7.65(dt,J=11.8、2.2 Hz,1H,H-2’),7.49~7.43 (m,1H,H-6’),7.39 (dt,J=15.0,7.5Hz,1H,H-4’),6.94 (ddd,J=8.1、2.5、1.8 Hz,1H,H-5’),2.48 (s,3H,-SCH3)。
在氮气保护和冰浴条件下,将4.55g(16.4mmol)化合物3和20g(65.6mmol)二(三氯甲基)碳酸酯溶于30mL干燥的四氢呋喃中,滴加5.7mL(32.8mmol)DIPEA,反应2h,TLC监测至反应完成后向反应液中加入50mL饱和氯化铵溶液,用乙酸乙酯(30mL×4)萃取,合并有机相,经无水硫酸钠干燥,过滤,滤液减压浓缩,残留物经柱层析纯化(乙酸乙酯/石油醚,1:1)得到淡黄色固体4.43g,收率88.9%,熔点139~140 ℃。ESI-MS m/z:305.5 [M+H]+;1H NMR (400MHz,DMSO-d6)δ:12.48(s,1H,H-7),8.93(s,1H,H-4),7.60~7.50(m,1H,H-4’),7.35~7.24(m,2H,H-2’,H-5’),7.21(d,J=7.9Hz,1H,H-6’),2.61(s,3H,-SCH3)。
在冰浴条件下,将4.43g(14.6mmol)化合物4溶于30mL干燥的二氯甲烷中,慢慢地向反应液中加入14.8g(73.0mmol)85%间氯过氧苯甲酸(m-CPBA),待固体溶解后,撤去冰浴,室温下反应12h,TLC监测至反应完成,向反应液中加入50mL饱和碳酸氢钠溶液,用二氯甲烷(30mL×4)萃取,合并有机相,经无水硫酸钠干燥,过滤,滤液减压浓缩,残留物经柱层析纯化(乙酸乙酯/石油醚,1:2)得到黄色固体3.45g,收率70.4%,熔点142~143 ℃。ESI-MS m/z:337.0 [M+H]+;1H NMR(400MHz,DMSO-d6)δ:13.18(s,1H,H-7),9.32(s,1H,H-4),7.58(dd,J=14.7、8.0 Hz,1H,H-4’),7.39~7.30 (m,1H,H-5’),7.26 (d,J=9.5Hz,1H,H-2’),7.22(d,J=8.0Hz,1H,H-6’),3.48(s,3H,-SO2CH3)。
在氮气保护下,将3.45g(10.3mmol)化合物5和2.85g(15.5mmol)3, 4-二甲氧基苯乙胺溶于10mL N, N-二甲基乙酰胺(DMA)中,80℃反应2h,TLC监测至反应完成,向反应液中加入30mL水淬灭反应,用二氯甲烷(30mL×4)萃取,合并有机相,经无水硫酸钠干燥,过滤,滤液减压浓缩,残留物经柱层析纯化(乙酸乙酯/石油醚,1:2)得到黄色固体3.50g,收率77.8%,熔点212~213 ℃。HRMS (ESI) m/z:C22H20FN5O4 [M+H]+,理论值437.4256,实测值437.4216;1H NMR (400MHz,DMSO-d6)δ:11.51 (s,1H,H-7),8.70 (s,1H,H-4),7.80 (s,1H,H-9″),7.51 (dd,J=15.1、7.7 Hz,1H,H-4’),7.29~7.10 (m,3H,H-2’,H-5’,H-6’),6.89 (d,J=8.4 Hz,2H,H-2″,H-5″),6.81 (d,J=7.3 Hz,1H,H-6″),3.79 (s,3H,-OCH3),3.77 (s,3H,-OCH3),3.67~3.61 (dd,J=7.1、6.8 Hz,2H,H-8″),2.87 (t,J=7.3Hz,2H,H-7″);13C NMR (101MHz,DMSO-d6)δ:173.70,167.52,162.23,161.61,160.16,150.38,149.83,148.82,147.15,137.91,127.82,123.96,122.33,117.25,116.98,113.26,112.81,103.86,57.82,57.73,45.62,33.96。
从目标化合物STL127705的1H NMR谱中观察到终产物存在互变异构现象,可能的互变异构位点为分子结构中的环内酰胺键,其可由酮式发生烯醇式互变(图式 3)。两种异构体之间存在一种动态平衡,在低温时这种互变速率比较慢,1H NMR可以检测出两套峰,当温度升高时,互变速率变快,就会融合成一套峰。因此测试了终产物的升温(80℃)1H NMR,图谱结果显示为一套峰,证实我们的推测。
中间体4和中间体5同样存在环内酰胺键,但是在1H NMR中并未观察到明显的两套峰现象,可能是由于两种异构体稳定性差距很大,那么平衡可能99%偏向于其中一个稳定结构,1H NMR就仅会显示一套峰。然而,目标化合物的结构发生了改变,会影响平衡的偏向,两个异构体的稳定性比较接近,在室温下观测到的1H NMR就会显示出二者的氢峰,所以是两套峰。
本文报道了一种简单、有效的DNA-PK抑制剂STL127705的合成方法。该方法以市售化工原料为起始原料,经过6步反应,以36.8%的收率得到目标产物,纯度达到95.5%。该制备方法条件温和,无需苛刻的反应条件,方法简单,技术可行性高,为新型的具有嘧啶并嘧啶二酮结构的DNA-PK抑制剂的合成研究提供了依据。
N Jette, S P Lees-Miller. Prog. Biophys. Mol. Biol., 2015, 117(2~3):194~205. doi: 10.1016/j.pbiomolbio.2014.12.003
FM Hsu, S Zhang, B P Chen. Transl. Cancer Res., 2012, 1(1):22~34. https://www.ncbi.nlm.nih.gov/pubmed/22943041
M B Chen, Z T Zhou, L Yang et al. Oncotarget, 2016, 7(13):17047~17059. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4941370/
L Cornell, J M Munck, C Alsinet et al. Clin. Cancer Res., 2015, 21(4):925~933. doi: 10.1158/1078-0432.CCR-14-0842
王傲雪, 马景昕.大连医科大学学报, 2018, 40(5):385~393. http://www.cnki.com.cn/Article/CJFDTotal-DLYK201805002.htm
隋江东, 王颖, 万跃.现代医药卫生, 2018, 34(23):3589~3593. doi: 10.3969/j.issn.1009-5519.2018.23.002
V J Weston, C E Oldreive, A Skowronska et al. Blood, 2010, 116(22):4578~4587. doi: 10.1182/blood-2010-01-265769
G Pratt, C Yap, C Oldreive et al. Brit. J. Haematol., 2018, 182(3):429~433. doi: 10.1111/bjh.14793
Y Chen, H Du. Biomed. Pharmacother., 2018, 99:552~560. doi: 10.1016/j.biopha.2018.01.094
E Wererings, A C Gallegos, L N Dominick et al. DNA Repair, 2016, 43:98~106. doi: 10.1016/j.dnarep.2016.03.014

图式 2 本文报道的目标化合物STL127705的合成路线
Scheme 2 Synthetic route of target compound STL127705 reported in this paper

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

 下载:
下载: