Citation:
Quanguo Jiang, Yushuai Qian, Yuqing Liu, Min Huang, Zhimin Ao. Strain boosts CO oxidation on Ni single-atom-catalyst supported by defective graphene[J]. Chinese Chemical Letters,
2023, 34(2): 107395.
doi:
10.1016/j.cclet.2022.03.118
Strain boosts CO oxidation on Ni single-atom-catalyst supported by defective graphene
English
Strain boosts CO oxidation on Ni single-atom-catalyst supported by defective graphene
College of Mechanics and Materials, Hohai University, Nanjing 210098, China
b.
Guangzhou Key Laboratory Environmental Catalysis and Pollution Control, Guangdong Key Laboratory of Environmental Catalysis and Health Risk Control, Institute of Environmental Health and Pollution Control, School of Environmental Science and Engineering, Guangdong University of Technology, Guangzhou 510006, China
Received Date:
12 January 2022 Accepted Date:
30 March 2022 Revised Date:
11 February 2022 Available Online:
15 February 2023
Abstract:
In order to realize the sulfur and water resistance and facilitate the CO oxidation reactions, the effects of strain on the adsorption of CO, O2, SO2 and H2O molecules on Ni single-atom-catalyst supported by single-carbon-vacancy graphene (Ni-SG) have been studied based on first principles calculations. It shows that the compressive strain increases the adsorption energies of all above mentioned molecules on Ni-SG, where SO2 is adsorbed more strongly on Ni-SG than CO. However, in the presence of tensile strain, the adsorption energies decreases significantly when the molecules (O2 and SO2) obtain electrons from Ni-SG, while the adsorption energies just slightly decrease when the molecules (CO and H2O) lose electrons to Ni-SG, which finally achieves the preferential adsorption of CO and O2 molecules on Ni-SG by tensile strain. In addition, with tensile strain increasing to 10%, the rate-limited energy barrier along Eley-Rideal (ER) path monotonically increases from 0.77 eV to 0.98 eV, while the rate-limited energy barrier along Langmuir-Hinshelwood (LH) path monotonically decreases from 0.54 eV to 0.44 eV, indicating that the tensile strain can facilitate the LH mechanism while imped the ER mechanism on Ni-SG. The Hirshfeld charge and orbital levels of O2 and CO molecules are modulated by the tensile strain, which plays an important role for the decreasing of energy barriers for CO oxidation. Overall, the tensile strain can enhance the sulfur and water resistance of Ni-SG, as well as boost the CO oxidation reactions.
The carbon monoxide is a dangerous gas due to its colorless and odorless, which can poison the human body imperceptibly when the CO concentration exceeds 0.1% in our daily life. The CO molecules in atmosphere mainly come from industrial waste gas and vehicle emissions, where the most efficient way for the treatment of CO is the catalytic oxidation of CO to CO2 [1-3]. Usually, noble metals show high activity towards CO oxidation reactions [4-8]. However, the noble metals are scarce in nature and need high operating temperature to overcome the relatively large reaction barrier for CO oxidation. To reduce the use of noble metals and enhance their catalytic activity [9], noble metal nanoparticles supported on suitable substrate are synthesized to lower the reaction barriers for the CO oxidation [10, 11]. When the nanoparticle contains only one atom, the so-called single atom catalyst (SAC) is formed [12-19], which exhibits excellent performance for the CO oxidation.
Two-dimensional materials have been studied for various applications as catalysts or supports [20-24]. Among varies substrates for supporting the single metal atom, graphene [25] has attracted much attention due to its outstanding structure stability and large surface-to-volume ratio. Carbon vacancy is always created to change the inert nature of pristine graphene and avoid the clustering problems. Single metal atom supported on graphene with single carbon vacancy (SG), such as Au- [26], Fe- [27], Cu- [28], Pt- [29], Al- [30], Co- [31], Ni- [32, 33] and Pd-SG [34] has exhibited high catalytic reactivity during CO oxidation. However, the SO2 and H2O molecules, which are often coexisted with CO molecules, sometimes are adsorbed on these single metal atoms too tightly, which impedes the adsorptions of O2 and CO molecules as well as the subsequent CO oxidation progress. To lower the adsorption energy of gas molecules and regulate their preferential adsorption on metal SAC, double carbon vacancy is introduced into graphene (DG) [35-38] and well water and sulfur resistance during CO oxidation is obtained [37, 38].
It is well known that strain is an effective method to change the chemical bond strength between adsorbates and substrate as discussed for the pristine two-dimensional materials [39, 40], but the effect of strain on the interaction between the gas molecules and single metal atom is more complicated because the bond strength for the metal atom and substrate is also changed by the strain [41-44]. Nickel is environmentally friendly and more abundant in earth compared with the noble metals, and Ni-SG has shown good performance during CO oxidation with relatively low energy barrier of 0.59 eV [32] (0.63 eV [33]). However, the adsorption of the SO2 and H2O molecules as well as their impact on the CO oxidation on Ni-SG is still lack.
In this work, we study the effect of strain on the competitive adsorption of gas molecules and CO oxidation on Ni-SG through first principles calculations to enhance the water and sulfur resistance of Ni-SG. The charge transfer and electronic orbitals for the molecule/Ni-SG system in the presence of different strain are further analyzed to understand the corresponding mechanisms.
The density functional theory (DFT) calculations in this work are mainly done by using Dmol3 code [45]. GGA with PBE [46] functional is adopted for the exchange-correlation functional. DSPPs are used for the core electrons and Grimme scheme [47] is used to deal with the van der Waals forces. The convergence tolerance of energy is 10−5 Hartree, and the maximal allowed force and displacement are 0.002 Hartree/Å and 0.005 Å, respectively. LST/QST [48] and NEB [49] tools are used to investigate the reaction pathway. 4 × 4 graphene supercell with vacuum width of 20 Å above the graphene layer is used to minimize the interlayer interaction and the k-point is set to 5 × 5 × 1 for structure relaxations. A finer k-point of 15 × 15 × 1 is used for the density of states (DOS) calculations. The electron orbitals are calculated in CASTEP code [50] with GGA-PBE functional, ultrasoft pseudopotentials and energy cutoff of 340 eV. The proper spin state for the Ni-SG system with the smallest total energy is obtained through comparing the total energy in different spin state.
The biaxial strain ε is applied by scaling the axial lattice constant of the relaxed Ni-SG. The adsorption energy Ead for the molecules on Ni-SG is determined by Eq. 1:
(1)
where EM/Ni-SG, ENi-SG and EM are total energies of the molecule/Ni-SG system, the isolate Ni-SG and an isolate molecule, respectively.
Before studying the molecule adsorption and CO oxidation progress, the electronic and atomic structures for Ni-SG are first studied in the following. After embedding Ni atom into the single carbon vacancy, the structure of Ni-SG is reconstructed and the relaxed structure is shown in Fig. 1a. The Ni atom moves out of the plane to get more space due to its relatively large atomic radius in single vacancy graphene [32, 33]. The C—Ni bond length lCnullNi is 1.80 Å, which is in good agreement with the reported result of 1.80 Å [32, 33]. The binding energy of the Ni atom in single vacancy graphene is −7.43 eV, which is similar to the literature results of −6.98 eV [32] (−7.57 eV [33]). To evaluate the clustering possibility of Ni atom, the diffusion of Ni to its adjacent position is studied in Fig. S1 (Supporting information), where the diffusion barrier is 4.21 eV, indicating that Ni-SG is stable without clustering problem due to its much larger barrier than the critical barrier Ecbar of 0.75 eV [51]. In the presence of strain, the binding energies of Ni atom on Ni-SG are −7.50, −7.52, −7.88, −8.14, −8.30, −8.40 and −8.42 eV with ε = −2%, 0%, 2%, 4%, 6%, 8% and 10% respectively. In addition, the diffusion barriers of Ni atom are 3.05, 4.21, 4.29, 4.40, 4.51, 4.59 and 4.63 eV with ε = −2%, 0%, 2%, 4%, 6%, 8% and 10% respectively. These results indicate that the tensile strain will enhance the stability of Ni atom on Ni-SG.
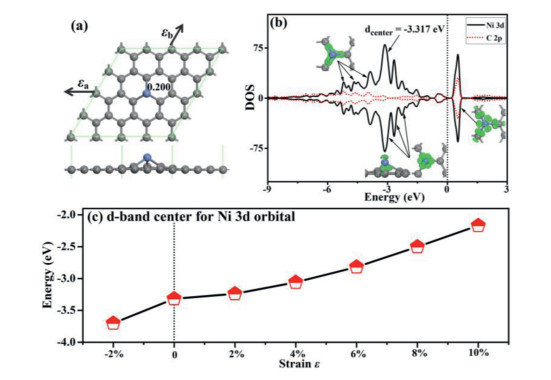
Figure 1
Figure 1.
(a) The atomic structure for Ni-SG, where the gray and blue balls represent C and Ni atoms. The directions for the applied strain and Hirshfeld charge are also given. (b) PDOS of Ni and C atoms for Ni-SG, where the corresponding molecular orbitals are plotted. (c) The d-band center for Ni 3d orbital in the presence of different strain.
The atomic charges obtained by Hirshfeld method near the dopant are also given in Fig. 1a, where the Ni atom transfers electron of 0.200 e to graphene to form an electron-depletion position. The strong interaction between the Ni atom and the nearby C atoms can be further confirmed by the partial density of states (PDOS) (Fig. 1b), where significant overlap for the Ni 3d and C 2p orbital is found in the range of −3 eV to −6 eV, which is confirmed by the charge density distributions in Fig. 1b. In addition, the Ni 3d orbital is mainly nonbinding above the energy level of −3 eV. In the presence of different strain, the electronic bands are significant upshift with strain increasing as shown in Fig. S2 (Supporting information), which is confirmed by the fact that the d-band center for Ni 3d orbital increases from −3.701 eV to −2.168 eV when the strain increases from −2% to 10% as shown in Fig. 1c. In addition, the Hirshfeld charge for Ni atom decreases from 0.229 e to 0.125 e when the strain increases from −2% to 10% as shown in Fig. S2, indicating that the charge transfer from Ni atom to defective graphene becomes less with the strain increasing. Therefore, the strain can tune the charge distributions and orbital levels for the single Ni atom on defective graphene substrate, which finally tune the gas adsorption and catalytic properties.
The performance for the CO oxidation catalysts usually degenerates in the presence of sulfur dioxide or moisture due to the adsorption of SO2 or H2O molecule [52, 53]. Therefore, the interactions between the gas molecules and the Ni-SG are important, which determine the preferential adsorption of gas molecules on the single metal atom catalyst. The most stable adsorption configurations for O2, CO, SO2 and H2O molecules on Ni-SG are shown in Fig. 2. It can be seen that all the above molecules are chemically adsorbed on the Ni-SG and the adsorption energies are shown in Table 1, where the corresponding adsorption energies are −1.68, −1.25, −1.30 and −0.94 eV for O2, CO, SO2 and H2O molecules, respectively. The adsorption energy for H2O molecule on Ni-SG is smaller than that for CO molecule. However, the adsorption energy for SO2 molecule on Ni-SG is larger than that for CO molecule. Therefore, the SO2 molecule may block the adsorption of CO molecule. It is well known that the strain is an efficient method to tune the selective adsorption towards gas molecules. Therefore, the adsorption behaviours for these molecules on Ni-SG in the presence of different strain are then studied in the following.
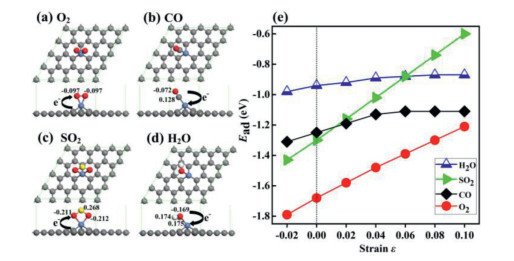
Figure 2
Figure 2.
The adsorption configurations for (a) O2, (b) CO, (c) SO2 and (d) H2O molecules on Ni-SG, and the corresponding adsorption energies in the presence of different strain are shown in panel (e).
Generally speaking, the adsorption energies for the four kinds of molecules on Ni-SG decrease with the increase of tensile strain, where the adsorption energy for SO2 and O2 molecules decreases more evidently, while the adsorption energy for CO and H2O molecules decreases slightly as shown in Fig. 2e. For O2 molecule adsorbed on Ni-SG, the adsorption energies are −1.79, −1.68, −1.58, −1.48, −1.39, −1.30 and −1.21 eV when ε = −2%, 0%, 2%, 4%, 6%, 8% and 10%, respectively, where the energy difference is 0.47 eV with the tensile strain increasing from 0% to 10% as shown in Table 1, indicating that the adsorption of O2 molecule on Ni-SG is sensitive to the tensile strain. For SO2 molecule adsorbed on Ni-SG, the adsorption energies are −1.43, −1.30, −1.16, −1.02, −0.88, −0.74 and −0.60 eV when ε = −2%, 0%, 2%, 4%, 6%, 8% and 10%, respectively, where the energy difference is 0.70 eV with the tensile strain increasing from 0% to 10% as shown in Table 1, indicating that the adsorption of SO2 on Ni-SG is even more sensitive to the tensile strain than O2 molecule.
For CO molecule adsorbed on Ni-SG, the adsorption energies are −1.31, −1.25, −1.19, −1.13 and −1.11 eV with ε = −2%, 0%, 2%, 4% and 6%, respectively, where the energy difference is 0.17 eV with the tensile strain increasing from 0% to 6%. When the tensile strain further increases, the adsorption energy no longer decreases, which is confirmed by the value of −1.11 eV with ε = 8% and 10%. For H2O molecule adsorbed on Ni-SG, the adsorption energies are −0.98, −0.94, −0.92, −0.89, −0.88 and −0.87 eV with ε = −2%, 0%, 2%, 4%, 6% and 8%, respectively, where the energy difference is 0.08 eV with the tensile strain increasing from 0% to 8%. When the tensile strain further increases, the adsorption energy no longer decreases, which is confirmed by the value of −0.87 eV with ε = 10%. Therefore, the adsorptions of CO and H2O molecules on Ni-SG are less sensitive to the tensile strain.
Based on the above discussions, the adsorptions of O2 and SO2 molecules on Ni-SG are more sensitive to the tensile strain, while the adsorptions of CO and H2O molecules on Ni-SG are less sensitive to the tensile strain. Interestingly, it is found that O2 and SO2 molecules obtain 0.254 and 0.155 e from Ni-SG, respectively, while CO and H2O molecules lose 0.044 and 0.183 e to Ni-SG based on Hirshfeld charge analysis, indicating that the charge transfer between gas molecules and Ni-SG may be a benchmark to estimate the adsorption sensitivity of gas molecules on Ni-SG to the tensile strain. In the presence of compressive strain, the adsorption energies of O2, CO, H2O and SO2 molecules are all increased, and the adsorption energy of SO2 is larger than that of CO, indicating that the compressive strain prevents the CO oxidation in the environment containing sulfur dioxide.
The PDOS are given in Fig. 3 to further understand the interactions between the molecules and Ni-SG in the presence of different strain. After the hybridization with the Ni atom, the molecular orbitals become difficult to recognize, thus the PDOS for molecule/Ni-SG system with different distance (hNi−M) between the gas molecule and Ni atom is first studied as shown in Figs. S3-S6 (Supporting information). For O2 molecule on Ni-SG, when hNi-O2 is 3.5 Å (Fig. S3a), the interaction between O2 and Ni atom is very weak and the O—O bond is 1.241 Å. When hNi-O2 is 2.5 Å (Fig. S3c), the hybridization between O2 5σ/1π orbitals and Ni 3d orbital becomes obvious and more electron enters the empty O2 2π* orbital near the Fermi level, where the O—O bond elongates to 1.280 Å (Fig. S3d). For the stable adsorption configuration (Fig. 3a and Fig. S3e), the hybridization between the O2 5σ/1π orbitals and Ni 3d orbital becomes more obvious and the O—O bond elongates to 1.383 Å with adsorption energy of −1.58 eV. When ε = 10%, the hybridization between the O2 5σ/1π orbitals and Ni 3d orbital becomes weaker, causing lower adsorption energy of −1.00 eV.
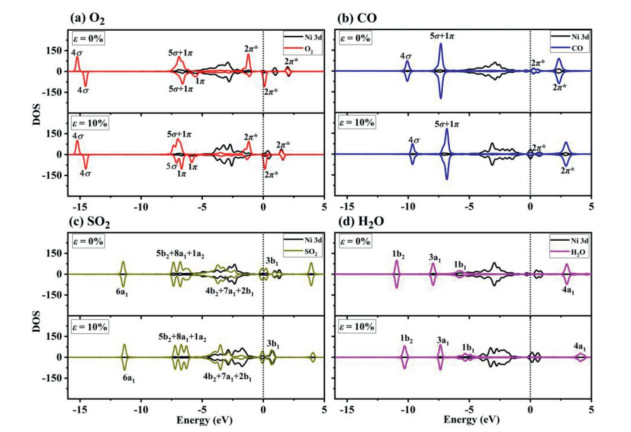
Figure 3
Figure 3.
PDOS for the adsorbed (a) O2, (b) CO, (c) SO2 and (d) H2O molecules on Ni-SG in the presence of different strain.
Similar method is applied to the PDOS analysis for other molecules on Ni-SG. For the CO molecule on Ni-SG (Fig. 3b), the hybridization between the CO 5σ orbital and Ni 3d orbital becomes more obvious and the C—O bond elongates to 1.153 Å with adsorption energy of −1.28 eV. When ε = 10%, the hybridization between the CO 5σ orbital and Ni 3d orbital changes slightly, causing only slightly lower adsorption energy of −1.11 eV. For SO2 molecule on Ni-SG (Fig. 3c), the hybridization between SO2 4b2, SO2 7a1, SO2 2b1, SO2 5b2, SO2 8a1 and SO2 1a2 orbitals and Ni 3d orbital becomes more obvious and the S—O bond elongates to 1.578 Å with adsorption energy of −1.30 eV. When ε = 10%, the hybridization between the above orbitals of SO2 and Ni 3d orbital becomes evidently weaker, causing much lower adsorption energy of −0.60 eV. For H2O molecule on Ni-SG (Fig. 3d), the hybridization between the H2O 1b1/3a1 orbitals and Ni 3d orbital becomes more obvious and the O—H bond slightly elongates to 0.979 Å with adsorption energy of −0.95 eV. When ε = 10%, the hybridization between the H2O 1b1/3a1 orbitals and Ni 3d orbital is similar to that without strain, causing only slightly lower adsorption energy of −0.87 eV.
Two classical reaction mechanisms have been established for the CO oxidation: LH mechanism and ER mechanism [26-34]. If the dissociation barrier of O2 molecule on Ni-SG is small, the LH mechanism can not happen due to the fact that the Ni site will be occupied by the two dissociated O atoms and the CO molecule can not adsorb on the Ni site. Therefore, the dissociation of O2 on Ni-G is studied in Fig. S8 (Supporting information), where the dissociation barrier is 2.02 eV. In the presence of strain, the dissociation barriers are 2.04, 2.02, 1.99, 1.89, 1.77, 1.75 and 1.69 eV with ε = −2%, 0%, 2%, 4%, 6%, 8% and 10% respectively, which are much larger than the critical barrier Ecbar of 0.75 eV [51]. Therefore, the O2 molecule will keep molecular state on Ni-SG.
In order to search the preferred reaction path on Ni-SG, the initial reaction step along both ER and LH mechanisms is studied in Fig. S8 (Supporting information), where the energy barrier for the first step along ER path is 0.77 eV (Fig. S8a). However, the energy barrier is only 0.04 eV for the co-adsorption of CO and O2 molecules (Fig. S8b), indicating a smooth progress due to its much lower barrier than the critical barrier Ecbar of 0.75 eV [51]. The effects of strain on the above two reactions on Ni-SG are further studied as shown in Fig. S8c. The reaction barrier of the first step along ER path increases from 0.72 eV to 0.98 eV with ε increasing from −2% to 10% as shown in the left panel of Fig. S8c, indicating that the tensile strain makes the CO oxidation along ER path more difficult to happen. Meanwhile, the barrier for the adsorption of CO molecule on Ni-SG with pre-adsorbed O2 molecule keeps almost constant as shown in the right of Fig. S8c, indicating that the co-adsorption of CO and O2 molecule is priority even in the presence of tensile strain. Thus the LH mechanism is mainly studied for the CO oxidation in the following.
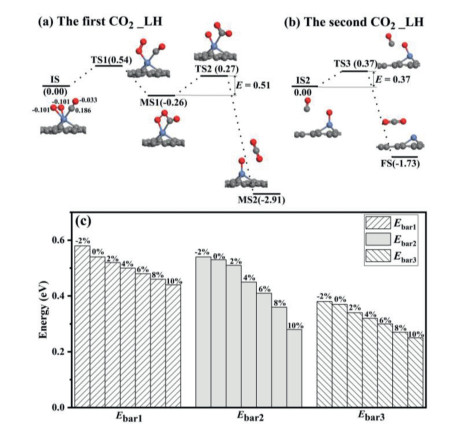
For CO oxidation on Ni-SG along LH path, the configuration with CO and O2 molecules co-adsorbed on Ni-SG is taken as the reactant (IS in Fig. 4a). At transition state TS1, one O—Ni bond is broken. After overcoming an energy barrier of 0.54 eV, an OOCO intermediate (MS1 in Fig. 4a) is formed. Then the first CO2 molecule (MS2) is formed on Ni-SG after overcoming an energy barrier of 0.51 eV (Fig. 4a), where the CO2 is physically adsorbed on Ni-SG and its adsorption energy is −0.20 eV. The reaction for this step can release a heat of 2.65 eV, which can sufficiently overcome the adsorption energy of CO2, and the first produced CO2 molecule would desorb from the Ni-SG efficiently. The subsequent CO molecule reacts with the remaining O atom to produce CO2 after surmounting an energy barrier of 0.37 eV as shown in Fig. 4b.The reaction for this step releases a heat of 1.73 eV, which can also surmount the adsorption energy of CO2 (−0.27 eV) on Ni-SG, and the second CO2 molecule desorbs from Ni-SG efficiently. The rate limiting step is the formation of OOCO intermediate with energy barrier of 0.54 eV. In addition, the CO oxidation reaction along the LH mechanism releases 0.26, 2.65, and 1.73 eV for steps 1–3, respectively, which indicates moderate thermodynamics for the CO oxidation on Ni-SG.
Figure 4
Figure 4.
The formation of the first CO2 molecule (a) and the second CO2 molecule (b) for the CO oxidation reaction on Ni-SG along LH mechanism. (c) The reaction barriers for CO oxidation on Ni-SG in the presence of different strain.
The effects of tensile strain on the CO oxidation reaction along LH path on Ni-SG are further studied as shown in Fig. 4c. The reaction barrier of the first step decreases from 0.58 eV to 0.44 eV with ε increasing from −2% to 10% as shown in the left panel of Fig. 4c. For the second step, the reaction barrier decreases from 0.54 eV to 0.28 eV with ε increasing from −2% to 10% as shown in the middle panel of Fig. 4c. For the third step, the reaction barrier decreases from 0.38 eV to 0.25 eV with ε increasing from −2% to 10% as shown in the right panel of Fig. 4c. The above results show that the tensile strain promotes the CO oxidation on Ni-SG along LH path. Therefore, the tensile strain makes the Ni-SG possess better catalytic performance during CO oxidation.
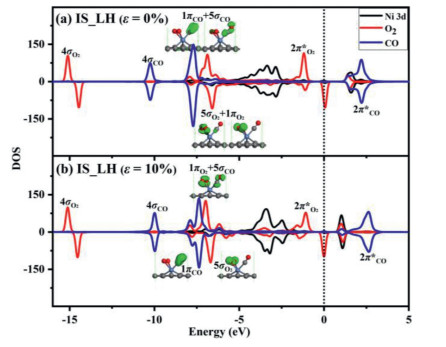
The Hirshfeld charge and electronic orbital are analysed to further reveal the origin of the high efficiency for the CO oxidation on Ni-SG along the LH path in the presence of tensile strain. The Hirshfeld charge for IS is shown in Fig. 4a, where the atomic charge for O2 molecule is −0.202 and −0.156 e when ε = 0% and ε = 10%, respectively, indicating that the charge transfer from Ni-SG to O2 molecule becomes less with the tensile strain increasing. Meanwhile, the atomic charge of CO molecule is 0.153 and 0.145 e when ε = 0% and ε = 10% as shown in Fig. 4a, respectively, indicating that the charge transfer from CO to Ni-SG is less affected by the tensile strain. Therefore, the absolute values for charge transfer of O2 and CO on Ni-SG are nearly the same when ε = 10%. The PDOS as well as the molecular orbital for the adsorbed O2 and CO molecules in IS state along LH path in different strain is shown in Fig. 5. Compared with Fig. 5a, it is found that the energy levels for 5σCO and 1πCO orbitals are upshift, which makes 5σCO/1πCO and 5σO2/1πO2 orbitals closer to each other as shown in Fig. 5b. In addition, the 5σCO and 1πO2 orbitals are even in the same energy levels as shown in inset of Fig. 5b, which finally facilitates the formation of new molecular orbital on Ni-SG and lower the reaction barrier for the formation of OCOO intermediate.
Figure 5
Figure 5.
The PDOS for Ni atom and adsorbed O2 and CO molecules in IS state along LH pathway (a) without strain and (b) in the presence of 10% strain. Insets are the corresponding molecular orbitals.
In summary, the adsorption of CO, O2, SO2 and H2O molecules on Ni-SG in the presence of different strain has been studied by using DFT calculations. The tensile strain makes Ni atom less attractive towards all above molecules, where the adsorption energy for SO2 decreases most significantly, which finally achieves the sulfur and water resistance for Ni-SG. In addition, the tensile strain facilitates the CO oxidation on Ni-SG along LH mechanism. The Hirshfeld charge and orbital levels of O2 and CO molecules are tuned by the tensile strain, which plays an important role for the CO oxidation. Overall, the tensile strain can enhance the sulfur and humidity resistance of Ni-SG as well as boost the performance towards CO oxidation on Ni-SG.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We acknowledge support by the Fundamental Research Funds for the Central Universities (No. B210202099), National Natural Science Foundation of China (Nos. 21703052, 22176041, 21777033), Science and Technology Planning Project of Guangdong Province (No. 2017B020216003).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.03.118.
[1]
J. Neugebohren, D. Borodin, H.W. Hahn, et al., Nature 558 (2018) 280–283. doi: 10.1038/s41586-018-0188-x
[2]
W. Zhan, J. Wang, H. Wang, et al., J. Am. Chem. Soc. 139 (2017) 8846–8854. doi: 10.1021/jacs.7b01784
G. Henkelman, H. Jonsson, J. Chem. Phys. 113 (2000) 9978. doi: 10.1063/1.1323224
[50]
M.D. Segall, P.L.D. Lindan, M.J. Probert, et al., J. Phys. : Condens. Matter 14 (2002) 2717. doi: 10.1088/0953-8984/14/11/301
[51]
D. Yan, Q. Li, H. Zhang, et al., Catal. Commun. 142 (2020) 1060.
[52]
C. Shang, Z.P. Liu, J. Am. Chem. Soc. 133 (2011) 9938–9947. doi: 10.1021/ja203468v
[53]
H. Shin, M. Baek, Y. Ro, et al., Appl. Surf. Sci. 429 (2018) 102–107. doi: 10.3390/su11010102
Figure 1
(a) The atomic structure for Ni-SG, where the gray and blue balls represent C and Ni atoms. The directions for the applied strain and Hirshfeld charge are also given. (b) PDOS of Ni and C atoms for Ni-SG, where the corresponding molecular orbitals are plotted. (c) The d-band center for Ni 3d orbital in the presence of different strain.
Figure 2
The adsorption configurations for (a) O2, (b) CO, (c) SO2 and (d) H2O molecules on Ni-SG, and the corresponding adsorption energies in the presence of different strain are shown in panel (e).
Figure 4
The formation of the first CO2 molecule (a) and the second CO2 molecule (b) for the CO oxidation reaction on Ni-SG along LH mechanism. (c) The reaction barriers for CO oxidation on Ni-SG in the presence of different strain.
Figure 5
The PDOS for Ni atom and adsorbed O2 and CO molecules in IS state along LH pathway (a) without strain and (b) in the presence of 10% strain. Insets are the corresponding molecular orbitals.

 DownLoad:
DownLoad:


 下载:
下载:






 下载:
下载:
