引用本文:
张复兴, 陶靖, 唐丹丹, 罗晶, 汤鹏, 邝代治, 冯泳兰, 朱小明. 三 (3, 5-二氟苄基) 氯化锡和四 (邻氯苄基) 锡的合成、晶体结构及量子化学[J]. 无机化学学报,
2017, 33(4): 644-650.
doi:
10.11862/CJIC.2017.073

Citation: ZHANG Fu-Xing, TAO Jing, TANG Dan-Dan, LUO Jing, TANG Peng, KUANG Dai-Zhi, FENG Yong-Lan, ZHU Xiao-Ming. Syntheses, Crystal Structures and Quantum Chemistry of Tri (3, 5-difluorobenzyl) tin Chloride and Tetra (o-chlorobenzyl) tin Chlorde and Tetra (o-chlorobenzyl) tin[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(4): 644-650. doi: 10.11862/CJIC.2017.073
Citation: ZHANG Fu-Xing, TAO Jing, TANG Dan-Dan, LUO Jing, TANG Peng, KUANG Dai-Zhi, FENG Yong-Lan, ZHU Xiao-Ming. Syntheses, Crystal Structures and Quantum Chemistry of Tri (3, 5-difluorobenzyl) tin Chloride and Tetra (o-chlorobenzyl) tin Chlorde and Tetra (o-chlorobenzyl) tin[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(4): 644-650. doi: 10.11862/CJIC.2017.073
三 (3, 5-二氟苄基) 氯化锡和四 (邻氯苄基) 锡的合成、晶体结构及量子化学
摘要:
3,5-二氟苄基氯和邻氯基苄基氯在适当的溶剂中与锡粉反应,合成了三(3,5-二氟苄基)氯化锡(1)和四(邻氯苄基)锡(2),经X射线衍射方法测定了新化合物的晶体结构。化合物1属单斜晶系,空间群为C2/c,晶体学参数:a=1.858 33(11)nm,b=1.140 98(7)nm,c=2.690 06(16)nm,β=109.288(10)°,V=5.383 6(6)nm3,Z=8,Dc=1.532 g·cm-3,μ(Mo Kα)=13.61 cm-1,F(000)=2 480,R1=0.085 1,wR2=0.168 1。化合物2属单斜晶系,空间群为P21/m,晶体学参数:a=0.585 54(5)nm,b=1.969 74(18)nm,c=0.857 86(8)nm,β=95.204 0(10)°,V=0.985 34(15)nm3,Z=2,Dc=1.805 g·cm-3,μ(Mo Kα)=14.91 cm-1,F(000)=524,R1=0.054 0,wR2=0.163 9;中心锡原子为畸变四面体构型。对其结构进行量子化学从头计算,探讨了化合物的稳定性、分子轨道能量以及部分前沿分子轨道的组成特征。测定了化合物的热稳定性。
-
关键词:
- 三 (3, 5-二氟苄基) 氯化锡
- / 四 (邻氯苄基) 锡
- / 合成
- / 晶体结构
- / 量子化学
English
Syntheses, Crystal Structures and Quantum Chemistry of Tri (3, 5-difluorobenzyl) tin Chloride and Tetra (o-chlorobenzyl) tin Chlorde and Tetra (o-chlorobenzyl) tin
Abstract:
Tri (3, 5-difluorobenzyl) tin chloride (1) and tetra (o-chlorobenzyl) tin (2) have been synthesized. The crystal structures of the compounds were determined by X-ray diffraction. The crystal 1 belongs to monoclinic space group C2/c with a=1.858 33(11) nm, b=1.140 98(7) nm, c=2.690 06 (16) nm, β=109.288(10)°, V=5.383 6(6) nm3, Z=8, Dc=1.532 g·cm-3, μ(Mo Kα)=13.61 cm-1, F(000)=2 480, R1=0.085 1, wR2=0.168 1. The crystal 2 belongs to monoclinic space group P21/m with a=0.585 54(5) nm, b=1.969 74(18) nm, c=0.857 86(8) nm, β=95.204 0(10)°, V=0.985 34(15) nm3, Z=2, Dc=1.805 g·cm-3, μ(Mo Kα)=14.91 cm-1, F(000)=524, R1=0.0540, wR2=0.163 9. The tin atoms have a distorted tetrahedral geometry. The study on two compounds have been performed with quantum chemistry calculation. The stabilities, some frontier molecular orbital energies and composition characteristics of some frontier molecular orbital of the compound have been investigated. The thermal stability of the compounds were tested.
-
0 引言
由于有机锡化合物结构丰富、反应多变和用途广泛,多年来一直引起人们的兴趣[1-6]。有机锡化合物具有多种类型,其中最基础的是烃基卤化锡,其它都可视为其衍生物。烃基卤化锡又有一烃基三卤化锡、二烃基二卤化锡和三烃基卤化锡之分,其中以二烃基二卤化锡和三烃基卤化锡为最重要和最常见。除此之外,还有四烃基锡。近年来人们利用取代苄基卤结构的多样性和独特的反应性,合成了一系列取代苄基锡化合物[7-9]。研究表明苯环上连有的取代基的性质和取代基的位置对取代苄基锡形成和结构有明显的影响[10-11],反应条件也是决定反应结果的重要因素[12-13]。为了更进一步探索反应条件和取代基对产物的影响,本文合成了三 (3, 5-二氟苄基) 氯化锡 (1) 和四 (邻氯苄基) 锡 (2)。通过元素分析、红外光谱对其结构进行了表征,用X射线单晶衍射测定了化合物的晶体结构,对其结构进行量子化学从头计算,探讨了化合物分子的稳定性、分子轨道能量以及一些前沿分子轨道的组成特征。测定了化合物的热稳定性。
1 实验部分
1.1 试剂和仪器
所有试剂均为化学纯。表征所用的仪器有日本岛津IRPrestige-21型红外光谱仪 (4 000~400 cm-1,KBr),PE-2400(Ⅱ) 型元素分析仪,Bruker SMART APEXⅡ型单晶衍射仪,X4型数字显微熔点仪,TGA Q50型热重分析仪 (空气氛,加热速度为20℃·min-1,气体流速为20 mL·min-1,40~800 ℃)。
1.2 化合物的合成
化合物1的合成:参照文献[12],在150 mL的三颈烧瓶中加入16.26 g (0.1 mol)3,5-二氟苄基氯,10 g (0.084 mol) 锡粉和80 mL正丁醇,少量碘,搅拌下将1 g镁条分批加入。快速搅拌回流6 h,趁热滤出未反应的锡粉,滤液倒入适量的稀盐酸溶液中,充分搅拌,冷却后分出水层。将正丁醇减压浓缩至适当的体积后放置,析出白色固体。分出固体,用苯重结晶得无色三 (3,5-二氟苄基) 氯化锡12.24 g,收率68.57%。m.p. 177~179 ℃。元素分析 (C21H15F6ClSn):实验值 (计算值,%):C 47.21 (47.06),H 2.81 (2.80)。红外光谱主要吸收峰 (cm-1):3 030 (w),2 932(w),1 611(s),1 584(s),1 479 (s),1 442 (m),556(w),519(w),438(m)。
化合物2的合成:参照文献[13],在三颈瓶中加入16.1 g (0.1 mol) 邻氯苄基氯,少量碘,60 mL正戊醇和40 mL邻二甲苯,回流搅拌下分批加入10.0 g (0.084 mol) 经活化处理的锡粉和1.0 g镁条,快速搅拌回流反应6 h。趁热过滤出未反应的锡粉,滤液倒入适量的稀盐酸溶液中,充分搅拌。冷却后分出水层,旋转蒸发部分溶剂后,放置析出白色固体。抽滤分离出固体,用少量冷无水乙醇洗涤固体,再用苯重结晶,得到无色晶体11.82g,收率76.16%。熔点:89~90 ℃。元素分析 (C28H24Cl4 Sn):实验值 (计算值,%):C53.96 (54.11),H 3.84 (3.86)。红外光谱主要吸收峰 (cm-1):3 049(w),2 931(w),1 585(m),1 470(s),1 438 (m),567(m)。
1.3 晶体结构分析
分别选取大小均为0.23 mm×0.21 mm×0.20 mm的晶体,在Bruker SMART APEX Ⅱ CCD单晶衍射仪上,采用经石墨单色化的Mo Kα射线 (λ= 0.071 073 nm),于296(2) K,以φ~ω扫描方式收集数据。可观察衍射点分别为2149个和5034个[I>2σ(I)]用于结构分析和精修。全部数据经Lp因子和经验吸收校正。晶体结构由直接法解出,非氢原子坐标通过数轮差值Fourier合成陆续确定,理论加氢法给出氢原子在晶胞中的位置坐标。对非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法修正。全部结构分析计算工作采用SHELX-97程序[14]系统完成。晶体学数据详见表 1。
 表 1
化合物的晶体学数据
Table 1.
Crystallographic data of the title compounds
表 1
化合物的晶体学数据
Table 1.
Crystallographic data of the title compounds
 表 1 化合物的晶体学数据
表 1 化合物的晶体学数据
Table 1. Crystallographic data of the title compoundsCCDC:1533663,1;1533662,2。
2 结果与讨论
2.1 晶体结构
化合物的主要键长和键角分别列于表 2、3,化合物的分子结构见图 1、图 2。
表 2
化合物的部分键长
Table 2.
Selected bond lengths (nm) of the compounds
1 Sn (1)-C (1)
Sn (1)-C (1)ⅰ0.214 7(6)
0.214 7(6)Sn (1)-C (8) 0.215 3(4) Sn (1)-Cl (1) 0.238 4(2) 2 Sn (1)-C (1)
Sn (1)-C (22)0.217 8(5)
0.217 8(6)Sn (1)-C (8) 0.217 0(6) Sn (1)-C (15) 0.218 4(6) Symmetry codes: ⅰ x, -y+1/2, z for 1 表 2 化合物的部分键长
Table 2. Selected bond lengths (nm) of the compounds
表 3
化合物的部分键角 (°)
Table 3.
Selected bond angles (°) of the compounds
1 C (1)-Sn (1)-C (1)ⅰ
C (1)-Sn (1)-Cl (1)117.20(4)
102.02(19)C (1)ⅰ-Sn (1)-Cl (1)
C (1)-Sn (1)-C (8)102.02(19)
114.87(18)C (1)ⅰ-Sn (1)-C (8)
C (8)-Sn (1)-Cl (1)114.87(18)
102.60(14)2 C (1)-Sn (1)-C (8)
C (8)-Sn (1)-C (22)110.0(2)
110.1(2)C (1)ⅰ-Sn (1)-C (22)
C ((8)-Sn (1)-C (15)106.6(2)
110.7(2)C (1)-Sn (1)-C (15)
C (15)-Sn (1)-C (22)111.9(2)
107.4(3)Symmetry codes: ⅰ x, -y+1/2, z for 1 表 3 化合物的部分键角 (°)
Table 3. Selected bond angles (°) of the compounds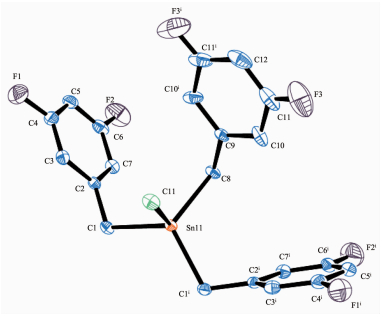 图 1
化合物1的分子结构图 (椭球概率20%)
Figure 1.
Molecular structure of compound 1 with the ellipsoids drawn at the 20% probability level
图 1
化合物1的分子结构图 (椭球概率20%)
Figure 1.
Molecular structure of compound 1 with the ellipsoids drawn at the 20% probability level
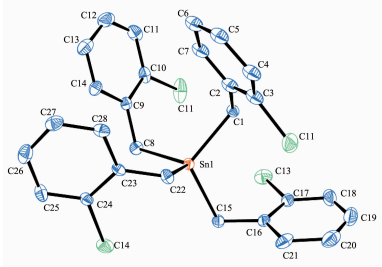 图 2
化合物2的分子结构图 (椭球概率20%)
Figure 2.
Molecular structure of compound 2 with the ellipsoids drawn at the 20% probability level
图 2
化合物2的分子结构图 (椭球概率20%)
Figure 2.
Molecular structure of compound 2 with the ellipsoids drawn at the 20% probability level
由分子结构图和结构参数可知:
化合物1为单锡核面对称分子,中心锡原子为四配位的四面体构型,与锡原子直接相连的是3个亚甲基碳和氯原子。分子的对称面是经过Cl、Sn、C (8)、C (9)、C (12) 五个原子的平面,扭转角∠Cl (1)-Sn (1)-C (8)-C (9) 为0°。分子中Sn (1)-C (1) 键和Sn (1)-C (1)ⅰ键键长均为0.214 7 nm、Sn (1)-C (8) 键键长为0.215 3 nm,Sn-Cl键键长为0.238 4 nm。由于3,5-二氟苄基的空间位阻大,使得亚甲基碳与中心锡原子间的键角均大于正四面体键角,分别为114.87°和117.20°;而氯原子和亚甲基碳与中心锡原子间的键角均小于正四面体键角,分别为102.02°和102.60°。因此,化合物中心锡原子为畸变的四面体构型。
化合物2为非对称分子,中心锡原子与4个亚甲基碳原子相连形成四面体构型。4个Sn-C键键长不完全相等,其中有2个相等为0.217 8 nm,另2个分别为0.217 0和0.218 4 nm,平均键长为0.217 75 nm。锡原子和与之相连的碳原子之间所构成的键角各不相等,在106.6°~111.9°之间,均偏离了正四面体角。分子中Sn-C键的平均键长要比四 (对氯苄基) 锡的Sn-C键平均键长 (0.217 5 nm)[10]稍长,这体现了取代基在苯环上的位置的影响。化合物2与按文献[8]的方法合成具有相同的分子结构的晶体相比,一是晶体的空间群不同,分别为P21/m和P21/c;二是Sn-C键的平均键长不同,分别为0.217 75和0.212 73 nm,相差0.005 02 nm。
3.2 量子化学研究
根据晶体结构的原子坐标,运用Gaussian 03W程序和B3ylp/lanl2dz基组水平,计算得到分子的总能量和前沿分子轨道能量。
化合物1:ET=-774.702 636 4 a.u., EHOMO=-0.265 03 a.u.,ELUMO=-0.064 26 a.u.,ΔELUMO-HOMO=0.200 77 a.u.。化合物2:ET=-1135.589 629 3 a.u., EHOMO=-0.308 30 a.u.,ELUMO=0.103 63 a.u.,ΔELUMO-HOMO=0.411 93 a.u.。从体系能量和前沿轨道的能量分析,2个化合物总能量和占有轨道能量均较低,表明2个化合物分子结构稳定。最高占据轨道与最低未占轨道的能量间隙ΔE均较大,从氧化还原转移的角度分析,均较难失去电子而被氧化。但相对而言化合物2比化合物1有更好的稳定性和更难被氧化。
为探索化合物的电子结构与成键特征,对化合物分子轨道进行分析,用参与组合的各类原子轨道系数的平方和来表示该部分在分子轨道中的贡献,并经归一化。
化合物1:把化合物原子分为6部分:(a) 锡原子Sn;(b) 苯环碳原子C(Ⅰ);(c) 亚甲基碳原子C(Ⅱ);(d) 氯原子Cl;(e) 氟原子F;(f) 氢原子H。前沿占有轨道和未占有轨道各取5个,计算结果如表 4和图 3所示。
表 4
化合物1的分子轨道组成
Table 4.
Some calculated frontier molecular orbitals composition of compound 1
% MO ε/Hartree Sn C(Ⅰ) C(Ⅱ) Cl F H 55 -0.393 75 12.586 35 64.564 42 8.681 32 4.896 32 9.246 95 0.024 64 56 -0.370 83 6.489 56 63.226 37 20.568 43 2.662 27 6.836 84 0.216 54 57 -0.365 04 33.121 25 20.369 25 8.472 63 20.569 37 16.897 64 0.569 87 58 -0.354 80 26.460 57 46.869 43 15.896 13 7.635 69 1.968 31 1.169 87 59HO -0.265 03 24.634 36 40.384 26 18.839 86 10.687 48 4.577 30 0.876 74 60MO -0.064 26 18.269 42 58.369 54 8.698 63 6.369 85 7.256 98 1.035 58 61 -0.019 09 38.623 16 40.598 83 15.521 63 2.208 53 2.235 16 0.812 6 62 0.082 28 4.325 96 65.026 58 4.126 84 13.683 68 11.872 15 0.964 79 63 0.090 01 40.524 55 25.174 14 15.307 24 7.930 02 10.082 02 0.982 03 64 0.108 74 30.259 16 15.851 23 38.633 45 6.13651 8.333 13 0.786 52 表 4 化合物1的分子轨道组成
Table 4. Some calculated frontier molecular orbitals composition of compound 1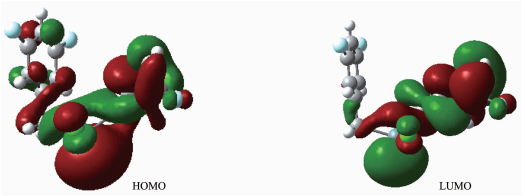 图 3
化合物1的前沿分子轨道示意图
Figure 3.
Schematic diagram of frontier MO for compound 1
图 3
化合物1的前沿分子轨道示意图
Figure 3.
Schematic diagram of frontier MO for compound 1
表 4和图 3显示化合物1分子的成键特征:① 前沿占有分子轨道中,苯环碳原子对分子轨道的贡最大,为40.38%,并且在深层次轨道中均有较大的贡献,说明苯环具有良好的共轭离域性和稳定性;② 前沿占有分子轨道中,锡原子、亚甲基碳原子和氯原子对分子轨道均有较大的贡献,分别为24.63%、18.84%和10.69%,说明Sn-C键和Sn-Cl键均较稳定;③ 比较HOMO与LUMO的各类原子轨道成份,可以看出,当电子从HOMO激发到LUMO轨道时,主要是锡原子、亚甲基碳原子和氯原子整体向苯环转移,从而使Sn-C键和Sn-Cl键强度减弱,化合物的稳定性降低。
化合物2:把化合物原子分为5部分:(a) 锡原子Sn;(b) 氯原子Cl;(c) 苯环碳原子C(Ⅱ);(d) 亚甲基碳原子C?;(e) 氢原子H。前沿占有轨道和未占有轨道各取5个,计算结果如表 5和图 4所示。
表 5
化合物2的分子轨道组成
Table 5.
Some calculated frontier molecular orbitals composition of compound 2
MO ε/Hartree Sn Cl C(Ⅰ) C(Ⅱ) H 108 -0.341 25 9.354 87 0.832 15 82.463 35 6.546 98 0.802 65 109 -0.334 20 4.112 42 2.124 51 84.321 26 8.231 63 1.210 18 110 -0.319 40 6.056 42 2.382 34 84.036 95 5.815 34 1.708 95 111 -0.312 37 10.589 1 1.915 16 70.758 14 15.523 12 1.214 48 112MO -0.308 30 25.623 59 1.425 64 50.252 17 22.214 36 0.484 24 113MO 0.103 63 0.563 21 3.742 44 80.015 53 14.128 40 1.550 42 114 0.107 32 3.584 30 4.985 47 84.405 63 5.810 28 1.214 32 115 0.111 56 22.397 46 1.46017 58.254 16 16.264 68 1.623 53 116 0.114 32 12.568 24 4.918 23 57.276 95 24.360 15 0.876 42 117 0.117 48 16.028 24 0.413 67 79.972 16 2.79213 0.793 81 表 5 化合物2的分子轨道组成
Table 5. Some calculated frontier molecular orbitals composition of compound 2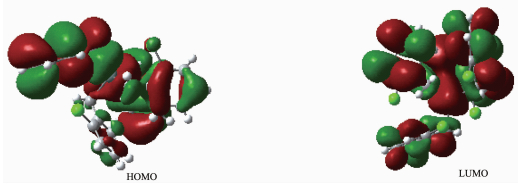 图 4
化合物2的前沿分子轨道示意图
Figure 4.
Schematic diagram of frontier MO for compound 2
图 4
化合物2的前沿分子轨道示意图
Figure 4.
Schematic diagram of frontier MO for compound 2
表 5和图 4显示化合物2分子的成键特征:① 前沿占有分子轨道中,苯环碳原子对分子轨道的贡最大,为50.25%,并且在深层次轨道中均有较大的贡献,表明苯环具有良好的共轭离域性和稳定性。② 前沿占有分子轨道中,锡原子和亚甲基碳原子均有较大的贡献,分别为25.62%和22.21%,表明结构中Sn-C键的强度较大,结构较稳定。③ 比较HOMO与LUMO的各类原子轨道成份,可以看出,当电子从HOMO激发到LUMO轨道时,主要是锡原子上的电子通过亚甲基苯环转移,电子转移的结果表明分子在激发态时Sn-C键的强度减弱,化合物的稳定性降低。
3.3 热稳定性分析
利用TG209F3热分析仪,在范围内对化合物进行热重测试,其结果如图 5所示:
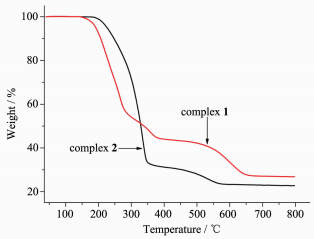 图 5
化合物1和2的热分析曲线
Figure 5.
Thermogravimetric analysis curves of compounds 1 and 2
图 5
化合物1和2的热分析曲线
Figure 5.
Thermogravimetric analysis curves of compounds 1 and 2
化合物1随温度的升高,发生失重的情况是:在40~150 ℃之间,化合物几乎没有失重;在150~380 ℃之间,化合物失重速度较快,且失重质量达到了近60%,这应该是化合物的Sn-C键的断裂,逐步失去苄基;在380~508 ℃之间,化合物的失重变得缓慢;从508 ℃开始失重速度再次加快,至660 ℃时失重基本停止,残留质量最后稳定在26.85%。总计失重73.15%,对应于化合物分子失去3个3,5-二氟苄基和氯,残余物可被假定为SnO2,与28.15%的计算值基本吻合。上述热分析结果表明该化合物结构在150 ℃之前是可以稳定存在的。
化合物2随温度的升高,发生失重的情况是:在40~175 ℃之间,配合物几乎没有失重;在175~355 ℃之间,配合物失重速度较快;在355~570 ℃之间,配合物失重速度变缓,在570 ℃以后几乎没有出现失重,最后残余物稳定在约22.75%。总计失重77.25%,对应于失去4个邻氯基苄基,残余物可被假定为SnO2,与24.27%的计算值基本吻合。上述热分析结果表明该配合物结构在175 ℃之前是可以稳定存在的。
-
-
[1]
Khan A, Komejan S, Patel A, et al. J. Organomet. Chem., 2015, 776:180-191 doi: 10.1016/j.jorganchem.2014.11.010
-
[2]
Wang F, Zhang R F, Cheng S, et al. J. Organomet. Chem., 2015, 789-790:46-51 doi: 10.1016/j.jorganchem.2015.05.003
-
[3]
Sirajuddin M, Ali S, Tahir M N. Inorg. Chim. Acta, 2016, 439:145-158 doi: 10.1016/j.ica.2015.10.017
-
[4]
Munoz-Flores B M, Santillán R, Farfán N, et al. J. Organomet. Chem., 2014, 769:64-71 doi: 10.1016/j.jorganchem.2014.07.002
-
[5]
Carraher Jr C E, Roner M R. J. Organomet. Chem., 2014, 751:67-82 doi: 10.1016/j.jorganchem.2013.05.033
-
[6]
Chandrasekhar V, Thirumoorthi R, Metre R K, et al. J. Organomet. Chem., 2011, 696(2):600-606 doi: 10.1016/j.jorganchem.2010.09.032
-
[7]
尹汉东, 高中军, 徐浩龙, 等.无机化学学报, 2006, 22(2):371~374 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20060236&journal_id=wjhxxbcnYIN Han-Dong, GAO Zhong-Jun, XU Hao-Long, et al. Chinese J. Inorg. Chem., 2006, 22(2):371~374 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20060236&journal_id=wjhxxbcn
-
[8]
Schurmann M, Silvestri A, Ruisi G, et al. J. Organomet. Chem., 1999, 584:293-300 doi: 10.1016/S0022-328X(99)00166-7
-
[9]
王剑秋, 张复兴, 邝代治, 等.有机化学, 2004, 24(1)80~84 http://www.cnki.com.cn/Article/CJFDTotal-YJHU200401015.htmWANG Jian-Qiu, ZHANG Fu-Xing, KUANG Dai-Zhi, et al. Chinese J. Org. Chem., 2004, 24(1)80~84 http://www.cnki.com.cn/Article/CJFDTotal-YJHU200401015.htm
-
[10]
张复兴, 王剑秋, 邝代治, 等.有机化学, 2003, 23(4):368~371 http://www.cnki.com.cn/Article/CJFDTotal-YJHU200304008.htmZHANG Fu-Xing, WANG Jian-Qiu, KUANG Dai-Zhi, et al. Chinese J. Org. Chem, 2003, 23(4):368~371 http://www.cnki.com.cn/Article/CJFDTotal-YJHU200304008.htm
-
[11]
张复兴, 邝代治, 许志锋, 等.无机化学学报, 2002, 18(8):854~858 http://www.cnki.com.cn/Article/CJFDTotal-WJHX200208021.htmZHANG Fu-Xing, KUANG Dai-Zhi, XU Zhi-Feng, et al. Chinese J. Inorg. Chem., 2002, 18(8):854~858 http://www.cnki.com.cn/Article/CJFDTotal-WJHX200208021.htm
-
[12]
王剑秋, 张复兴, 邝代治, 等.无机化学学报, 2015, 31(2):237-242 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150204&journal_id=wjhxxbcnWANG Jian-Qiu, ZHANG Fu-Xing, KUANG Dai-Zhi, et al. Chinese J. Inorg. Chem., 2015, 31(2):237-242 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150204&journal_id=wjhxxbcn
-
[13]
张复兴, 邝代治, 冯泳兰, 等.无机化学学报, 2015, 31(6):1194-1200 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150619&journal_id=wjhxxbcnZHANG Fu-Xing, KUANG Dai-Zhi, FENG Yong-Lan, et al. Chinese J. Inorg. Chem., 2015, 31(6):1194-1200 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150619&journal_id=wjhxxbcn
-
[14]
Sheldrick G M. SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997.
-
[1]
-

图 1 化合物1的分子结构图 (椭球概率20%)
Figure 1 Molecular structure of compound 1 with the ellipsoids drawn at the 20% probability level
Symmetry codes: ⅰ x, -y+1/2, z

图 2 化合物2的分子结构图 (椭球概率20%)
Figure 2 Molecular structure of compound 2 with the ellipsoids drawn at the 20% probability level
表 2 化合物的部分键长
Table 2. Selected bond lengths (nm) of the compounds
1 Sn (1)-C (1)
Sn (1)-C (1)ⅰ0.214 7(6)
0.214 7(6)Sn (1)-C (8) 0.215 3(4) Sn (1)-Cl (1) 0.238 4(2) 2 Sn (1)-C (1)
Sn (1)-C (22)0.217 8(5)
0.217 8(6)Sn (1)-C (8) 0.217 0(6) Sn (1)-C (15) 0.218 4(6) Symmetry codes: ⅰ x, -y+1/2, z for 1  下载: 导出CSV
下载: 导出CSV
表 3 化合物的部分键角 (°)
Table 3. Selected bond angles (°) of the compounds
1 C (1)-Sn (1)-C (1)ⅰ
C (1)-Sn (1)-Cl (1)117.20(4)
102.02(19)C (1)ⅰ-Sn (1)-Cl (1)
C (1)-Sn (1)-C (8)102.02(19)
114.87(18)C (1)ⅰ-Sn (1)-C (8)
C (8)-Sn (1)-Cl (1)114.87(18)
102.60(14)2 C (1)-Sn (1)-C (8)
C (8)-Sn (1)-C (22)110.0(2)
110.1(2)C (1)ⅰ-Sn (1)-C (22)
C ((8)-Sn (1)-C (15)106.6(2)
110.7(2)C (1)-Sn (1)-C (15)
C (15)-Sn (1)-C (22)111.9(2)
107.4(3)Symmetry codes: ⅰ x, -y+1/2, z for 1
下载: 导出CSV
表 4 化合物1的分子轨道组成
Table 4. Some calculated frontier molecular orbitals composition of compound 1
% MO ε/Hartree Sn C(Ⅰ) C(Ⅱ) Cl F H 55 -0.393 75 12.586 35 64.564 42 8.681 32 4.896 32 9.246 95 0.024 64 56 -0.370 83 6.489 56 63.226 37 20.568 43 2.662 27 6.836 84 0.216 54 57 -0.365 04 33.121 25 20.369 25 8.472 63 20.569 37 16.897 64 0.569 87 58 -0.354 80 26.460 57 46.869 43 15.896 13 7.635 69 1.968 31 1.169 87 59HO -0.265 03 24.634 36 40.384 26 18.839 86 10.687 48 4.577 30 0.876 74 60MO -0.064 26 18.269 42 58.369 54 8.698 63 6.369 85 7.256 98 1.035 58 61 -0.019 09 38.623 16 40.598 83 15.521 63 2.208 53 2.235 16 0.812 6 62 0.082 28 4.325 96 65.026 58 4.126 84 13.683 68 11.872 15 0.964 79 63 0.090 01 40.524 55 25.174 14 15.307 24 7.930 02 10.082 02 0.982 03 64 0.108 74 30.259 16 15.851 23 38.633 45 6.13651 8.333 13 0.786 52
下载: 导出CSV
表 5 化合物2的分子轨道组成
Table 5. Some calculated frontier molecular orbitals composition of compound 2
MO ε/Hartree Sn Cl C(Ⅰ) C(Ⅱ) H 108 -0.341 25 9.354 87 0.832 15 82.463 35 6.546 98 0.802 65 109 -0.334 20 4.112 42 2.124 51 84.321 26 8.231 63 1.210 18 110 -0.319 40 6.056 42 2.382 34 84.036 95 5.815 34 1.708 95 111 -0.312 37 10.589 1 1.915 16 70.758 14 15.523 12 1.214 48 112MO -0.308 30 25.623 59 1.425 64 50.252 17 22.214 36 0.484 24 113MO 0.103 63 0.563 21 3.742 44 80.015 53 14.128 40 1.550 42 114 0.107 32 3.584 30 4.985 47 84.405 63 5.810 28 1.214 32 115 0.111 56 22.397 46 1.46017 58.254 16 16.264 68 1.623 53 116 0.114 32 12.568 24 4.918 23 57.276 95 24.360 15 0.876 42 117 0.117 48 16.028 24 0.413 67 79.972 16 2.79213 0.793 81
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 3
- 文章访问数: 420
- HTML全文浏览量: 30
 下载:
下载:
