
图式 1.
CuAAC反应及其可能机理
Scheme 1.
CuAAC reaction and its possible mechanism

1963年, Huisgen[1]对高温条件下叠氮与炔烃的1, 3-偶极环加成反应进行了研究, 然而由于反应条件苛刻且区域选择性不理想, 很大程度上限制了该反应的应用. 2002年, Meldal和Sharpless小组[2]分别独立报道了铜催化的叠氮与炔烃的环加成反应(简称CuAAC).他们发现, 使用一价铜作为催化剂, 能够单一地得到1, 4-取代的1, 2, 3-三氮唑类化合物.由于其具有区域选择性高、底物普适性广、反应效率高且条件温和等优点, 符合2001年Sharpless等[3]提出的“点击化学”的概念.至此, CuAAC反应得到了广泛研究, 并作为点击化学的精华在生物化学、超分子化学和材料化学等研究领域得到了广泛应用[4].
CuAAC反应的机理研究因此得到了关注, 早期研究认为可能是单核铜催化的过程[5a], 然而随后的理论计算和动力学实验表明, 反应可能涉及多核铜络合物中间体. 2013年, Fokin等[5b]基于同位素交叉等实验, 提出了双铜协同催化的机理, 认为两个铜原子在活化炔烃和叠氮的同时, 即时组织两者以类似于分子内反应的方式进行反应, 从而高选择地得到1, 4-三氮唑产物(Scheme 1).
尽管CuAAC反应本身不产生手性, 但是通过对具有潜手性的炔烃或叠氮的去对称化以及外消旋叠氮或炔烃化合物的(动态)动力学拆分, 均可以发展不对称催化的CuAAC反应(Scheme 2), 进而实现含手性的三氮唑化合物的高效合成.由于该类结构单元具有和肽键类似的电子特性和结构特点, 同时不易水解, 是一类重要的药效基团, 在药物和生物活性分子中普遍存在[6]; 此外, 通过动力学拆分还能够为具有叠氮或者末端炔烃官能团的手性化合物提供新的不对称催化合成方法, 具有上述两种官能团的化合物均是重要的手性合成子, 能够发生多种转化, 然而其合成方法并不多[7].
尽管不对称CuAAC反应可用于合成含三氮唑和具有末端炔基或叠氮基的手性化合物, 但是其研究相较于CuAAC反应在众多领域的成功应用十分滞后.直到2005年, Fokin和Finn等[8]才利用去对称化和动力学拆分两种策略进行了尝试, 但是结果均不理想, 基于外消旋叠氮的动力学拆分仅能取得8的拆分因子(s-factor) (Scheme 3a).令人吃惊的是基于偕二叠氮的去对称化反应仅能取得中等对映选择性, 且基本以双三氮唑副产物为主(Scheme 3b).他们对此现象做了进一步研究[9], 发现在无配体条件下, 双叠氮参与的CuAAC反应, 尽管使用10 equiv.叠氮8与炔烃2反应, 仍然是双三氮唑产物10为主, 仅得到痕量单三氮唑9 (Scheme 4).他们认为三氮唑铜络合物I能进一步与炔烃作用(II或III), 使其易与分子中的另一叠氮发生反应.此外, 单三氮唑化合物9也可能进一步发生反应得到双三氮唑化合物.双炔体系的反应同样是得到双三氮唑为主的产物.这一发现, 充分说明发展去对称化的CuAAC反应十分困难.
上述早期探索说明发展不对称CuAAC反应具有很高的挑战性, 从而导致随后8年这一研究陷入沉寂, 未见任何报道.由于CuAAC反应可能经历双核铜催化机理, 且末端炔烃和叠氮均为线型结构, 因此其与手性铜络合物的识别模式, 较常见的醛、酮、亚胺等单齿或双齿配位型底物有所不同.此外, 无论是通过去对称化还是动力学拆分发展不对称CuAAC反应, 底物的潜手性中心或已有的手性中心, 均与反应位点距离较远, 而通过“远程控制”来实现理想的立体选择性对手性催化剂要求很高.其次, 反应过程中可能存在的一系列副反应同样限制了不对称CuAAC反应的发展.由Scheme 1所示机理可知, 反应经历炔基铜和三氮唑铜中间体, 而手性配体的引入引起的手性铜催化剂的空间环境和电性的改变, 有可能导致诸如Glaser偶联副反应的发生; 此外, 发展去对称化CuAAC反应还需要在取得优异选择性的时候, 抑制非手性的双三氮唑生成.这些是发展高选择性的不对称CuAAC反应必须克服的问题.
2013年, 周剑小组[10]实现了首例高对映选择性的不对称CuAAC反应.至此, 该研究重新引起了化学家的关注, 近年来逐渐实现了一系列含三氮唑、炔基或叠氮基团的碳或磷中心手性、轴手性以及平面手性化合物的合成.尽管Fossey等[11]在2016年对不对称CuAAC反应做了亮点介绍, 但是迄今已有四年, 不能反映最新进展.因此, 本文将对该领域的研究进行全面的综述, 按照去对称化的和动力学拆分的CuAAC反应进行分类, 而前者进一步按手性中心、手性轴以及手性面等的分类[12]进行介绍, 后者则进一步根据原料种类进行综述.
利用去对称化策略, 从具有潜手性元素的对称双炔或双叠氮化合物出发, 发展不对称CuAAC反应具有下述特点[13].由于反应位点通常远离潜手性中心, 虽然需要远程控制手性中心的构建, 但受位阻效应的影响不大, 对于构建季碳手性中心较为有利, 其理论收率可以达到100%;另外, 所得手性三氮唑产物还同时具有炔基或叠氮基团, 可方便进行多种后续衍生化.当然, 如前所述, 如何在实现理想立体选择性控制的同时, 抑制非手性双三氮唑的生成来提高单/双三氮唑的比例(M/D), 是实现这类反应的关键.可喜的是, 通过开发新型具有较深的手性口袋的配体, 结合反应条件的调控, 已发展了一系列成功的去对称化CuAAC反应, 并用于含三氮唑结构的(碳或磷)中心手性、轴手性和面手性化合物的高立体选择性合成.
2013年, 周剑等[10]提出利用潜手性双炔体系来发展不对称催化的CuAAC反应(Scheme 5).基于该课题组对季碳氧化吲哚构建的兴趣[14], 作者首先设计了3, 3-双炔丙基取代的氧化吲哚11来进行反应研究.幸运的是在手性PyBOX 13/CuCl络合物催化下, 使用2, 5-己二酮这种特殊溶剂, 能够有效抑制非手性双三氮唑的产生并有利于提高对映选择性; 进一步通过调节炔烃11和叠氮12的物质的量之比, 最终能够以高达82%的收率和98%的ee值得到含有炔基和三氮唑基团的全碳季碳氧化吲哚14.这既是首例高对映选择性的不对称CuAAC反应, 也是首例成功的基于双炔体系的分子间去对称化反应[15].产物可进行多样性转化得到季碳氧化吲哚18~21.此外, 观察到的明显的负的非线性效应为CuAAC反应的双核铜催化机理提供了新的佐证.
利用双炔体系的去对称化来发展不对称CuAAC反应随后得到进一步发展. Stephenson小组[16]尝试了α-双炔基取代的氰基乙酸甲酯类化合物22与苄基叠氮12a的反应(Scheme 6).作者筛选了BOX配体26、PHOX类单膦配体27和BINAP等双膦配体, 发现采用手性双膦配体23与CuI形成的配合物能以6%的转化率和18%的ee值到目标化合物24.

2015年, 徐利文小组[17]设计合成了一种新型的大位阻多齿手性膦配体Tao-PHOS 31.相较于BINOL衍生的34和PyBOX 13等手性配体, Tao-PHOS与CuF2形成的络合物作为催化剂, 在基于马来酰胺骨架的双炔29与叠氮化合物30的CuAAC反应中, 以60%~80%的产率和70%~99% ee值得到手性三氮唑32 (Scheme 7).这个反应的特点在于所有反应例子的单/双三氮唑比例均大于12/1, 只有痕量非手性双三氮唑生成.这说明具有手性深腔的新型配体, 有望抑制生成双三氮唑的过度反应.但这类配体的手性控制的能力还需进一步改善, 因为在所报道的26个例子中, 仅两例能取得大于90%的ee值, 进一步研究表明Tao-PHOS配体中的叔膦、羟基和三甲基硅基(TMS)对反应的活性和立体选择性均有着至关重要的影响[18], 当配体上的叔膦换成膦氧基团时, 反应的ee值为零; 而当羟基上的氢原子被氘替换时, 只能得到痕量产物; 使用不含TMS的配体33时, 反应仅取得46% ee.因此, 作者认为配体通过叔膦与铜配位, 羟基与双炔29的羰基存在氢键作用, 而大位阻TMS的存在有利于提高反应的手性控制.此外, 非线性效应和质谱研究结果也支持反应经历了双核铜催化的机理.
2018年, 他们[19]进一步使用Tao-PHOS配体31与纳米Cu2O形成的手性催化剂, 对吡唑啉酮衍生的双炔37与叠氮30的去对称化CuAAC反应进行了研究(Scheme 8), 最终以高达70%的产率和90%的ee值得到了目标化合物38, 单/双三氮唑比例均大于20/1.双核铜催化的机理再次得到非线性效应和质谱研究的支持.值得一提的是, 该工作是首例非均相的不对称CuAAC反应, 手性催化剂能够重新回收利用.

具有磷手性中心的光学活性膦氧化合物, 在医药、农药和手性催化剂等领域应用很广[20], 其不对称催化合成近年来受到了广泛关注[21], 但可用于模块化合成磷手性砌块的方法依然很少.最近, 周剑小组[22]实现了潜手性双炔基膦氧化合物的不对称CuAAC反应, 高对映选择性地合成了乙炔基取代的磷手性膦氧化合物(Scheme 9).作者设计合成的吡啶C-4位带有大位阻柔性基团的PyBOX配体40与CuBr形成的络合物, 能顺利催化双乙炔基膦氧39与叠氮12的反应, 以高达85%的产率和96%的ee值得到目标产物41.
相较于母体PyBOX配体13, 吡啶环C-4位大位阻取代基的引入可以有效提高对映选择性, 并显著抑制非手性双三氮唑的生成, 使得单/双三氮唑比例最高能达23/1.根据可能的双核铜催化机理, 作者认为吡啶环的C-4位大位阻基团有助于催化剂形成更好的手性口袋, 不但提高反应的立体选择性, 而且使得潜手性双炔原料39的炔基比起位阻更大的手性产物41的炔基, 更容易被催化剂识别与活化, 从而抑制双三氮唑的生成.
进一步对反应的单/双三氮唑的比值和产物ee值随时间的变化进行监测, 发现反应涉及去对称化和动力学拆分的协同过程.在去对称化过程中, 形成R构型产物的反应速度快于生成S构型的速度(k1>k2); 与此同时, R构型的手性单三氮唑产物会通过动力学拆分, 进一步提高自身的ee值, 但是反应的单/双三氮唑比例也逐渐降低.而新发展的手性配体在上述两个过程中均能取得更为优秀的立体选择性.此外, 所得手性产物中同时含乙炔基和三氮唑基团, 能够作为手性膦的合成子, 进一步转化为手性叔膦44a和手性膦氧46a (Scheme 9).
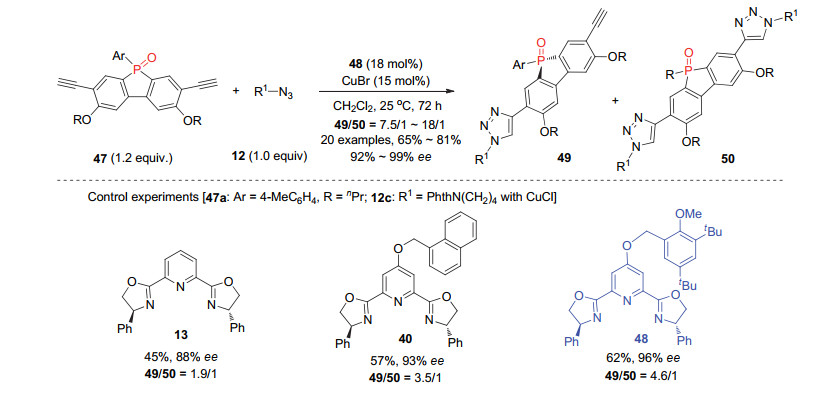
利用这一方法进一步实现了手性膦杂茂化合物的合成[23].膦杂茂化合物在有机光电材料研究中具有重要的应用价值, 然而尚无手性合成方法. C-4位含有大位阻芳基取代的PyBOX配体48/CuBr配合物催化剂能顺利催化二苯并膦杂茂二炔47与叠氮12的不对称CuAAC反应, 以65%~81%的产率和92%~99% ee值得到结构新颖的磷手性膦杂茂化合物49 (Scheme 10), 双三氮唑副产物可以明显得到抑制, 49/50的比例最高达到18/1.对所得手性膦杂茂衍生物的光电性质进行研究发现, 其紫外吸收和荧光发射光谱可以很容易得到调节.
上述反应中, 二苯并膦杂茂二炔47中的乙炔基反应位点与潜手性磷原子相隔四根共价键, 这在远程控制实现的高对映选择性去对称化反应中很少见.而PyBOX配体C-4位大位阻基团在远程控制立体选择性和抑制非手性双三氮唑形成方面再次展示了明显的作用.基于这一点, 周剑课题组[24]进一步利用这类配体成功实现了高对映选择性的潜手性双叠氮化合物的去对称化CuAAC反应.手性PyBOX配体54与CuCl形成的配合物催化剂, 顺利地催化双叠氮取代的叔醇51与末端炔烃52的反应, 取得中等的收率以及中等到优秀的对映选择性.进一步研究发现, 对于碘代的非末端炔烃53, 反应同样可以顺利进行, 以高达68%的收率和97%的ee值得到含碘取代的三氮唑基团的β-叠氮手性叔醇56 (Scheme 11).这也是首例不对称铜催化的叠氮和碘代炔烃的环加成反应.通过对叠氮或羟基的转化, 所得手性叠氮醇55a能够顺利衍生为手性叔醇57~59或噁唑啉硫酮61.该反应同样涉及去对称化和动力学拆分的协同作用, 产物的ee值随反应进行逐渐升高, 而单/双三氮唑的比例随之下降.尽管两类反应中均能取得良好到优秀的对映选择性, 但是单双比最高仅为3/1.这进一步说明发展去对称化CuAAC反应还需在配体设计方面继续努力.
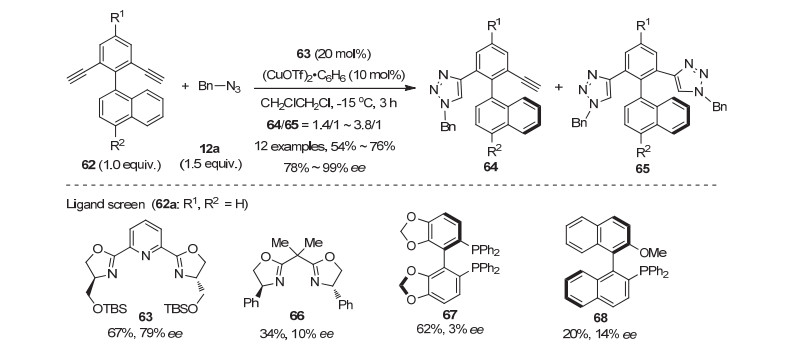
轴手性存在于许多生物活性分子以及手性配体和催化剂中, 发展构建轴手性的新方法是不对称催化的研究热点[25]. 2014年, Uozumi等[26]首次通过去对称化的CuAAC反应合成了轴手性的联芳基化合物(Scheme 12).考察各类配体后发现PyBOX配体63和(CuOTf)2•C6H6生成的络合物催化剂, 在潜手性联芳基双炔62与苄基叠氮12a的反应中, 以最高达76%的产率和99%的ee值得到了含三氮唑结构的轴手性联二芳基化合物64.反应的单/双三氮唑的比例并不理想, 最高只有3.8/1.作者还发现当苄基叠氮的用量由1 equiv.增加到2 equiv.时, 产物的ee值从80%上升到97%, 而单双比则由6/1下降到1/2.8.据此, 作者首次发现在不对称去对称化的CuAAC反应中, 对映选择性的控制可能是去对称化和动力学拆分协同作用的结果[27].
不对称CuAAC反应也可以用于构建平面手性[28]. 2019年, Stephenson等[29]报道了二茂铁衍生的双炔化合物69与苄基叠氮12a的去对称CuAAC反应(Scheme 13).他们发现在如Scheme 13所示的反应条件下, PyBOX 70、BOX 66和面手性双膦配体73均能够取得优异的对映选择性, 而使用双膦配体73所得产物的绝对构型截然相反.此外, 二茂铁双炔底物中的羟基对反应的活性和立体选择性有至关重要的影响, 将羟基改为酯基时, 目标产物的产率和ee值均大幅度下降.作者也发现该反应是去对称化和动力学拆分协同进行的过程.尽管该反应中只有一个底物能取得优秀的立体选择性, 且伴随几乎等物质的量双三氮唑副产物的生成, 但是它显示不对称CuAAC反应可以为炔基或叠氮基取代的具有面手性的化合物的构建提供了一种新方法.
基于外消旋叠氮或炔烃的(动态)动力学拆分是实现不对称催化CuAAC反应的另一重要途径, 这两种方法各有其特点[30].理想的动力学拆分能通过一步反应同时得到两种手性化合物:含手性的三氮唑以及含手性的炔烃(或叠氮), 虽然回收率和产率最高各只有50%, 但是对于多样性合成手性化合物有帮助; 而动态动力学拆分反应则能够以100%的理论产率得到含手性的三氮唑化合物.
除了Fokin等[8]的早期探索外, Fossey等[31]在2015年利用周剑小组所发展的反应条件, 尝试了对外消旋3-炔丙基氧化吲哚74的动力学拆分(Scheme 14a), 但是结果并不理想.当转化率在50%时, 原料75能以81% ee值回收; 而转化率在仅为8%时, 产物76才能取得80%的ee值.随后, 他们使用BINOL衍生的硼酸酯80作为手性辅基, 研究了手性底物诱导的动力学拆分, 但是选择性因子最高也仅为4.1 (Scheme 14b)[32].他们还尝试了外消旋炔烃74和外消旋叠氮1的不对称CuAAC反应.当转化率为36%时, 能以84:16的dr值及90%的ee值得到目标化合物85, 显示能通过不对称CuAAC反应构建两个手性中心(Scheme 14c)[33].

2019年Topczewski等[34]使用1.25 mol%的PyBOX 88/Cu(Ⅰ)络合物为催化剂, 在外消旋二级叠氮86与末端炔烃87的不对称CuAAC反应中, 能以高达97%的产率和94% ee值得到含手性的三氮唑89 (Scheme 15).具有环状结构的外消旋叠氮化合物的反应结果优于非环状的, 然而回收原料的ee值仍不理想.

例如, 作者尝试了克级规模的反应, 能以92%的产率和78%的ee值制备1.2 g含手性的三氮唑89a, 同时以52% ee值回收手性叠氮86a.需要指出的是该方法的立体选择性仍有很大的提升空间, 在所报道的30个例子中, 仅有两例的对映选择性超过90%.
在基于膦氧双炔的去对称化CuAAC反应的基础上, 周剑小组进一步发展了对外消旋乙炔基膦氧的动力学拆分CuAAC反应, 通过简单的改变吡啶环C-4位含大位阻的PyBOX型配体的取代基, 使用配体54或92与CuBr的络合物催化剂, 可以高效地催化外消旋单乙炔基膦氧90与叠氮12的CuAAC反应[22], 以良好到优秀的对映选择性分别得到具有磷手性的单乙炔基膦氧(S)-91和三氮唑膦氧(R)-93 (Scheme 16).所得手性乙炔基膦氧91b经Sonogashira偶联、HSiCl3还原, 即可得到手性保持的磷手性叔膦94b, 其作为亲核催化剂可顺利地催化烯酮95和联烯酸酯96的不对称环加成反应, 并取得中等的产率和对映选择性.

高效的动态动力学拆分[35]的不对称CuAAC反应能够把外消旋底物全部转换为单一构型的含手性的三氮唑化合物, 产率最高可达100%.发展合适的方法使外消旋底物发生消旋化, 且能够与动力学拆分过程相结合, 是实现动态动力学拆分的关键. 2019年, Topczewski等[36]在这一领域实现了突破, 他们结合烯丙基叠氮的3, 3-Winstein重排首次实现了动态动力学拆分的CuAAC反应(Scheme 17).利用PyBOX 100/Cu(Ⅰ)络合物催化剂, 通过调节温度来平衡叠氮的消旋化和动力学拆分的反应速率, 最终能够以高达99%的产率和99%的对映选择性得到烯丙基取代的手性三氮唑化合物101.研究发现, PyBOX配体相对其他类型的氮配体或膦配体, 能实现更优异的立体选择性和催化活性, 催化剂用量仅需1.25 mol%.反应的底物普适性较好, 具有五元、六元、七元结构的环状叠氮或开链状叠氮, 如101a~101e, 以及含有复杂药物分子结构的端炔, 如101f, 均具有良好的普适性, 充分展现了该方法的实用价值.
综上所述, 过去几年, 不对称CuAAC反应的研究取得了长足进步.基于去对称化或(动态)动力学拆分两种策略, 已成功发展出了一系列不对称CuAAC反应, 用于含三氮唑、炔烃或叠氮的碳(磷)中心手性、轴手性或面手性化合物的高对映选择性合成, 有效地拓宽了CuAAC反应的应用范围.
尽管如此, 该领域的研究工作仍然有巨大的发展空间.首先, 现有方法的底物普适性都不高, 距离将不对称催化的CuAAC反应发展为多样性和高选择性制备含叠氮、炔基或三氮唑取代基的手性化合物的目标还很遥远.其次, 在发展去对称化的不对称CuAAC反应时, 如何在取得高对映选择性的同时抑制过度反应生成非手性双三氮唑依旧是一个难题.再次, 如何利用不对称CuAAC反应来实现小单元的链接, 合成手性高分子材料或生物功能材料, 尚待开发.解决这些问题, 从现有结果来看, 关键在于发展具有可调手性深腔的新型手性催化剂.因此, 通过设计开发新型催化剂来拓展反应的类型, 探索其在有机合成、材料、药物研发等领域的潜在应用价值, 无疑是这一领域的发展方向.我们相信不对称催化的CuAAC反应必将迎来更大的发展, 并开拓出更加广阔的应用前景.
Huisgen, R. Angew. Chem., Int. Ed. 1963, 2, 565. doi: 10.1002/anie.196305651
(a) Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057.
(b) Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596.
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5
(a) Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952.
(b) Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302.
(a) Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V. V.; Noodleman, L.; Sharpless, K. B.; Fokin, V. V. J. Am. Chem. Soc. 2005, 127, 210.
(b) Worrell, B. T.; Malik, J. A.; Fokin, V. V. Science 2013, 340, 457.
Kolb, H. C.; Sharpless, K. B. Drug Discovery Today 2003, 8, 1128. doi: 10.1016/S1359-6446(03)02933-7
(a) Trost, B. M.; Weiss, A. H. Adv. Synth. Catal. 2009, 351, 963.
(b) Ding, P. G.; Hu, X. S.; Zhou, F.; Zhou, J. Org. Chem. Front. 2018, 5, 1542.
Meng, J.-C.; Fokin, V. V.; Finn, M. G. Tetrahedron Lett. 2005, 46, 4543. doi: 10.1016/j.tetlet.2005.05.019
Rodionov, V. O.; Fokin, V. V.; Finn, M. G. Angew. Chem., Int. Ed. 2005, 44, 2210. doi: 10.1002/anie.200461496
Zhou, F.; Tan, C.; Tang, J.; Zhang, Y. Y.; Gao, W. M.; Wu, H. H.; Yu, Y. H.; Zhou, J. J. Am. Chem. Soc. 2013, 135, 10994. doi: 10.1021/ja4066656
Brittain, W. D. G.; Buckley, B. R.; Fossey, J. S. ACS Catal. 2016, 6, 3629. doi: 10.1021/acscatal.6b00996
Cahn, R. S.; Ingold, C.; Prelog, V. Angew. Chem., Int. Ed. 1966, 5, 385. doi: 10.1002/anie.196603851
Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Chem. Rev. 2016, 116, 7330. doi: 10.1021/acs.chemrev.6b00094
Cao, Z.-Y.; Zhou, F.; Zhou, J. Acc. Chem. Res. 2018, 51, 1443. doi: 10.1021/acs.accounts.8b00097
(a) Wilking, M.; Mück-Lichtenfeld, C.; Daniliuc, C. G.; Hennecke, U. J. Am. Chem. Soc. 2013, 135, 8133.
(b) Mourad, A. K.; Leutzow, J.; Czekelius, C. Angew. Chem., Int. Ed. 2012, 51, 11149.
(c) Sato, Y.; Nishimata, T.; Mori, M. J. Org. Chem. 1994, 59, 6133.
(d) Tanaka, K.; Fu, G. C. J. Am. Chem. Soc. 2002, 124, 10296.
Stephenson, G. R.; Buttress, J. P.; Deschamps, D.; Lancelot, M.; Martin, J. P.; Sheldon, A. I. G.; Alayrac, C.; Gaumont, A.-C.; Page, P. C. B. Synlett 2013, 24, 2723. doi: 10.1055/s-0033-1340152
Song, T.; Li, L.; Zhou, W.; Zheng, Z.-J.; Deng, Y.; Xu, Z.; Xu, L.-W. Chem.-Eur. J. 2015, 21, 554. doi: 10.1002/chem.201405420
Chen, M.-Y.; Song, T.; Zheng, Z.-J.; Xu, Z.; Cui, Y.-M.; Xu, L.-W. RSC Adv. 2016, 6, 58698. doi: 10.1039/C6RA13687G
Chen, M.-Y.; Xu, Z.; Chen, L.; Song, T.; Zhen, Z.-J.; Cao, J.; Cui, Y.-M.; Xu, L.-W. ChemCatChem 2018, 10, 280. doi: 10.1002/cctc.201701336
(a) Yao, Q.; Wang, A.; Pu, J.; Tang, Y. Chin. J. Org. Chem. 2014, 34, 292(in Chinese).
(姚秋丽, 王安俊, 蒲家志, 唐瑜敏, 有机化学, 2014, 34, 292.)
(b) Grabulosa, A.; Granell, J.; Muller, G. Coord. Chem. Rev. 2007, 251, 25.
(c) Wozniak, L. A.; Okruszek, A. Chem. Soc. Rev. 2003, 32, 158.
(d) Pietrusiewicz, K. M.; Zablocka, M. Chem. Rev. 1994, 94, 1375.
(a) Harvey, J. S.; Gouverneur, V. Chem. Commun. 2010, 46, 7477.
(b) Glueck, D. S. Chem.-Eur. J. 2008, 14, 7108.
(c) Glueck, D. S. Synlett 2007, 2627.
Zhu, R.-Y.; Chen, L.; Hu, X.-S.; Zhou, F.; Zhou, J. Chem. Sci. 2020, 11, 97. doi: 10.1039/C9SC04938J
(a) Xi, W.; Scott, T. F.; Kloxin, C. J.; Bowman, C. N. Adv. Funct. Mater. 2014, 24, 2572.
(b) Chu, C.; Liu, R. Chem. Soc. Rev. 2011, 40, 2177.
(c) Golas, P. L.; Matyjaszewski, K. Chem. Soc. Rev. 2010, 39, 1338.
Wang, C.; Zhu, R.-Y.; Liao, K.; Zhou, F.; Zhou, J. Org. Lett. 2020, 22, 1270. doi: 10.1021/acs.orglett.9b04522
(a) Wang, Y.-B.; Tan, B. Acc. Chem. Res. 2018, 51, 534.
(b) Kozlowski, M. C.; Morgan, B. J.; Linton, E. C. Chem. Soc. Rev. 2009, 38, 3193.
(c) Bringmann, G.; Mortimer, A. J. P.; Keller, P. A.; Gresser, M. J.; Garner, J.; Breuning, M. Angew. Chem., Int. Ed. 2005, 44, 5384.
(d) Bringmann, G.; Gulder, T.; Gulder, T. A. M.; Breuning, M. Chem. Rev. 2011, 111, 563.
Osako, T.; Uozumi, Y. Org. Lett. 2014, 16, 5866. doi: 10.1021/ol502778j
Osako, T.; Uozumi, Y. Synlett 2015, 26, 1475. doi: 10.1055/s-0034-1380534
(a) Yasue, R.; Kazuhiro, Y. Chem.-Eur. J. 2018, 24, 18575.
(b) Ferbera, B.; Kagan, H. B. Adv. Synth. Catal. 2007, 349, 493.
(c) Dai, L.-X.; Tu, T.; You, S.-L.; Deng, W.-P.; Hou, X.-L. Acc. Chem. Res. 2003, 36, 659.
Wright, A. J.; Hughes, D. L.; Page, P. C. B.; Stephenson, G. R. Eur. J. Org. Chem. 2019, 7218.
(a) Vedejs, E.; Jure, M. Angew. Chem., Int. Ed. 2005, 44, 3974.
(b) Keith, J. M.; Larrow, J. F.; Jacobsen, E. N. Adv. Synth. Catal. 2001, 343, 5.
Brittain, W. D. G.; Buckley, B. R.; Fossey, J. S. Chem. Commun. 2015, 51, 17217. doi: 10.1039/C5CC04886A
Brittain, W. D. G.; Chapin, B. M.; Zhai, W.; Lynch, V. M.; Buckley, B. R.; Anslyn, E. V.; Fossey, J. S. Org. Biomol. Chem. 2016, 14, 10778. doi: 10.1039/C6OB01623E
Brittain, W. D. G.; Dalling, A. G.; Sun, Z.; Duf, C. S. L.; Male, L.; Buckley, B. R.; Fossey, J. S. Sci. Rep. 2019, 9, 15086. doi: 10.1038/s41598-019-50940-4
Alexander, J. R.; Ott, A. A.; Liu, E.-C.; Topczewski, J. J. Org. Lett. 2019, 21, 4355. doi: 10.1021/acs.orglett.9b01556
(a) Bhat, V.; Welin, E. R.; Guo, X.; Stoltz, B. M. Chem. Rev. 2017, 117, 4528.
(b) Pellissier, H. Tetrahedron 2008, 64, 3769.
(c) Huerta, F. F.; Minidis, A. B. E.; Bäckvall, J.-E. Chem. Soc. Rev. 2001, 30, 321.
Liu, E.-C.; Topczewski, J. J. J. Am. Chem. Soc. 2019, 141, 5135. doi: 10.1021/jacs.9b01091

图式 6 氰基乙酸甲酯类双炔的去对称化CuAAC反应
Scheme 6 Desymmetric CuAAC of methyl cyanoacetate-based dialkynes

图式 14 外消旋氧化吲哚类炔烃的动力学拆分CuAAC反应
Scheme 14 CuAAC based on kinetic resolutions of oxindole-based racemic alkynes

图式 15 外消旋二级叠氮的动力学拆分CuAAC反应
Scheme 15 CuAAC based on kinetic resolutions of racemic secondary azides

图式 16 外消旋单乙炔基膦氧的动力学拆分CuAAC反应
Scheme 16 CuAAC based on kinetic resolutions of racemic ethynylphosphine oxides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:










 下载:
下载:












 下载:
下载: