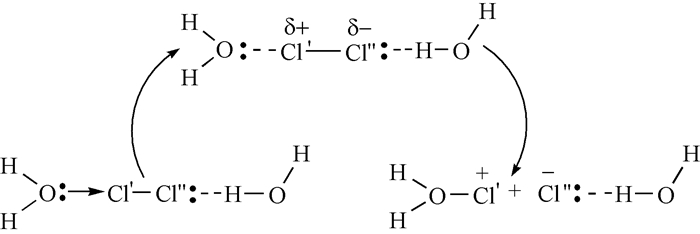
 图 图式1
H2O诱导下Cl2的离子化
Figure 图式1.
Cl2 ionization induced by H2O
图 图式1
H2O诱导下Cl2的离子化
Figure 图式1.
Cl2 ionization induced by H2O
引用本文:
王稼国, 荆西平. 共价键的离子化[J]. 化学通报,
2017, 80(4): 400-407.

Citation: Wang Jiaguo, Jing Xiping. Ionization of Covalent Bond[J]. Chemistry, 2017, 80(4): 400-407.
Citation: Wang Jiaguo, Jing Xiping. Ionization of Covalent Bond[J]. Chemistry, 2017, 80(4): 400-407.
共价键的离子化
English
Ionization of Covalent Bond
Abstract:
Several ionization patterns of covalent bond, including induced-ionization of covalent bond in metals and nonmetals, induced-ionization of covalent bond in hydrogen compounds and metal compounds, lowering temperature and self-induced ionization of compound, and so on, were deduced and analyzed by using chemical bond theory. The trend of ionization was also analyzed on energy changing and several important applications of the ionization of covalent bond were discussed.
-
Key words:
- Covalent bond
- / ionization
- / Coordination bond
-
在现行的国内外化学教科书中,离子键和共价键都是核心内容,分别以独立章节专门介绍其形成和作用方式,而它们之间的联系和转化却没有进行系统地介绍[1~3]。这导致学生对离子化合物和共价化合物的认识大都停留在记忆的积累,缺少深入系统地理解。学习过程中一些问题时常让学生感到困惑:为什么非金属元素的原子之间也能形成离子键?高氧化态的金属化合物形成的共价化合物为什么也能离子化?本文从实验现象出发,归纳和分析了共价键向离子键转化的几种方式和转化前后的能量变化,提供了一种理解共价键向离子键转化的思路,并将这一分析思路用于对更多实验现象的解释。
1 共价键的离子化方式
以分子RA-DR′中的成键情况为例进行讨论。其中A为电负性小的原子,D为电负性大的原子(下同),R和R′分别为连接在A和D上的诱导原子或原子团,A-D间的化学键为共价键。A-D间的共价键通常有两种方式断裂:
即反应(1) 的均裂自由基方式和反应(2) 的异裂离子化方式。一般来说,物质受光照或受热的情况下容易发生均裂,即生成自由基,对于键能较小且极性较小的共价键更是如此。如由卤素或过氧化物参与的化学反应,由于X-X(X表示卤素)和过氧链O—O的共价键均弱而易断裂,在光或热的作用下容易发生生成自由基的均裂。共价键的离子化不易通过升温或光照来实现,这是因为升温和光照都是将能量低的分子轨道上的电子激发到高能量的分子轨道,而高能量的分子轨道在成分上往往以电负性小的原子的轨道为主,因而以上过程更易导致共价键的均裂。共价键的离子化通常通过其他方式实现,如降温可以实现共价键的离子化,而更常见的离子化方式则是通过其他原子、分子或离子的诱导来实现,也是本文重点讨论的内容。
共价键的离子化是一个典型的化学过程,要发生则必须在能量上有利,即ΔG < 0,根据吉布斯-亥姆霍兹公式:
其中,ΔH项由离子化前后化学键的变化决定,而ΔS则是离子化前后的熵变。如果离子化过程没有溶剂参与,离子化趋势主要由ΔH项决定;而如果离子化过程有溶剂参与,则熵变往往也成为主要决定因素。离子化反应最终也会建立平衡,可以用平衡常数的大小来直接表示离子化趋势的大小。离子化过程也会体现动力学控制,此时就只与反应物的组成和结构有关了,尤其与形成共价键的A、D原子及其化学键有关。
2 单质中共价键的离子化
单质包括非金属单质和金属单质,其中,非金属单质可以分为小分子非金属单质和巨型分子非金属单质,它们的离子化行为稍有不同。由于形成共价键的原子相同,因此单质中的共价键几乎一样,即绝大多数情况下都是非极性键,因此象光、温度和压力这些外界条件很难使其离子化,只有通过其他反应性的分子或离子的诱导才能发生离子化。
2.1 非金属单质的离子化
以Cl2为例,该双原子分子只形成一条共价键,即Cl-Cl,其中每个Cl上有3对孤电子,且还有能量相对较低的非键3d空轨道,因此它既可以向缺电子原子或原子团提供孤电子对,又可以接受富电子原子或原子团提供的孤电子对。
分析Cl2在水溶液中的离子化时,考虑H2O为诱导分子,其中O上有孤电子对,而H是缺电子原子。Cl2分子中的一个Cl(标识为Cl′)通过3d轨道接受H2O的电子对,则Cl′原子由于电子增多而表现为富电子,从而将Cl2中的共用电子对推向另一Cl原子(标识为Cl″),而Cl″原子也会与H2O中的缺电子的H作用,即将孤电子对与H作用形成氢卤键,氢卤键的形成会将Cl″原子上的电子拉走,因此也增强了Cl″原子拉共用电子对的能力,结果导致Cl′原子带上正电荷,而Cl″原子带上负电荷,见图式 1,如果此时共价键断裂,则离子化就完成,生成的产物进一步与水作用:
图 图式1
H2O诱导下Cl2的离子化
Figure 图式1.
Cl2 ionization induced by H2O
此过程中的能量因素包括Cl-Cl化学键的强弱、新形成的O-Cl键强弱、H2O与H+形成配位键的强弱和水合氯离子的稳定性、反应物和产物在溶剂中的分散则提供熵效应,这些都是影响离子化反应的主要的热力学因素。H2O对Cl的配位能力、Cl2的变形能力和氢卤键的强弱等则是影响离子化反应动力学的主要因素。
如果将H2O换成供电子能力更强的OH-,则在与Cl″作用后推电子能力更强,使Cl-Cl间的共用电子对偏离更大,从而使离子化反应变得更容易。同时OH-也能与反应(3) 中的离子化产物进一步反应,促进离子化反应的平衡正向移动,使总反应变为:
如果将Cl2换成I2,由于I2相对更容易变形,且I-I共价键更弱,因此其离子化的动力学活性更高。但由于I2的非键空轨道5d能量也高,与H2O中O上孤电子对所在的轨道能级差异更大,即H2O-I间的配位键相对更弱,且其氢卤键也因I的半径大也弱,导致I2在水中的离子化总趋势反而不如Cl2。因此,I-I共价键的离子化在酸性和中性水溶液中趋势很小,只有在碱性条件下才变得彻底。
如果将Cl2换成电负性比H小的非金属元素单质,如Si,在水中的离子化反应变为:
由于Si表面存在一层致密的SiO2氧化膜,因此在水中反应难以进行。但如果在碱性溶液中,碱会与SiO2反应而使Si表面裸露出来,则除了会发生反应(6) 外,还会有如下后续反应:
即电负性相对小的Si″上的电子会向H2O上的H转移,从而还原成H2,见反应(8);而碱的存在使失电子Si′最终转化为可溶性SiO32-,从而实现表面不断更新,使反应持续向正向进行。
考虑将Cl2换成N2的情况:N2中能量最低的空轨道为π*,由于分子中的π键很强,因此π*轨道的能量很高,难以接受孤电子对,而且N2分子中形成的是叁键,稳定性高,极难变形,所以N2不易发生离子化。
非金属单质发生离子化的难易程度分为三类:(1) H2、N2、O2和F2单质分子中没有空的低能量价轨道,这几种单质不发生离子化;(2) C (石墨)、P、S、Cl2、Se、Br2和I2单质中均有能量相对较低的空轨道,元素的电负性均大于H,在诱导下可以发生离子化,在碱性条件下易发生歧化反应;(3) B、Si、As和Te单质中均有能量相对较低的空轨道,元素的电负性均小于H,在碱或更强配体作用下发生离子化,并放出氢气。
2.2 金属单质的离子化
金属单质常以巨型分子存在,在成键方面存在以下一些特点:金属中相邻原子之间的金属键可以看成共价键,金属原子的缺电子性导致金属晶体拥有大量低能量的空轨道,因而金属可以被看作路易斯酸;金属的原子半径普遍较大,相对容易变形;金属表面原子由于只有朝内方向能形成金属键,而朝外方向没有化学键,因此表面原子形成的键少,但其方向性更强,拉电子能力更强,电负性更大,形成的化学键也更强,这些原子周围在离子化过程中更易积累电子而带负电荷;成键数多的金属原子的电负性相对较小,更易失去电子而形成阳离子,相当于积累正电荷。因而,表面形貌不规则的金属晶粒容易在诱导下产生正电荷积累区和负电荷积累区。
基于以上金属的成键特点,我们可以讨论金属单质在水溶液中的离子化过程。以M代表金属原子(下同),H2O中O上的孤电子对会对缺电子的金属原子的空轨道进行配位,使该金属原子上的电子密度增大,则该金属原子将金属键上的电子推离到其他金属原子上,导致与O配位的金属原子带正电荷,而接受推送过来电子的金属原子带负电荷,见图式 2。与O配位的金属原子被阳离子化,进一步与更多的H2O形成配位键,从而被拉离金属母体Mm而进入溶液形成水合阳离子;富电子区金属将负电荷与H2O上电正性的H作用,即将水吸附在其表面。如果电子没有移出体系,则金属带负电,而溶液带正电,在金属和溶液界面上产生电势差。需要特别强调的是,由于金属的缺电子性,金属晶体中拥有很多低能量的空轨道,这些轨道更适合电子占据,因此在金属单质存在的情况下,如果没有容易得到电子的氧化剂存在,电子不容易进入溶液,即不容易形成溶剂化电子。
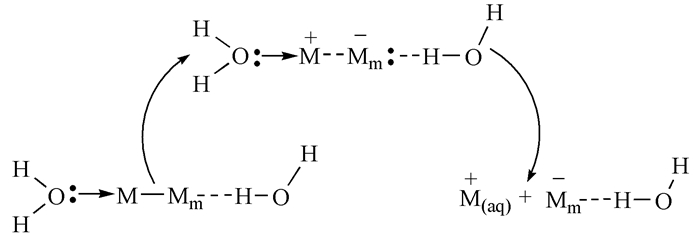 图 图式2
H2O诱导下金属单质的离子化
Figure 图式2.
Ionization of metal elemental induced by H2O
图 图式2
H2O诱导下金属单质的离子化
Figure 图式2.
Ionization of metal elemental induced by H2O
如果水溶液中存在氧化剂,则缺电子的氧化剂会比H2O更易吸附在金属的负电荷区。这时,电子会向氧化剂传递而发生氧化还原反应,即电子被消耗,从而使金属进一步溶解,导致金属腐蚀。如果溶液是酸性的,则在富电子区,水合氢离子H3O+上的H会作用到带负电荷的金属母体M-m上,由于金属的电子密度较高(金属较活泼),电子会从金属上转移到H3O+上而放出氢气,即发生析氢腐蚀:
如果溶液中溶解有氧气,则溶解氧替代H2O更易吸附到富电子的金属表面,也会发生由金属母体Mm到O2间的电子转移,只是需要转移的电子更多,即发生吸氧腐蚀:
在富电子区生成OH-,再通过金属水合阳离子和OH-的扩散并作用生成金属氢氧化物。
如果金属中含有少量其他惰性金属或非金属原子,即合金材料,则惰性金属和非金属原子的电负性显然要大于相对活泼的金属,这些惰性金属或非金属原子的聚集区就成为富电子区,而活泼金属原子区则电子被拉走,是缺电子区。在缺电子区,H2O中O上的孤电子对通过配位不断将金属离子拉出并溶解形成水合离子,而在富电子区,氧化剂则不断消耗电子,即形成了原电池,导致金属被快速腐蚀。
如果换成配位能力比水强的配体,由于形成配合物的趋势增加,则金属键的离子化更加容易,从而导致金属富电子区的电子密度更高,还原能力会增强,即金属更容易被氧化腐蚀。如金单质在水溶中非常稳定,不可能被溶解氧所腐蚀,但在NaCN溶液中,由于CN-的配位能力远强于H2O,与Au+形成非常稳定的配合物[Au(CN)2]-,从而使富电子区的电子密度显著提高,即金的还原能力显著增强,可以被溶解氧腐蚀:
金属虽然通常情况下表现为失去电子而形成阳离子,但某些金属的最外层轨道也可以得到电子而使轨道上电子排布更稳定,如碱金属由ns1得电子变为ns2,铜族元素的原子也有类似倾向,从而形成金属阴离子。在没有氧化剂存在的情况下,金属阴离子上的电子无法传递,就能与金属阳离子形成离子键而稳定下来,相当于金属单质发生了歧化反应。此类盐的形成也要求在配体诱导下才能形成。
以金属Cs为例,由于Cs原子最外层只有1个电子,单质形成金属键必然是缺电子集团,即拥有大量的低能量的空轨道,相邻原子之间可以看成共价键。若以L表示供电子的配体(下同),配体L在金属表面某处靠近时,配位原子将电子提供给作用的Cs原子,导致该Cs原子上的电子富集,则必然会将Cs-Cs之间的共用电子推向远处,该Cs原子变为阳离子并被配体稳定后离开金属表面。而带负电荷的部分也会受到配位阳离子的静电吸引,由于铯原子间的金属键很弱,带负电的部分被配位阳离子吸引后会以Cs-的形式离开金属表面,其离子化过程可以表示为:
在这里,配体L起至关重要的作用。一方面要求它能提供电子对,即通常是氮族或氧族原子作为配位原子,且能将Cs+和Cs-两种离子隔开;另一方面又要保证强还原性的Cs-不被氧化,即配体上不能存在有氧化性的原子和原子团。满足这一条件的配体有冠醚、穴醚(硫醚)、氮杂穴醚和穴胺(叔胺)等。这类碱金属阴离子盐绝大多数稳定性较差,往往只有低温下才能存在。近来,一些室温下稳定的金属阴离子盐也已被合成出来[4, 5]。与非金属单质的离子化稍有不同,由于金属通常是缺电子原子,因此其离子化主要靠诱导配体的供电子形成配位键来稳定其中的阳离子,而阴离子只有通过同阳离子形成的离子键来稳定。所以,配体的配位能力越强,离子化趋势越大;金属原子的变形能力越强、金属键越弱,离子化趋势越大;动力学上活化能越低、反应速率越快。如原子的变形大小比较,有:
配体也在形成碱金属阴离子盐时有决定作用,如穴状配体的孔体积决定了可以进入的金属阳离子大小,要合成Li和Cs的阴离子盐,如果使用小孔配体,则成盐时Li+入孔穴,而外围为Cs-(图 1,为氮杂穴醚[2.1.1]形成的金属阴离子盐Li+(C211) Cs-的晶体结构图[5]);反之,使用大孔配体,则Cs+入孔穴,而外围为Li-。
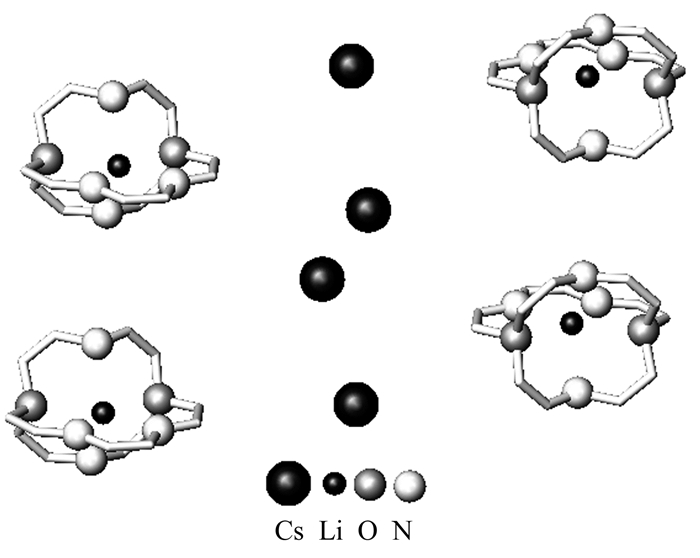 图 1
碱金属阴离子盐Li+(C211) Cs-的结构图
Figure 1.
Structure of alkali anion salt Li + (C211) Cs-
图 1
碱金属阴离子盐Li+(C211) Cs-的结构图
Figure 1.
Structure of alkali anion salt Li + (C211) Cs-
如果此时环境中存在氧化剂,如在含有活泼氢的醇、水、胺类溶剂中,金属阴离子将不会稳定存在。此时电正性的活泼H原子会与金属阴离子相互作用,后者会将电子传递给活泼氢,从而发生氧化还原反应而放出氢气。因此,这类金属阴离子盐只有在严格无氧、无水、低温且无其他氧化剂存在的情况下才能合成。
从以上分析可以看出,单质中的共价键只有在外来分子或离子的诱导下,尤其是配体诱导下才能离子化。外来分子或离子的诱导的本质是通过另一个或多个化学反应来推动离子化反应的进行,即让一个能量上不利的离子化反应通过耦合能量上有利的反应来实现总反应的正向进行。由于金属不论是离子态还是单质态都具有低能量的空轨道,且金属键相对较弱,因此既容易接受配体的孤电子对形成稳定的配离子,又可以通过化学键的离域而分散电子,因此诱导离子化比非金属单质更加容易。
3 化合物中共价键的离子化
如果形成共价键的原子A和D不同,由于存在电负性差值,则共价键A-D属于极性共价键,其中电负性小的原子带部分正电荷,而电负性大的原子带部分负电荷。与单质比较,化合物中的共价键相对更易离子化。电负性差值越大则共价键的固有极性越大,越容易离子化。这一规则尤其适用于周期表中同周期元素间的比较,如对于非金属元素的氢化物而言,离子化趋势有以下顺序:
不同周期元素之间则不能简单地只考虑成键原子间的电负性差异,此时共价键的强弱则是更重要的因素。一般来说,共价键越弱的离子化趋势越大。如同族元素氢化物之间的比较,离子化趋势为:
如果成键原子A和D之间的电负性差值很大,则形成的化学键离子键成分很高,此时无需诱导也能离子化。活泼金属和活泼非金属之间形成的化学键离子成分很高。O、F和Cl与碱金属、重碱土金属以及f区元素形成的键大都是离子键,如NaCl和CaO就是典型的离子化合物,它们甚至在高温熔融状态下都能离子化。
尽管由电负性差异引起离子化趋势增大,但对共价成分高,尤其是键能很大的共价键来说,离子化仍然很不容易;要使其离子化往往需要外来分子或离子的诱导,这也相当于通过耦合能量上有利的反应以使整体反应在能量上有利,从而实现共价键的离子化。
对于以共价键为主的化学键,要实现离子化可以有多种方式。
3.1 分子内离子化
分子内离子化的例子不多,通常发生在降温过程中,此过程涉及到一种不稳定的结构向稳定结构转变,即离子化后的体系在能量上更有利。该类型的离子化往往出现在负氧化态的成键原子相对容易变形的分子中,或是中心原子的空间位阻大,这样形成的极性共价键较弱;还有些体系在降温过程中发生成键方式或成键类型变化,主要表现为化学键增强或是化学键数量增加,离子化在这两种情况下能量上有利。
典型的大位阻的例子如PBr5,它在降温过程中离子化[6]:
P和Br之间存在较明显的电负性差,Br原子的半径大易变形,再由于P原子的半径相对较小,周围容纳5个较大体积的Br原子较困难,因此拥挤的空间导致三角双锥结构的PBr5中的P-Br化学键变长而较弱,离子化后生成四面体结构的PBr4+位阻小,有4条短的P-Br键,同时PBr4+和Br-之间还存在离子键,使体系总能量更低。
降温过程中化学键转化并增强的体系也易离子化。如N2O5在降温过程中离子化[7]:
气体分子中的π710离域键相当于键级为3,转化为晶体后发生离子化,NO2+中的两个π34和NO3-中的π46,总键级仍然是3,但增加了离子键,所以体系稳定性提高。相似地,Cl2O6离子化为[ClO2]+[ClO4]-,离子化过程中伴随歧化反应[8]。
3.2 分子间离子化
化合物中共价键的诱导离子化极为普遍,此类诱导离子化又可分为同种分子间相互诱导离子化与外来分子的诱导离子化两类;前者相当于自耦电离,在孤电子对和低能量的空的价轨道同时存在的情况下较易发生;后者是指在路易斯碱或路易斯酸的诱导下离子化,更为常见,又可以分为含氢共价键的诱导离子化和非氢共价键的诱导离子化。
3.2.1 同种分子间相互诱导离子化
降温过程中同种分子间在相互靠近时首先通过分子间作用力发生相互作用,再将分子间作用力转化为配位键,并与原共价键平均化,进一步引发离子化,即同种分子间通过相互诱导而离子化。分子间离子化过程主要表现为总化学键键能增大或化学键数量增加,甚至将分子间作用力转化为化学键。
典型的例子有PCl5、SbCl5、BiBr、Ga2X3、MI和M2X4(M=Ga、In、Tl,X=Br、I)等。这些化合物在降温过程中都会通过分子间相互作用而离子化,其晶体结构中均可以显示出其离子化结果。在这些化合物中虽然离子化产物稍有差异,但离子化过程存在很多类似性。
PCl5在降温过程中发生如下离子化:
P上有低能量的3d非键空轨道,而Cl上有孤电子对,两个PCl5分子靠近后,一个分子中的Cl将孤电子对配位到另一分子中的P上,离子化前后其共价键总数量没有改变,即其中的一个PCl5基团给出一个Cl-而转化为PCl4+,此时形成4条短的P-Cl键,而另一PCl5接受一个Cl-形成PCl6-,其P-Cl键增长了。由于体系中增加了离子键,因此总能量是有利的。图 2是PCl5晶体结构[9],可以清楚地显示其离子化后的微粒。
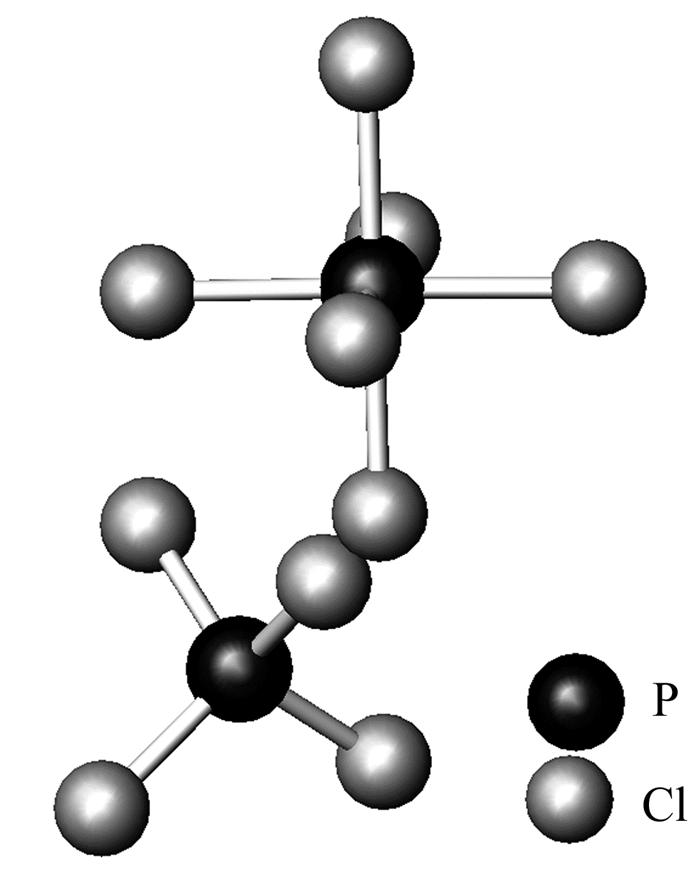 图 2
PCl5的晶体结构示意图
Figure 2.
Crystal structure diagram of PCl5
图 2
PCl5的晶体结构示意图
Figure 2.
Crystal structure diagram of PCl5
BiBr晶体结构显示,该化合物降温离子化后形成Bi+[BiBr2]-,[BiBr2]-通过共用配位原子Br而形成平面四边形BiBr4单元,该单元相互连接形成无限长链,长链排列形成层状结构,而Bi+处于层间,见图 3。晶体结构[10]中[BiBr2]-中的Bi-Br键长为290~300 pm,而层间Bi原子与Br原子最短距离大于355pm。BiBr的离子化过程可以理解为Br上的孤电子对与一部分Bi的价层空轨道相互作用形成配位键,即形成[BiBr2]-链,而使另一部分Bi与Br之间的共价键减弱并离子化为Bi+和Br-,即将体系中原有的共价键转化为配位键+离子键,离子化后体系的化学键数量增加,因此能量上有利。
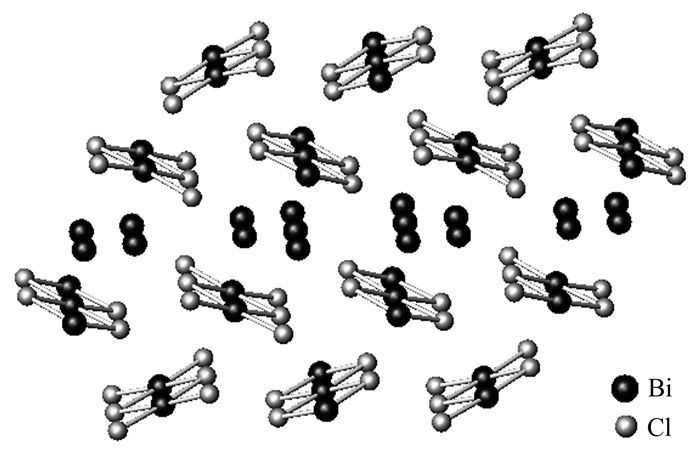 图 3
BiBr的晶体结构
Figure 3.
Crystal structure of BiBr
图 3
BiBr的晶体结构
Figure 3.
Crystal structure of BiBr
在含氢化合物中,如果与氢成键的原子电负性大于H且其上还含有孤电子对,则可以通过氢键和配位键之间的转化而发生质子传递,产生自耦电离。这类含氢化合物包括H2O、HF、NH3、H2SO4等,如H2SO4分子之间相互诱导发生如下自耦离子化:
3.2.2 含氢共价键的诱导离子化
含H共价键的离子化是最常见的离子化方式之一。此类共价键的离子化的基本要求是,体系中含有具有富电子特性的原子或原子团,通常是指有孤电子对或至少是π电子对。这些富电子原子或基团通过向H+提供电子形成配位键,从而将原有的含H共价键断裂,并形成阴离子和阳离子。此过程中,破坏了一条共价键,形成了一条配位键,产生了新的离子键。如果体系中原有共价键强度大,则离子化过程在能量上不利,即体现弱离子化趋势;反之,如果原共价键较弱,则离子化过程在能量上有利,离子化趋势大,甚至完全离子化。虽然有些含H共价键可以在降温过程中通过分子间相互作用实现离子化,如HBr[11]中的H-Br共价键,但更多情况下含H共价键的离子化过程需要通过其他分子的诱导来实现。
电负性比H小的原子A与H形成共价键时,共价键H-A中,A是缺电子原子,而H为富电子原子,配体L将与缺电子的原子A作用,导致与A成键的H上电子密度进一步增大,并使之更容易失去电子。这类氢化物包括由B、Si、As等非金属元素和其他主族金属元素与H形成共价键的化合物,它们的共性是这些元素都有低能量的空轨道,易受路易斯碱诱导而离子化。这里以SiH4与碱作用为例进行讨论。Si电负性小于H,因而SiH4中Si带正电荷而H带负电荷;OH-中O上的孤电子对向缺电子的Si的3d空轨道配位,生成的产物进一步脱水生成SiO32-;而H2O上的H缺电子,与Si-H上的富电子H作用产生H2:
即电负性小的元素与H形成的共价键,在与水或碱作用时放出氢气,类似的有B2H6、LiH等。如果溶液中存在氧化性强于H2O的氧化剂,H-A上的H会将电子传递给更强的氧化剂。
电负性比H大的原子D与H形成共价键时,共价键H-D中,D是富电子原子,而H是缺电子原子。如果用路易斯碱(配体L)来诱导,将向缺电子的原子H供电子,导致H-D间的电子对更偏向D,从而通过形成LH+和D-离子而离子化,此时相当于质子传递,即H-D的离子化就是质子酸性的体现,如:
如果用路易斯酸来诱导,将接受富电子的原子D的孤电子对,也会导致H-D间的电子对更偏向D,使H+更易电离。如:
这类氢化物包括由F、Cl、Br、I、O、S、Se、N和P与H形成共价键的化合物,它们的共性是这些与H成键的元素的电负性都大于H,且成键后还有孤电子对,因此还可以看作路易斯碱。如果表现路易斯碱性时配位能力比L还强,则离子化方式将会改变。如NH3溶解在H2O中,NH3的供电子能力强于H2O,因此前者是配体,诱导后者离子化;而HF溶解在H2O中,HF的配位能力弱于H2O,后者是配体,诱导前者离子化。
质子化趋势的大小与质子酸的组成和结构有关。在元素周期表中,不同周期比较,D的原子半径越大,则H-D键越弱,质子化趋势越大,即酸性越强;同周期比较,D的电负性越大,则与H形成的键极性越强,质子化趋势也越大;如果在D原子上还引入了诱导基团,则诱导基团的拉电子能力越强,质子化趋势越大。对酸性强弱的比较,有如下的顺序:
质子化趋势还与提供孤电子对的配体的强弱有关。配体供电子能力越弱,则酸的质子化趋势越弱;配位原子的原子半径越大,则形成的配位键越弱,酸的质子化趋势越弱。如HF在如下溶剂中体现的酸性强弱顺序为:
由于强酸具有很强的供质子能力,常用于醇、醚、酯、酰胺和酮醛等参与的化学反应的催化剂。这些都是基于O或N上孤电子对与质子发生作用后化学键强弱发生改变或是电子密度发生改变。乙醇在强酸作用下接受质子会生成多种活性中间体:
含双键和叁键的分子,由于π键的存在,其电子裸露程度虽然不如孤电子对,但仍然具有一定的配位能力,也能作为配体使H-D共价键离子化,即H-D异裂成H+和D-,而H+首先加在双键上形成正离子,同时生成D-离子,这即为氢化物对双键的加成反应机理见,图 4。对于一些大型离域体系,如苯环和共轭烯烃,也能通过π电子来接受质子,并可以使H+在形成π键的原子上游走。
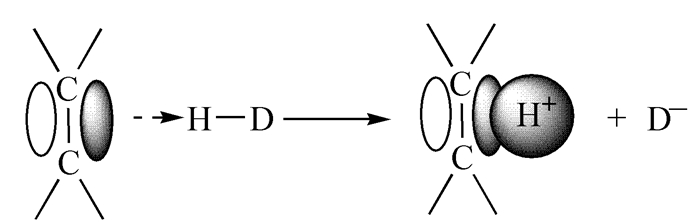 图 4
双键的质子化
Figure 4.
Protonation of the double bond
图 4
双键的质子化
Figure 4.
Protonation of the double bond
3.2.3 非氢共价键的诱导离子化
共价键的诱导离子化是常见的化学反应,对于共价键A-D来说,如果用外来富电子的配体诱导,则向缺电子的A提供电子,按如下反应进行:
以上反应方程可以理解为发生了亲核反应而离子化,如:
显而易见,配体L的供电子能力是该类离子化反应的主要动力,另外,离子化产物的稳定性也对离子化趋势有一定影响。
如果离子化过程需要外来缺电子的路易斯酸诱导(用Q表示),则要求富电子的D上有孤电子对,以便向酸Q提供电子对:
反应式(29) 可以理解亲电反应,这也是很常见的一类反应,如:
这类反应中缺电子的金属化合物都是路易斯酸,都能催化电负性原子上有孤电子对的共价键离子化。在这类反应中路易斯酸酸性越强,催化效果越好。在下列氯化物中,路易斯酸性由弱到强的顺序为:NaCl < MgCl2 < AlCl3,可以通过金属最低能量空轨道的能量高低来判断酸性强弱。空轨道能量越低则路易斯酸的酸性越强。
如果路易斯酸和路易斯碱同时使用,则同时发生亲电反应和亲核反应。如:
反应(31) 是在酸碱共同诱导下,离子化反应更加容易,趋势更大。
在无机物中,金属与非金属形成的共价键(M-D,D为非金属原子)非常普遍,这类化合物由于金属原子M上缺电子,因此本身就是路易斯酸。这些化合物包括金属卤化物MX、含氧酸盐、金属的醇盐或酚盐(ROM)、硫醇盐(RSM)、硒醇盐(RSeM)和胺基盐(RNHM)等,它们也能在其他配位体或缺电子物质的作用下发生离子化。M-D共价键的离子化方式也有两种:
反应(32) 实际上就是金属离子的配位反应,即配体L对金属中心发生亲核进攻,并取代配体D-。金属盐类在水中或其他配位溶剂中的溶解过程即属于这一类:
反应(34) 分多步完成,且生成的产物[Fe(OH2)6]3+还会与水作用发生质子传递,相当于水解。反应(35) 中,由于醇的位阻大于水,导致乙醇的配位能力弱于水,因此与在水中相比,化合物在醇中溶解度有所下降。因此,配体的配位能力越强,则离子化趋势越大;反过来理解,如果离子化后金属的氧化态(电荷)越高,则与配体的作用力能力越强,离子化反应在能量上越有利,离子化趋势也越大。从共价键中金属原子的电子结构方面来说,具有低能量的内层d空轨道来形成配位键,则配位键越强,离子化趋势越大,也就是说过渡金属共价键更易离子化。有效核电荷越高,也越易离子化,即同周期元素从左到右形成配位键的能力增强。反应(33) 常见于两种金属化合物之间的反应中,尤其在熔盐中更常见,如:
反应(36) 中,FeCl2的路易斯酸性帮助LiCl离子化,如果换成酸性更强的FeCl3,则LiCl的离子化更加容易。这类离子化能促进相互溶解。
4 总结
共价键的离子化在化学反应中是普遍存在的现象。通过分析可以得出,离子化过程一般需要富电子原子或原子团对缺电子原子或原子团的诱导,或是通过缺电子原子或原子团对富电子原子或原子团的诱导,从而促进共价键中的共用电子对显著偏移,并最终离子化。共价键的离子化应用非常广泛,可以用来解释金属的腐蚀、亲核反应、亲电反应、配合物的形成、酸碱类催化反应以及物质的配位溶解等。因此,对共价键离子化方式的理解可以加深对化学原理的认识。
-
-
[1]
北京师范大学等校编. 《无机化学》第四版, 高等教育出版社, 2002.
-
[2]
C E Housecroft, A G Sharpe. Inorganic Chemistry, 4th Ed. 2012.
-
[3]
J C Kotz, P M Treichel, J R Townsend. Chemistry & Chemical Reactivity, 8thEd. 2011.
-
[4]
J Kim, A S Ichimura, R H Huang et al. J. Am. Chem. Soc., 1999, 121:10666~10667. https://www.researchgate.net/publication/231532592_Crystalline_Salts_of_Na-_and_K-_(Alkalides)_that_Are_Stable_at_Room_Temperature?ev=prf_cit
-
[5]
A S Ichimura, R H Huang, Q S Xie et al. J. Phys. Chem. B, 2006, 110:12293~12301. https://www.researchgate.net/publication/6983198_One-dimensional_zigzag_chains_of_Cs-_the_structures_and_properties_of_Li_(cryptand2.1.1)Cs-_and_Cs_(cryptand2.2.2)Cs-?ev=auth_pub
-
[6]
W Gabes, K Olie. Acta Crystallogr. B, 1970, 26: 443~444. https://www.researchgate.net/publication/234163170_Refinement_of_the_crystal_structure_of_phosphorus_pentabromide_PBr5
-
[7]
E Grison, K Eriks, J L de Vries. Acta Crystallogr., 1950, 3: 290~294. https://www.researchgate.net/publication/250910379_Structure_cristalline_de_l'anhydride_azotique_N_2_O_5
-
[8]
K M Tobias, M Jansen. Z. Anorg. Allg. Chem., 1987, 550: 16~26. https://www.researchgate.net/publication/229876297_Untersuchungen_an_festem_Dichlorhexoxid_Erste_Kristallstrukturbestimmung_an_einem_Chloroxid
-
[9]
H Preiss. Z. Anorg. Allg. Chem., 1971, 380: 51~56. https://www.researchgate.net/publication/230468522_Strukturverfeinerung_und_Untersuchung_der_thermischen_Schwingungen_am_festen_Phosphor(V)chlorid
-
[10]
H von Benda, A Simon, W Bauhofer. Z. Anorg. Allg. Chem., 1978, 438: 53~67. https://www.researchgate.net/publication/230377296_zur_kenntnis_von_bibr_und_bibr1167
-
[11]
J K Cockcroft, A Simon, K R A Ziebeck. Z Krist-Cryst. Mater., 1988, 184: 229~246. https://www.researchgate.net/publication/244746982_The_structures_of_crystalline_hydrogen_bromide
-
[1]
-


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 50
- 文章访问数: 4158
- HTML全文浏览量: 1535
 下载:
下载:
