表 1
化合物的实验及预测活性值
Table 1.
Experimental and predicted activity values of compounds

整合酶作为HIV-1复制位点的必需酶,可以介导病毒DNA与宿主细胞内DNA的结合,使其基因得以表达[1],但在人类细胞中并未发现整合酶抑制剂的功能类似物,因此,该酶被认为是设计抗HIV药物的理想治疗靶标。HIV整合酶介导的整合反应可以分为两个催化步棸,3′加工(3′Processing)和链转移(Strand transfer)[2]。目前临床上用于抗HIV病毒的二酮类衍生化合物是一类成熟的抗逆转录病毒药物,因其抑制酶促反应的DNA链转移步骤,被称为整合酶链转移抑制剂(INSTIs)[3, 4]。然而,在使用INSTIs治疗的过程中,整合酶会发生突变,使HIV病毒出现耐药性,耐药突变是抗病毒治疗的常见问题,其影响病毒对HIV-1整合酶抑制剂的易感性。因此,进一步开发出抗突变性、抗耐药、安全有效的新型HIV INSTIs愈发迫切[5]。
Zhao等[6]设计合成的一系列1, 8-二氮杂萘-3-甲酰胺衍生物能够抑制链转移过程,且选择性比二酮类衍生物好。他们对该系列化合物4位和7位进行修饰,同时探索改变6位的取代基,实验结果显示,在6位引入不同的取代基可以增强化合物的抗病毒效应和抗耐药性,但这一类化合物的结构和活性之间的关系尚未报道。本研究拟采用3D-QSAR、分子对接和分子动力学模拟来研究这一系列化合物结构与活性的关系,为开发新的有效HIV INSTIs提供参考。
本文研究的32个1, 8-二氮杂萘-3-甲酰胺类衍生物的分子结构及活性数据均来源文献[6]。基于化合物的结构多样性和活性范围的综合考虑,将这32个1, 8-二氮杂萘-3-甲酰胺类化合物中的27个作为训练集,另外5个作为测试集。这些化合物的结构文件是通过ChemBioDraw11.0软件构建[7],并且运用Sybyl-X 2.0中Minimize分子力学程序进行能量最小化,得到低能稳定构象;最后采用Tripos力场,在加载Gasteiger-Hückel电荷的条件下,以Powell能量梯度法对这些小分子进行1000次优化,能量变化收敛条件设定为0.005kJ·(mol·Å)-1,其他参数均为默认值[8]。所有的化合物的实验和预测的pIC50值见表 1。
 下载:
导出CSV
下载:
导出CSV
| No. | R4 | R6 | R7 | pIC50 (实验) |
pIC50(预测) CoMFA |
pIC50(预测) CoMSIA |
 |
||||||
| 1* | OH | H | H | 7.259 | 7.761 | 7.764 |
| 2 | OH | H | OCH3 | 7.481 | 7.607 | 7.568 |
| 3* | OH | H | piperidinyl | 4.155 | 5.122 | 5.265 |
| 4* | OH | H | morpholino | 5.721 | 5.122 | 5.113 |
| 5 | NH | H | H | 7.721 | 7.797 | 7.686 |
| 6 | NH2 | H | OCH3 | 7.959 | 7.795 | 7.607 |
| 7 | NH2 | H | piperidinyl | 4.268 | 4.272 | 4.278 |
| 8 | NH2 | H | morpholino | 4.268 | 4.295 | 4.297 |
| 9 | NHCH2CO2CH3 | H | H | 7.677 | 7.660 | 7.631 |
| 10 | NHCH2CO2CH3 | H | OCH3 | 7.376 | 7.415 | 7.505 |
| 11 | NHCH2CO2CH3 | H | piperidinyl | 5.202 | 5.19 | 5.214 |
| 12 | NHCH2CO2CH3 | H | morpholino | 5.208 | 5.211 | 5.232 |
| 13 | OH | (CH2)5OH | H | 7.921 | 7.915 | 7.900 |
| 14 | NH2 | (CH2)5OH | H | 8.568 | 8.491 | 8.812 |
| 15 | NH2 | (CH2)3OAc | H | 8.000 | 8.032 | 8.054 |
| 16 | NH2 | (CH2)6OBz | H | 5.481 | 5.438 | 5.419 |
| 17 | NH2 | (CH2)3Hex | H | 5.481 | 5.431 | 5.911 |
| 18 | NH2 | (CH2)4Ph | H | 5.699 | 5.712 | 5.611 |
| 19 | NH2 | (CH)2Ph | H | 7.854 | 7.900 | 7.331 |
| 20 | NH2 | (CH2)3N(CH3)2 | H | 7.854 | 7.908 | 8.066 |
| 21* | NH2 | (CH2)3O(CH2)2)OH | H | 8.698 | 7.424 | 7.302 |
| 22 | NH2 | (CH2)2COOH | H | 8.398 | 8.429 | 8.516 |
| 23 | NH2 | (CH2) 2CON(CH3)2 | H | 8.097 | 8.144 | 8.516 |
| 24 | NH2 | (CH)2CON(CH3)2 | H | 8.301 | 8.272 | 8.571 |
| 25* | NH2 | (CH2)2CONHPr | H | 8.699 | 9.103 | 9.089 |
| 26 | NH2 | (CH)2CONHPr | H | 8.222 | 8.281 | 8.063 |
| 27 | NH2 | (CH2)2CONH(CH)2OH | H | 8.699 | 8.707 | 8.701 |
| 28 | NH2 | (CH)2CONH(CH)OH | H | 8.155 | 8.235 | 8.384 |
| 29 | NH2 | (CH2)2CO2CH3 | H | 8.508 | 8.407 | 7.986 |
| 30 | NHCH2CO2CH3 | (CH2)5OH | H | 8.398 | 8.430 | 8.606 |
| 31 | NH(CH2)2OH | (CH2)5OH | H | 8.602 | 8.616 | 8.608 |
| 32 | NH(CH2)2OH | (CH2)2CO2CH3 | H | 8.509 | 8.497 | 8.621 |
| *代表测试集化合物 | ||||||
在3D-QSAR中,分子叠合对于建立稳定可靠的模型至关重要。选取数据集中活性较好的化合物29作为模板分子,再将化合物基于模板分子29进行分子叠加,叠加模型以及模板分子如图 1所示。

为了得到更加可靠的模型,本文的3D-QSAR模型是采用Syby1-X 2.0软件的QSAR模块计算。将叠合好的化合物置于3D立方晶格中,网格间距为2.0nm[9]。使用具有范德华半径为1.52Å和+1.0电荷的sp3杂化碳原子作为探针来得到CoMFA描述符,再用Lennard-Jones势和库仑势分别计算每个化合物在CoMFA模型中的立体场(S)和静电场(E),最后用30kcal·mol-1的截止值进行能量计算。在CoMSIA中,探针原子除了计算立体场(S)和静电场(E)外,还会计算疏水性场(H)、氢键受体场(A)和氢键供体场(D)描述符。CoMSIA描述符来自与CoMFA中使用的相同的格子盒,最小柱过滤和衰减因子分别设定为2.0kcal·mol-1和0.3[10]。将偏最小二乘法(PLS)用于3D-QSAR模型方程的构建[11]。要以CoMFA和CoMSIA描述符作为独立变量,pIC50值作为PLS分析中的因变量进行模型的建立。通过留一法(LOO)交叉验证以确定交叉验证的相关系数(q2)和最佳组分数(N)[12]。使用先前获得的最佳组成分数N值进行非交叉验证以估计常规确定系数(r2)。评判模型的交叉验证系数q2用下面方程计算得到[13]。
|
$ q^{2}=1-\frac{\sum_{i=1}^{n}\left(Y_{\text { pred }}-Y_{\text { actual }}\right)^{2}}{\sum_{i=1}^{n}\left(Y_{\text { actual }}-Y_{\text { mean }}\right)^{2}} $ |
其中,Ypred是pIC50预测值,Yactual是pIC50实验值,Ymean是pIC50平均值。为检验所建立的3D-QSAR模型的预测能力,对测试集分子进行外部预测,可用预测相关系数rpred2来表征模型的预测能力。rpred2通过下面公式计算得到[14],SD表示其测试集与训练集的pIC50实验值的平均值之间的平方偏差之和。PRESS表示测试集的实验值和预测值pIC50的平方偏差之和。
|
$ r_{\mathrm{pred}}^{2}=\frac{\mathrm{SD}-\mathrm{PRESS}}{\mathrm{SD}} $ |
本文使用Molegro Virtual Docker 5.5软件进行对接研究。INSTIs的蛋白晶体来自于Protein Data Bank数据库[15],PDB蛋白代码为5MMB。在将分子对接之前,需要将蛋白晶体中的原配体和水分子除去[16],检查氨基酸序列一级结构的完整性和准确性。先用自身对接来验证对接的可靠性,然后再将这一系列化合物与INSTIs蛋白受体逐一进行对接。将对接参数设置为最大迭代次数500,导出构象30个,其他参数均为默认值。
为进一步解释配体和受体的相互作用,本文利用Gromacs5.0.4软件包对化合物和INSTIs的复合物体系进行20ns的分子动力学模拟[17~20]。INSTIs蛋白和小分子配体力场均采用GROMOS96力场,拓扑文件和小分子的部分电荷是由PRODRG2程序产生。为了给复合物创造溶剂环境,将蛋白配体复合物模拟放置于边长为1cm的十二边形盒子中。为了使整个模拟体系呈电中性,使用verlet切断法选择性加入Na2+或者Cl-来平衡体系的电荷[21]。再对溶剂环境进行优化,整个系统先用最陡下降法优化500步,再用500步共轭梯度法消除高能碰撞。将模拟温度从0K增加到300K,在恒温恒溶条件下,给体系增加1000kJ·mol-1·nm-2的限制力,进行100ps的分子动力学模拟用于平衡溶剂分子。之后在101.325kPa、300K下恒温恒压平衡体系100ps,最后进行20ns的分子动力学模拟。
由27个训练集分子的活性数据构建CoMFA和CoMSIA模型,再用5个测试集分子的活性数据来检验模型的可靠性,得到的CoMFA和CoMSIA模型的相关统计参数见表 2。
下载:
导出CSV
| CoMFA | CoMSIA | |
| PLS | ||
| N | 8 | 5 |
| q2 | 0.809 | 0.816 |
| r2 | 0.998 | 0.981 |
| SEE | 0.069 | 0.216 |
| F | 1378.014 | 220.522 |
| rpred2 | 0.785 | 0.745 |
| 贡献值 | ||
| 立体场 | 0.693 | 0.243 |
| 静电场 | 0.361 | |
| 疏水场 | 0.229 | |
| 氢键供体场 | 0.240 | |
| 氢键受体场 | 0.288 |
一般认为,q2>0.5、r2>0.5,表明该模型是可靠的且具有较好的预测能力[22]。从表中可知,CoMFA模型的最佳组成分是8,交叉验证系数q2为0.809(>0.5)。而由最佳组成分8建立的CoMFA模型的非交叉验证相关系数r2为0.998,标准偏差(SEE)是0.069,立体场和静电场对模型的贡献值分别为0.693和0.361。说明基团的空间效应和电性分布对活性均有一定影响,且立体场效应比静电场效应对化合物生物活性的影响大。
对于CoMSIA模型,需要考虑立体场(S)、静电场(E)、疏水场(H)、氢键供体场(D)、氢键受体场(A),将这5个力场进行组合分别考察每个组合所构建模型的可靠性和预测能力。选择CoMSIA(SHDA)模型进行分析,其q2=0.816、r2=0.981、SEE=0.216、N=5、F=220.522。立体场、疏水场、氢键供体场和氢键受体场的贡献值分别为0.243、0.229、0.288和0.240。由于CoMSIA模型考虑了疏水场和氢键场,因此三维构效模型更优。散点图用于描述训练集和测试集的实验与预测pIC50之间的相关性。对训练集和测试集的实验与预测pIC50分别进行线性回归,由图 2可知,CoMFA和CoMSIA的实验值和预测值均分布在线性回归趋势线的附近。CoMFA和CoMSIA的外部验证相关系数分别为0.785和0.745,表明该模型具有良好的预测能力。

等势图解释了化合物结构特征与生物活性之间的关系。为更加方便对3D-QSAR模型等势图的分析讨论,选取化合物29作为模板。CoMFA模型的立体场和静电场等势图如图 3所示。在立体场中(图 3(a)),绿色区域表示引入大体积基团有利于提高化合物的生物活性,黄色区域则表示引入大体积基团不利于化合物的活性。从图中得知,绿色区域主要集中在6位,这表明在此区域引入较大基团有利于提高化合物活性。化合物5在6位没有取代基,而化合物14在6位引入了取代基,从而使得化合物14的活性明显高于化合物5。然而在6位取代基的末端出现了一块黄色区域,则表明在此位置引入大基团会影响化合物活性,化合物29(pIC50=8.508)在末端引入的是小基团甲基,其活性明显高于引入更大基团的化合物16(pIC50=5.481)、17(pIC50=5.481)、18(pIC50=5.699)。
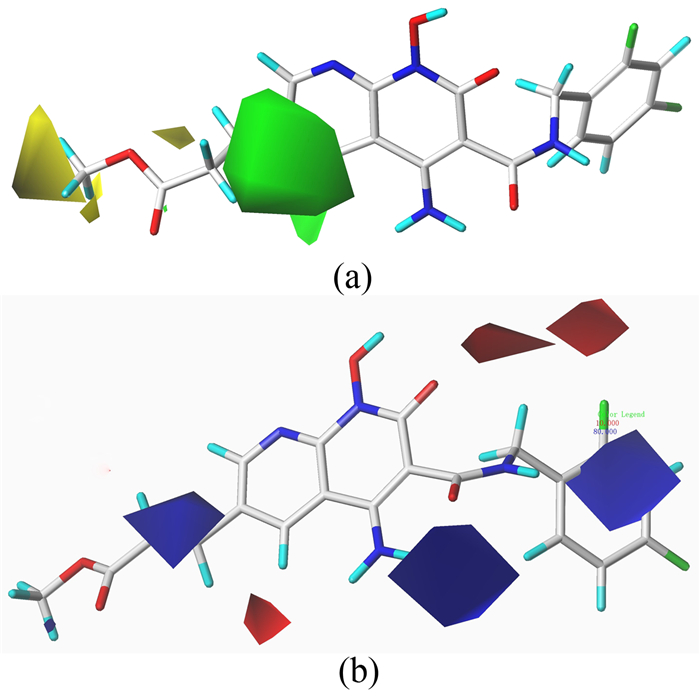
在CoMFA的静电等势图中,红色区域表示引入负电性基团有利于提高化合物的活性,蓝色则表示引入正电性基团有利于化合物的生物活性提高。图 3(b)中,有一大块蓝色占据了4位区域,表明在4位引入正电性基团能提高化合物的活性。在4位引入羟基的化合物10(pIC50=7.376)的活性小于引入氨基的化合物5(pIC50=7.721)的活性可以说明这一情况。而红色区域主要集中在6、7位,化合物3(pIC50=4.155)在7位引入了一个吡啶环,而化合物4(pIC50=5.721)在这个位置引入了一个吗啉基团,因为化合物4引入的负电性基团所以导致这两个化合物的活性相差较大。
CoMSIA的立体场、静电场的等势图与CoMFA模型类似,在此不再赘述。而在CoMSIA的疏水场等势图 4(a)中,黄色区域表示在此位置引入疏水基团有利于化合物活性的增强,灰色区域则刚好相反。在等势图 4(a)中可以明显看到有一大块黄色分布在4位,说明在这个位置引入疏水基团有利于增强化合物的活性。化合物1和化合物5分别在4位引入了亲水基团羟基和氨基,而化合物9在此位置引入的则是疏水基团酯基,其活性明显高于化合物1和5。图 4(b)为氢键供体等势图,蓝色区域表示氢键供体是有利于活性的增强,而紫色区域则相反。有两块蓝色区域分别覆盖在1位的羟基位置和6位酯基的氧原子位置,说明这两基团可能都是作为氢键供体增强化合物的活性。从对接结果可知,两个位置的氧原子分别与氨基酸残基形成了氢键,进一步验证了结果。图 4(c)是氢键受体场等势图,在氢键受体场中,紫红色区域表示增加氢键受体有利于活性,红色区域则不利于活性。可以看到紫红色区域分布在6位酯基的位置,说明氧原子与结合位点中的氨基酸残基形成的氢键有利于活性。红色区域主要分布在4位的氨基位置,意味着这个区域的氢键受体不利于化合物活性的提高。

为了方便解释,选择模板分子29与INSTIs蛋白晶体的对接结果来分析这一类化合物与INSTIs蛋白的结合特点。化合物29呈“V”字型镶嵌在由氨基酸Asp185、Gln186、Tyr212、Pro214、Glu221、Gln215、Tyr129、Arg329、Asp128和Gly187形成的结合口袋中。从图 5中可知,6位取代基伸入到空间体积较大的活性口袋,说明在此区域引入大基团有利于化合物的活性。然而在6位取代基上如果引入过大的基团时则容易与氨基酸残Gln186和Tyr212产生空间位阻,反而会影响化合物的活性。所以在末端甲基位置又不宜引入过大基团。这与CoMFA的结果一致。化合物29的4、6、7取代基均处于Asp185、Gln186、Tyr212、Glu221、Tyr129、Arg329和Gly187等氨基酸残基构成的疏水口袋。说明这些位置都是以疏水相互作用为主,这与CoMSIA的结果也是相一致的。除了疏水作用外,氢键作用在配体与受体的相互结合中也起着很重要的作用。化合物29的6位取代酯基上的两个氧原子分别与Tyr212和GIn186形成氢键,母核1.8-二氮杂萘啶环上的羟基与Asp128和Glu221形成了双氢键。这些氢键作用都增强了小分子与靶标蛋白酶结合的稳定性,有利于提高抑制剂的生物活性。
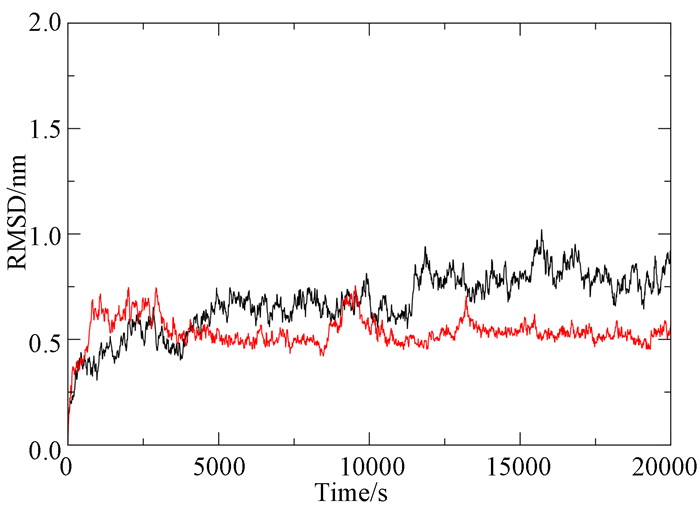
为进一步研究受体与配体的结合模式,并验证对接结果的准确性,对化合物29、11和INSTIs蛋白的复合物进行了20ns的分子动力学模拟。均方偏根差(RMSD)可用来评价分子动力学模拟平衡的稳定性。由图 6可以看出,化合物29与INST蛋白的复合物(29-5MMB)在14000ps之前震荡幅度比较大,而在14000ps之后达到了稳定状态,且RMSD值为0.53nm,化合物11与INSTIs蛋白的复合物(11-5MMB)则在整个过程中一直处于一种波动转态。这说明化合物29在INSTIs蛋白的结合口袋中能够形成更加强的相互作用,同时也符合化合物11的活性比较低这一现象。
均方根涨落(RMSF)是用来评价分子动力学模拟过程中INSTIs蛋白中氨基酸的稳定性。图 7中描述了受体蛋白上各个氨基酸的运动状态,从图中可以看到,在0~100时两个化合物的RMSF值波动比较大,后面就趋于比较稳定的状态,这说明由Asp185、Gln186、Tyr212、Pro214、Glu221、Gln215、Tyr129、Arg329、Asp128和Gly187组成的INSTIs酶的结合口袋是相对稳定的,并且这两个化合物均占据了这个结合口袋。从图中还可以看到,29-5MMB的RMSF值比11-5MMB的RMSF值要低,且化合物29与四个氨基酸Tyr212、Gln128、Asp128和Glu221形成了氢键相互作用,这说明化合物29与氨基酸的结合比化合物11更加稳定,这与之前的对接结果一致。

为了进一步阐述复合物体系的稳定性,本文利用MM-PBSA方法计算复合物体系的结合自由能。截取复合物体系在分子动力学模拟的14~20 ns相对稳定状态区间,在每100ps时截取200个快照。得到的复合物体系的不同能量贡献值见表 3,结合自由能越低说明复合物体系越稳定。从图中发现,化合物29的自由结合能明显比化合物11的低,说明化合物29的复合物体系更加稳定。这与之前得出的实验数据是相一致的。同时还可以看到,范德华相互作用贡献比其他的能量的贡献更大,说明范德华力对于结合自由能有着重要作用,也提示疏水作用对抑制剂与INSTIs蛋白复合物的稳定性起着关键作用。
下载:
导出CSV
| No | Van der Waals energy /(kJ·mol-1) | Electrostattic energy /(kJ·mol-1) | Polar solvation energy /(kJ·mol-1) | Non-Polar solvation energy /(kJ·mol-1) | Binding energy /(kJ·mol-1) |
| 11 | -144.891 | 228.824 | 275.351 | -13.421 | 346.391 |
| +/- 3.826 | +/- 10.104 | +/- 9.697 | +/- 0.349 | +/- 2.863 | |
| 29 | -167.503 | -109.193 | 243.741 | -15.169 | -48.611 |
| +/- 23.550 | +/- 20.250 | +/- 35.201 | +/- 1.708 | +/- 10.172 |
为探寻抑制剂分子与INSTIs靶蛋白的作用方式,寻找新的INSTIs分子。本文对萘啶类化合物进行了3D-QSAR模型的建立、分子对接和分子动力学的研究,构建的CoMFA和CoMSIA模型是稳定、可靠的,且具有较好的预测能力和拟合能力。通过等势图,我们能更好地分析结构与活性的关系,获得了影响这一系列化合物活性的关键结构。在母核的R6和R7位置引入大基团取代基有利于提高化合物的活性,但是在R6取代基的酯基位置又不宜引入过大基团,因为基团过大易与周围的氨基酸残基发生碰撞,反而会降低化合物的活性。同时,等势图也显示疏水作用和氢键作用对化合物活性也有很大的影响。分子对接和分子动力学则说明配体与受体的结合主要是通过疏水相互作用和氢键作用。化合物大多可以与INSTIs靶蛋白的关键氨基酸残基Asp128和Glu221形成氢键,增强了化合物与活性口袋的结合稳定性,有利于提高抑制剂的生物活性。通过分子动力学模拟可知,结合位点的关键氨基酸Asp185、Gln186、Tyr212、Glu221、Tyr129、Arg329和Gly187都显较低的RMSF值,表明如果萘啶类化合物与这些氨基酸形成疏水相互作用和氢键作用将有利于活性的提高。本文的研究结果可以为开发新的HIV INSTIs的结构提供理论依据。
V Summa, A Petrocchi, F Bonelli et al. J. Med. Chem., 2008, 51(18): 5843~5855. doi: 10.1021/jm800245z
J F Mouscadet, O Delelis, A G Marcelin et al. Drug Resist. Updat., 2010, 13(4~5): 139~150.
T Masuda. Front. Microbiol., 2011, 2(1): 1~5.
X Fan, F H Zhang, R I Al-Safi et al. Bioorg. Med. Chem., 2011, 19(16): 4935~4952. doi: 10.1016/j.bmc.2011.06.058
R Dolezal, J Korabecny, D Malinak et al. J. Mol. Graph. Model., 2015, 56: 113~129. doi: 10.1016/j.jmgm.2014.11.010
X Z Zhao, S J Smith, D P Maskell et al. J. Med. Chem., 2017, 60(17): 7315~7332. doi: 10.1021/acs.jmedchem.7b00596
G W A Milne. Chem. Inf. Model., 2010, 50(11): 2053~2053. doi: 10.1021/ci100385n
S Goyal, S Grover, J K Dhanjal et al. J. Mol. Graph. Model., 2014, 51: 64~72. doi: 10.1016/j.jmgm.2014.04.015
R D I Cramer, D E Patterson, J D Bunce. J. Am. Chem. Soc., 1988, 110(18): 5959~5967. doi: 10.1021/ja00226a005
G Klebe, U Abraham, T Mietzner. J. Med. Chem., 1994, 37(24): 4130~4146. doi: 10.1021/jm00050a010
张贝娜, 武涛, 宰小丽等.化学研究与应用, 2017, 29(4): 492~498. http://www.cnki.com.cn/Article/CJFDTOTAL-HXYJ201704009.htm
J Caballero. J. Mol. Graph. Model., 2010, 29(3): 363~371. doi: 10.1016/j.jmgm.2010.08.005
吴建军, 马玉卓, 戴雪娥等.化学通报, 2015, 78(8): 709~718. http://www.hxtb.org/ch/reader/create_pdf.aspx?file_no=20150318002&year_id=2015&quarter_id=8&falg=1
熊迪, 马玉卓, 赵钟祥等.结构化学, 2017, 36(4): 575~588. http://www.cnki.com.cn/Article/CJFDTotal-JGHX201704003.htm
A Brooun, K S Gajiwala, Y L Deng et al. Nat. Commun., 2016, 7: 11384. doi: 10.1038/ncomms11384
X Q Yan, Z C Wang, Z Li et al. Bioorg. Med. Chem. Lett., 2015, 25(20): 4664~4671. doi: 10.1016/j.bmcl.2015.08.026
A W Schüttelkopf, D M F Van Aalten. Acta. Crystallogr. D, 2004, 60(8): 1355~1363. doi: 10.1107/S0907444904011679
C Oostenbrink, A Villa, A E Mark et al. J. Comput. Chem., 2004, 25(13): 1656~1676. doi: 10.1002/jcc.20090
P Padma Kumar, A G Kalinichev, R J Kirkpatrick et al. J. Phys. Chem B, 2006, 110(9): 3841~3844. doi: 10.1021/jp057069j
B Hess, H Bekker, H J C Berendsen et al. J. Chem. Theory Comput., 2008, 4(1): 1463~1472.
蔡晓力, 马玉卓, 赵钟祥等.结构化学, 2018, 37(6): 839~853.
S Cao. J. Mol. Struct., 2012, 1020(8): 167~176.

图 1 模板分子29(a)和基于公共骨架的叠加图(b)
Figure 1 Docking template compound 29 (a) and common substructure-based alignment of the dataset (b)

图 2 CoMFA和CoMSIA中训练集与测试集化合物的pIC50实验值和预测值之间的相关性
Figure 2 Plots of predicted pIC50 values versus experimental pIC50 values for the CoMFA and CoMSIA models

图 3 CoMFA模型的立体场等势图(a)和静电场等势图(b)
Figure 3 Steric contour maps (a) and electrostetic contour maps (b) for CoMFA

图 4 CoMSIA模型的疏水场等势图(a)、氢键供体场等势图(b)和氢键受体场等势图(c)
Figure 4 hydrophobic(a), H-bond donor(b) and H-bond acceptor(c)contour maps for CoMSIA

图 6 29-5MMB(红)和11-5MMB(黑)的RMSD值随模拟时间变化图
Figure 6 RMSD of the 29-5MMB (red) and 11-5MMB (black) versus simulation time

图 7 模拟过程中29-5MMB(红)和11-5MMB(黑)的RMSF
Figure 7 RMSF of the 29-5MMB (red) and 11-5MMB (black) during simulation process
表 1 化合物的实验及预测活性值
Table 1. Experimental and predicted activity values of compounds
| No. | R4 | R6 | R7 | pIC50 (实验) |
pIC50(预测) CoMFA |
pIC50(预测) CoMSIA |
| |
||||||
| 1* | OH | H | H | 7.259 | 7.761 | 7.764 |
| 2 | OH | H | OCH3 | 7.481 | 7.607 | 7.568 |
| 3* | OH | H | piperidinyl | 4.155 | 5.122 | 5.265 |
| 4* | OH | H | morpholino | 5.721 | 5.122 | 5.113 |
| 5 | NH | H | H | 7.721 | 7.797 | 7.686 |
| 6 | NH2 | H | OCH3 | 7.959 | 7.795 | 7.607 |
| 7 | NH2 | H | piperidinyl | 4.268 | 4.272 | 4.278 |
| 8 | NH2 | H | morpholino | 4.268 | 4.295 | 4.297 |
| 9 | NHCH2CO2CH3 | H | H | 7.677 | 7.660 | 7.631 |
| 10 | NHCH2CO2CH3 | H | OCH3 | 7.376 | 7.415 | 7.505 |
| 11 | NHCH2CO2CH3 | H | piperidinyl | 5.202 | 5.19 | 5.214 |
| 12 | NHCH2CO2CH3 | H | morpholino | 5.208 | 5.211 | 5.232 |
| 13 | OH | (CH2)5OH | H | 7.921 | 7.915 | 7.900 |
| 14 | NH2 | (CH2)5OH | H | 8.568 | 8.491 | 8.812 |
| 15 | NH2 | (CH2)3OAc | H | 8.000 | 8.032 | 8.054 |
| 16 | NH2 | (CH2)6OBz | H | 5.481 | 5.438 | 5.419 |
| 17 | NH2 | (CH2)3Hex | H | 5.481 | 5.431 | 5.911 |
| 18 | NH2 | (CH2)4Ph | H | 5.699 | 5.712 | 5.611 |
| 19 | NH2 | (CH)2Ph | H | 7.854 | 7.900 | 7.331 |
| 20 | NH2 | (CH2)3N(CH3)2 | H | 7.854 | 7.908 | 8.066 |
| 21* | NH2 | (CH2)3O(CH2)2)OH | H | 8.698 | 7.424 | 7.302 |
| 22 | NH2 | (CH2)2COOH | H | 8.398 | 8.429 | 8.516 |
| 23 | NH2 | (CH2) 2CON(CH3)2 | H | 8.097 | 8.144 | 8.516 |
| 24 | NH2 | (CH)2CON(CH3)2 | H | 8.301 | 8.272 | 8.571 |
| 25* | NH2 | (CH2)2CONHPr | H | 8.699 | 9.103 | 9.089 |
| 26 | NH2 | (CH)2CONHPr | H | 8.222 | 8.281 | 8.063 |
| 27 | NH2 | (CH2)2CONH(CH)2OH | H | 8.699 | 8.707 | 8.701 |
| 28 | NH2 | (CH)2CONH(CH)OH | H | 8.155 | 8.235 | 8.384 |
| 29 | NH2 | (CH2)2CO2CH3 | H | 8.508 | 8.407 | 7.986 |
| 30 | NHCH2CO2CH3 | (CH2)5OH | H | 8.398 | 8.430 | 8.606 |
| 31 | NH(CH2)2OH | (CH2)5OH | H | 8.602 | 8.616 | 8.608 |
| 32 | NH(CH2)2OH | (CH2)2CO2CH3 | H | 8.509 | 8.497 | 8.621 |
| *代表测试集化合物 | ||||||
 下载: 导出CSV
下载: 导出CSV
表 2 由PLS分析CoMFA和CoMSIA模型的统计学数据
Table 2. The statistical data of CoMFA and CoMSIA models by PLS analysis
| CoMFA | CoMSIA | |
| PLS | ||
| N | 8 | 5 |
| q2 | 0.809 | 0.816 |
| r2 | 0.998 | 0.981 |
| SEE | 0.069 | 0.216 |
| F | 1378.014 | 220.522 |
| rpred2 | 0.785 | 0.745 |
| 贡献值 | ||
| 立体场 | 0.693 | 0.243 |
| 静电场 | 0.361 | |
| 疏水场 | 0.229 | |
| 氢键供体场 | 0.240 | |
| 氢键受体场 | 0.288 |
下载: 导出CSV
表 3 化合物29和11与INSTIs蛋白的复合物在分子动力学过程中的结合自由能
Table 3. Binding free energy of complexes of compounds 29 and 11 with INSTIs proteins in molecular dynamics
| No | Van der Waals energy /(kJ·mol-1) | Electrostattic energy /(kJ·mol-1) | Polar solvation energy /(kJ·mol-1) | Non-Polar solvation energy /(kJ·mol-1) | Binding energy /(kJ·mol-1) |
| 11 | -144.891 | 228.824 | 275.351 | -13.421 | 346.391 |
| +/- 3.826 | +/- 10.104 | +/- 9.697 | +/- 0.349 | +/- 2.863 | |
| 29 | -167.503 | -109.193 | 243.741 | -15.169 | -48.611 |
| +/- 23.550 | +/- 20.250 | +/- 35.201 | +/- 1.708 | +/- 10.172 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: