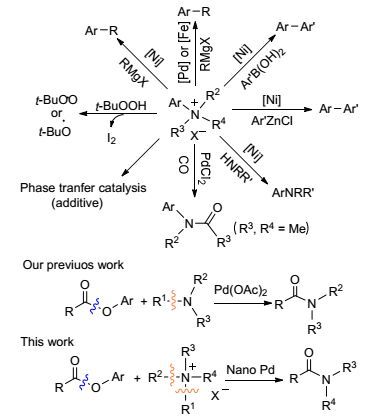
 Figure Scheme 1.
Application of quaternary ammonium salt
Figure Scheme 1.
Application of quaternary ammonium salt
Citation:
Wang Lili, Li Pengshuai, Jia Meilin, Bao Yongsheng. Aminolysis of Esters Using Quaternary Ammonium Salts as Amine Sources via Twice C-N Bond Activations[J]. Chinese Journal of Organic Chemistry,
2017, 37(5): 1220-1230.
doi:
10.6023/cjoc201612014

季铵盐作为胺源经由两次C-N键活化的酯的胺解反应
English
Aminolysis of Esters Using Quaternary Ammonium Salts as Amine Sources via Twice C-N Bond Activations
Abstract:
Catalyzed by supported palladium nanoparticles, an aminolysis reaction between various aryl esters and quaternary ammonium salts via twice C-N bond activations has been developed for selectively synthesis of amides. The Pd/γ-Al2O3 catalyst exhibited an excellent catalytic activity and reusability of at least five recycles in air for the reaction. The experiment results indicated that the first C-N cleavage of quaternary ammonium salt affords the tertiary amine and halohydrocarbon, and the second C-N cleavage proceeds via the formation of an iminium intermediate.
-
Key words:
- quaternary ammonium salts
- / aminolysis
- / palladium
- / C-N activation
- / aryl esters
-
The amide bond is one of the most abundant functional groups contained in many natural products, polymers, pharmaceuticals, biological systems and fine chemicals.[1] Owing to the importance of amide bonds, intensive efforts have been made to develop methods for the construction of the amide bonds. The most common routes toward amides are the aminolysis reactions of amines with the carboxylic acids (or their derivatives). Following the development of catalytic chemistry, using primary and secondary amines as amine sources various oxidative aminolysis reactions of aldehydes, [2] ketones[3] and alcohols[4] to synthesize amides were reported. In recent years, transition-metal catalyzed C—N bond activation or cleavage is becoming a hot topic.[5] Catalyzed by transition metal catalysts, tertiary amines and formamides were also served as amine sources to couple with esters, [6] aldehydes, [7] alcohols, [8] toluenes[9] or methylamines[10] to form amides via C—N bond activation. Despite the utility of such processes, there is still a great demand for the development of milder, environmentally friendly and efficient amine sources to access these biologically important amide scaffolds.
The quaternary ammonium salt was commonly used as phase transfer catalyst[11] and additive[12] to promote the organic reaction. Presently, the metal-free radical-initiated catalytic system "n-Bu4NI-TBHP" has rapidly emerged as a very active area of research.[13] But in fact, the quaternary ammonium salt is one of the most common N-containing substrates in organic synthesis (Scheme 1).[14] The aryltri-methylammonium salts could be applied to Kumada-Corriu cross-coupling, [15] Suzuki cross-coupling, [16] Negi-shi cross-coupling, [17] and Buchwald-Hartwig amina-tion.[18] Meanwhile, the quaternary ammonium halides could be applied to the catalytic carbonylation for the synthesis of tertiary amides in the presence of Pd catalysts and CO, but the substrates scope was limited in methylammonium halides.[19] These transformations share a common mechanism: an oxidative addition of the transition metalcatalyst to the C—N bond cleaves the C—N bond of quaternary ammonium salts and affords the active intermediate and tertiary amine. Our previous work has confirmed that aryl esters can generate an activated acyl intermediate via C—O bond activation and perform aminolysis reaction with tertiary amines to form amides under Pd(OAc)2 catalyzed conditions (Scheme 1).[6] Based on these developments, we envisioned that if the tertiary amines generated by C—N bond activation of quaternary ammonium salts in situ might react with esters, amide formation is expected. Given that quaternary ammonium salts are more suitable for aqueous system than other amines, the synthesis of amides from quaternary ammonium salts offers new opportunities for amide bond formation. Herein, we report the first example of a supported PdNPs catalytic system for the aminolysis of aryl esters using quaternary ammonium salts as amine sources.
Figure Scheme 1.
Application of quaternary ammonium salt
1 Results and discussion
Our initial attempt was executed toward the aminolysis of perfluorophenyl furan-2-carboxylate (1a) using tetrabutylammonium iodide (TBAI, 2a) as amine source, chlorobenzene (PhCl) as solvent, and 3 wt% Pd/γ-Al2O3 as catalyst. To our delight, after 24 h of refluxing in the solvent of PhCl in the presence of 3 wt% Pd/γ-Al2O3, the conversion of perfluorophenyl furan-2-carboxylate (1a) was above to 88%, the selectivity of desired product N, N-dibutylfuran-2-carboxamide (3aa) was above to 99% (Table 1, Entry 1). Other PdNPs on different supports including Nd2O3, Sm2O3, ZrO2 and SiO2 were also evaluated (Entries 2~5). These catalysts exhibited much lower catalytic activity compared with 3 wt% Pd/γ-Al2O3 due to the higher surface area and open porosity with γ-Al2O3.[20] We also examined the effect of different Pd loadings on the reaction, and 3 wt% Pd/γ-Al2O3 was the most effective catalyst (Entries 6, 7). Other solvents, such as ethylbenzene, isopropylbenzene and p-xylene led to lower yields of amide 3aa (Entries 8~10). Because that the quaternary ammonium salts were the common phase transfer catalysts, the feasibility of the catalytic aminolysis reaction in water was detected. Disappointedly, no product can be observed from the reaction (Entry 11). But when two-phase solvent [V(PhCl):V(H2O)=2.5:1] was used in the aminolysis reaction, the conversion of 1a reached 73%. This result indicated that the aminolysis reaction suit for heterogeneous aqueous system and quaternary ammonium salts would be more attractive amine sources than other amines. Both a decrease and an increase in the reaction temperature (100 and 130 ℃) reduced the conversion of 2-phenylpyridine (Entries 13, 14). To compare homogeneous-and heterogeneous-catalyzed systems, PdCl2, PdCl2/L-lysine and Pd(O-Ac)2 were used as the catalyst to perform the aminolysis reaction. The experiment results showed that the TON with the homogeneous catalyzed reaction system was much less than that with supported PdNPs (Entries 15~17). It was noteworthy that no reaction was observed in the absence of palladium under otherwise identical conditions (Entry 18).
 Table 1.
Optimization of reaction conditionsa
Table 1.
Optimization of reaction conditionsa

Entry Catalyst Solvent Conv./% Select./% TON 1 3wt% Pd/γ-Al2O3 PhCl 88 99 31 2 3wt% Pd/Nd2O3 PhCl 24 76 8 3 3wt% Pd/Sm2O3 PhCl 44 91 15 4 3wt% Pd/ZrO2 PhCl 72 88 25 5 3wt% Pd/SiO2 PhCl 50 92 17 6 5wt% Pd/γ-Al2O3 PhCl 75 91 26 7 1wt% Pd/γ-Al2O3 PhCl 45 91 16 8 3wt% Pd/γ-Al2O3 Ethylbenzene 52 86 18 9 3wt% Pd/γ-Al2O3 Isopropylbenzene 42 89 14 10 3wt% Pd/γ-Al2O3 p-Xylene 31 79 10 11b 3wt% Pd/γ-Al2O3 Water — — — 12 3wt% Pd/γ-Al2O3 V(PhCl):V(water)=2.5:1 73 99 13c 3wt% Pd/γ-Al2O3 PhCl 47 97 16 14d 3wt% Pd/γ-Al2O3 PhCl 76 95 27 15 PdCl2 PhCl 85 99 8.5 16 PdCl2/L-Lysine PhCl 80 99 8 17e Pd(OAc)2 PhCl 60 93 6 a Reaction conditions: 1a (0.2 mmol), 2a (2 equiv.), catalyst (20 mg), air, 120℃, 24 h. Conversion and selectivity were based on 1a, determined by GC. b Water (100 ℃). c The reaction temperature was 100 ℃. d The reaction temperature was 130 ℃. e Catalyst (10 mol%). Table 1. Optimization of reaction conditionsaWith the optimized condition in hand (Table 1, Entry 1), the aminolysis reaction has been extended to the various aryl esters and quaternary ammonium salts in the presence of 3 wt% Pd/γ-Al2O3. As shown in Table 2, every perfluorophenyl carboxylate, including picolinate, pyrazine-2-carboxylate, indole-2-carboxylate, benzoate and naphthoate, reacts with TBAI 2a to give the corresponding amide in 45%~86% yields (Entries 1~5). Our experiment indicated that the aminolysis reaction has good generality and tolerates various functional groups, such as cyano-, bromo-and nitro-groups, and compared with perfluorophenyl benzoate (1e) these aryl esters which bear electron-withdrawing groups on the benzoyl part increased the yield of the amides (Entries 6~8). Excitingly, perfluorophenyl 2-(1H-indol-2-yl)acetate (1j), which is classified as fatty acid ester, is still very effective in our catalytic system and gives a good result (Entry 9). Next, the scope of this reaction was investigated with respect to multiple quaternary ammonium salts under the optimized conditions (Table 2, Entries 10~17). The experiment results showed that tetrabutylammonium bromide (2b) and tetrabutyl-ammonium chloride (2c) both performed the aminolysis reaction with 1b to afford the same product as TBAI in 73% and 61% yields, respectively (Entries 10, 11). Obviously, with the strengthening of ionic bond, the C—N bond activation becomes much more difficult. Compared with halide salts, tetrabutylammonium hydrogen sulphate affords the lowest yield of product 3ba. The regioselectivity was observed in the reaction of 1b with benzyltributyl-ammonium bromide (2e) to exclusively afford the product 3ba without other amides (Entry 13). Likewise, the benzyltriethylammonium bromide (2f) exclusively affords the product N, N-diethylpicolinamide (3bf) in higher yield than tetraethylammonium iodide (2h) (Entries 14, 15). These results indicated that C(benzyl)—N bond is more easily to cleavage than C(alkyl)—N bond in quaternary ammonium salts. But when tetramethylammonium iodide (2i) was used instead of TBAI to react with 1b, no product was isolated (Entry 17). According the references, [15~18] the aryltrimethylammonium salts always give the Ar-R products in cross-coupling reactions. These results indicated that the C(methyl)—N bond of quanternary ammonium salts is more stable, so the twice C—N bond cleavages of tetramethylammonium iodide 2i could be hard to achieve.
Table 2.
Substrate scope of various perfluorophenyl carboxylates 1 with different quaternary ammonium salts 2a

Entry Ester Quaternary ammonium salt Product Yieldb/% 1 
2a 
86 2 2a 58 3 2a 72 4 2a 46 5 2a 45 6 2a 83 7 2a 81 8 2a 65 9 2a 69 10 1b 
3ba 73 11 1b 3ba 61 12 1b 3ba 31 13 1b 3ba 63 14 1b 
53 15 1b 54 16 1b 3bf 46 a Reaction conditions: 1 (0.2 mmol), 2 (2 equiv.), 3 wt% Pd/γ-Al2O3 (20 mg), air, 120 ℃, 24 h. bIsolated yield. Table 2. Substrate scope of various perfluorophenyl carboxylates 1 with different quaternary ammonium salts 2aTo demonstrate the synthetic utility of the method, other aryl or alkyl carboxylates were used instead of perfluorophenyl carboxylate to react with TBAI 2a. As shown in Table 3, although the 2, 3-dichlorophenyl picolinate (1k) and 4-chlorophenyl picolinate (1l) also underwent this aminolysis reaction, but the yields were decreased with the decreasing of electron-withdrawing effect in aryl part of ester. When 2, 3-dichlorophenyl furan-2-carboxylate (1m) and 4-chlorophenyl furan-2-carboxylate (1n) were used as the ester source to compare with perfluorophenyl furan-2-carboxylate (1a), the same result was obtained. Interestingly, quinolin-6-yl picolinate (1p) could also undergo the aminolysis reaction smoothly to afford a higher yield of product compared with naphthalen-2-yl picolinate (1o). But when ethyl picolinate 1r reacts with 2a, no product was detected. Even if strong electron deficient alkyl ester, such as 2, 2, 2-trichloroethyl picolinate (1s) and 2, 2, 2-tri-fluoroethyl picolinate (1t), also could not perform the aminolysis reaction. The result indicated that benzene ring in carboxylates 1 is necessary in the aminolysis reaction, and could control the acyl C—O bond cleavage with the aid of palladium. Correlating DFT calculation of Pd-cata-lyzed C(acyl)—O bond activation of aryl ester has been reported recently by Newman et al.[21]
Table 3.
Substrate aryl group of ester 1a
 Table 3. Substrate aryl group of ester 1a
Table 3. Substrate aryl group of ester 1aTo obtain good knowledge about the catalyst, the fresh and used (recovered after 5th cycle) PdNPs on γ-Al2O3catalysts were comparative studied by various characterization methods. X-ray photoelectron spectroscopy (XPS) measurement is the most widely employed technique to reveal information about the surface oxidation states of metal components.[22] The XPS analysis of the fresh and used catalysts showed that PdNPs on the support exist in the metallic state, which was confirmed by XPS signal appeared at binding energies for 335.27 eV of Pd 3d5/2 and 340.62 eV of Pd 3d3/2 over fresh catalyst, and 335.22 eV of Pd 3d5/2 and 340.47 eV of Pd 3d3/2 over used catalyst (Figure 1a). The binding energies of Pd0 in the literature are 335.20 eV of Pd 3d5/2 and 340.50 eV of Pd 3d3/2, respectively, whereas the values of PdⅡ are 336.70 eV of Pd 3d5/2 and 342.00 eV of Pd 3d3/2, respectively.[23] It is shown that Pd0 as the active center completes the catalytic cycle. The XRD patterns of the fresh and used Pd/γ-Al2O3 catalysts are shown in Figure 1b. No evident peaks from palladium cystals were observed in all samples compared with γ-Al2O3. It is shown that PdNPs disperse uniformly on γ-Al2O3.
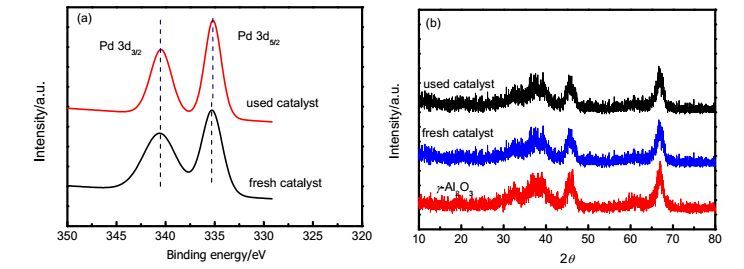 Figure 1.
XPS spectra (a) and XRD patterns (b) of fresh and used 3 wt% Pd/γ-Al2O3
Figure 1.
XPS spectra (a) and XRD patterns (b) of fresh and used 3 wt% Pd/γ-Al2O3
The particle size and surface morphology of the fresh and used 3 wt% Pd/γ-Al2O3catalysts was studied by trans-mission electron microscopy (TEM) (Figure 2). It can be seen that there is uniform dispersion of Pd nanoparticles on the support. The mean diameters of the PdNPs are 3.49 nm for the fresh catalyst and 4.06 nm for the used catalyst, respectively. The used catalyst had a slightly larger Pd particle size, but PdNPs still distributed evenly on the γ-Al2O3 surface. No apparent agglomeration was observed.
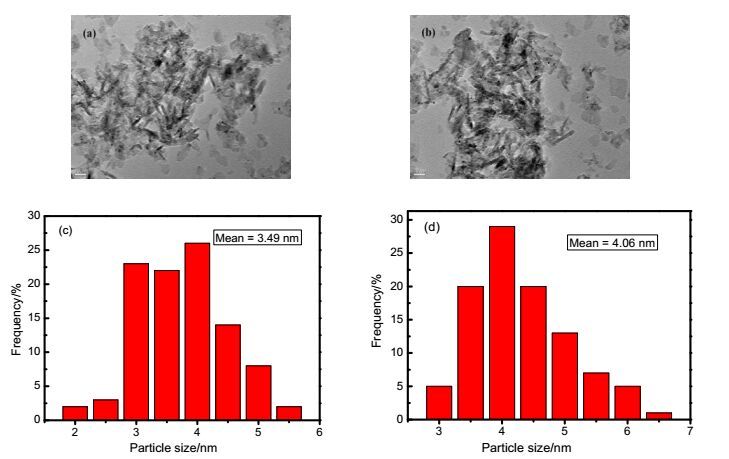 Figure 2.
TEM images of fresh 3 wt% Pd/γ-Al2O3 (a) and used 3 wt% Pd/γ-Al2O3 (b), respectively, and PdNP size distributions of fresh (c) and used catalysts (d)
Figure 2.
TEM images of fresh 3 wt% Pd/γ-Al2O3 (a) and used 3 wt% Pd/γ-Al2O3 (b), respectively, and PdNP size distributions of fresh (c) and used catalysts (d)
The Brauner-Emmet-Teller (BET) specific surface areas of the catalysts were derived from N2 physical sorption data of the samples using the BET model (Table 4). The γ-Al2O3 is a kind of support which has a large specific surface area, and it does not cause obvious change in the specific surface area of these catalysts after loading a small amount of the PdNPs. The amounts of Pd loading in the samples were determined by atomic absorption spectrophotometer (AAS), and the Pd content of the fresh catalyst was approximately 3 wt%. We did note a slight decrease in the Pd content after being cycled 5 times (2.56%, Table 4), which can decrease the catalytic activity on the basis of available Pd on the support surface.
Table 4.
Characterization results of BET and AAS of different Pd/γ-Al2O3 catalysts
Sample SBET/(m2•g-1) Pd loading/wt% γ-Al2O3 101 — 3 wt% Pd/γ-Al2O3 (fresh) 112 2.91 Table 4. Characterization results of BET and AAS of different Pd/γ-Al2O3 catalystsThe stability and reusability of the catalyst was investigated in the reaction of 1a and 2a as provided in the Experimental section. As shown in Figure 3, the catalyst can be reused for five cycles with 25% decline of activity after the 5th recycle. The decreased catalytic activity of the PdNPs may result from the slight increase in the average size of the particles (Figure 2d) and a slight decrease in the Pd content (Table 4) recovered after the 5th cycle.
 Figure 3.
Reusability of catalyst
Figure 3.
Reusability of catalyst
To identify which was formed by the cleavage of the C—N bond of quaternary ammonium salt and which was formed by the cleavage of the C—O bond of esters, the reaction solution of 1a with 2a was detected with GCMS after the end of reaction (see the Supporting Information). The GCMS data showed that along with the desired product 3aa, pentafluorophenol and tributylamine were obtained, which indicated that the alkoxyl group of esters eliminated to phenols and the C—N bond cleavage of quaternary ammonium salt affords the tertiary amine in this reaction. The GCMS analysis of the reaction solution of 1b with benzyltributylammonium bromide (2e) (the higher molecular weight quaternary ammonium salt) showed that not only benzyl bromide and tributylamine were obtained but also a smaller amount of benzaldehyde and N-benzyl-N-butylbutan-1-amine was obtained (Scheme 2 and the Supporting Information). These results indicated that the first C—N cleavage of quaternary ammonium salt affords the tertiary amine and halohydrocarbon, [19] and the second C—N cleavage proceeds via the formation of an iminium intermediate, which would cause formation of an aldehyde.[6b, 24]
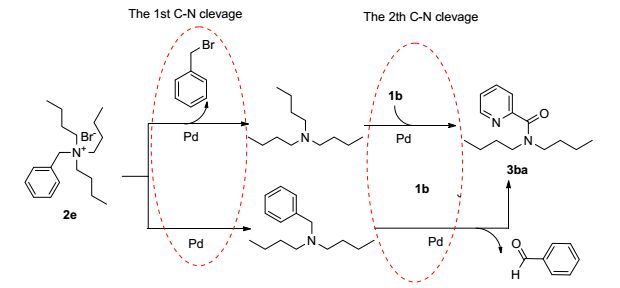 Figure Scheme 2.
GCMS analysis of the reaction solution of 1b with 2e
Figure Scheme 2.
GCMS analysis of the reaction solution of 1b with 2e
In order to prove whether the present aminolysis proceeded with a heterogeneous catalyst or homogeneous catalyst, the hot filtration test of the model reaction mixture was executed. GC data are given in Table 5. The conversion of 1a and TOF value indicated that after filtration the substrate turnover ceases. This is intended to preclude the possibility of homogeneous catalysis.
Table 5.
Results of hot filtration test
Time/h Conv.a/% TOF 0 0 0 2 27.21 4.80 aGC data. Table 5. Results of hot filtration testOn the basis of the experiments and previously reported literatures, the possible mechanism and intermediates I—V in the aminolysis pathway are postulated as follows (Scheme 3). Initially, Pd0coordinates with aryl ester 1 to form intermediate Ⅰ. Then palladium performs an oxidative addition with the acyl C—O bond in aryl ester 1, generating a PdⅡ intermediate Ⅱ. And then, the tertiary amine readily prepared via C—N activation of quaternary ammonium salt 2 catalyzed by Pd0, coordinates to Ⅱ to give intermediate Ⅲ. Subsequently, the alkoxide attack on the alpha position of the tertiary amine, results in the phenol, with generating iminium type intermediate Ⅳ via the general accepted mechanism.[25]Intermediate Ⅳ is then hydrolyzed to be converted into the intermediate V by elimination of aldehyde.[26] Reductive elimination of V results in the desired tertiary amide 3 and regenerates Pd0 to complete the catalytic cycle.
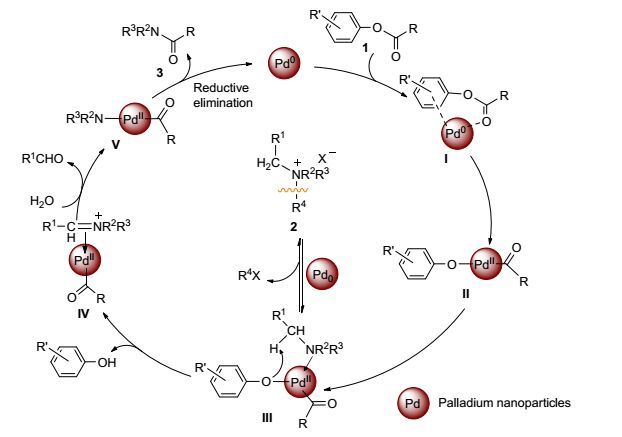 Figure Scheme 3.
Plausible reaction mechanism
Figure Scheme 3.
Plausible reaction mechanism
2 Conclutions
In conclusion, we report a supported PdNPs catalysis system for the aminolysis of aryl esters using quaternary ammonium salts as amine sources via twice C—N bond activations. Using 3% Pd/γ-Al2O3 as heterogeneous cata-lyst, a broad scope of aryl esters, including furan-2-car-boxylate, picolinate, pyrazine-2-carboxylate, indole-2-carboxylate, 2-(indol-2-yl) acetate, benzoate and naphthoate, react with various quaternary ammonium salts to synthesize the corresponding amides in yields up to 87%. Also, electron-withdrawing groups in either the alkyl part or the acyl part of the esters are more beneficial for this reaction. In addition, the catalyst can be readily recovered and reused for five cycles with only 25% decline of activity after the 5th recycle. The hot filtration test suggests that the present reaction would proceed via heterogeneous catalytic process. Compared with homogeneous catalysis, the reaction is more efficient and green, especially suited for heterogeneous aqueous catalytic system. This study not only increased our understanding of the character of C—N bond activation of quaternary ammonium salts but also shed important light on how to further expand its scope and utility.
3 Experimental section
3.1 Catalyst preparation
The PdNPs on γ-Al2O3 and other supports were prepared by a modified impregnation-reduction method.[27] For example, 3 wt% Pd/γ-Al2O3 catalyst was prepared by the following procedure: 0.97 g of γ-Al2O3 powder was suspended in 50 mL of distilled water, followed by adding 28.2 mL of 0.01 mol/L of PdCl2 aqueous solution and stirred at room temperature. Then 1 mL of 0.03 mol/L of L-lysine aqueous solution was adding into the mixture above. Subsequently, 0.1 mol/L NaOH aqueous solution was added into the mixture to adjust the pH to 7. Whereafter, 4.5 mL of 0.35 mol/L NaBH4 aqueous solution was added dropwise within 10 min while vigorous stirring. The mixture was aged for 24 h and the solid was separated by centrifuging, washed with distilled water for 4 times and ethanol for once, and dried at 80 ℃. The dried solid was used directly as the catalyst.
3.2 Catalyst characterization
The transmission electron microscopy (TEM) images of catalysts were obtained on a Tecnai G2 F20 microscope operating at 200 kV. Samples were treated by ultrasonic dispersion in ethanol and deposited on a copper grid coated with a carbon layer for analysis. X-ray photoelectron spectroscopy (XPS) measurements of the catalysts were carried out on a Thermo ESCALAB 250Xi spectrometer using monochromatic Al Kα radiation of energy 1486.6 eV. The binding energy (BE) was acquired by utilizing C1s line as a reference with energy of 284.8 eV. X-ray powder diffraction (XRD) patterns were measured on a Rigaku Ultima ⅣX-ray diffractometer operating at 40 kV and 40 mA by using a Cu Kα radiation source and scanned at a rate of 8 /min over the range 10°~80°. Brunauer-Emmett-Teller (BET) surface areas of the samples were measured by nitrogen sorption using a ASAP-2020 accelerated surface area and a porosity analyzer of American Micromeritics Company. The exact Pd loading was recorded on a Z-8000-type polarized Zeeman atomic absorption spectrophotometer (AAS) of Hitachi Company.
3.3 Preparation of esters 1a~1r
A mixture of carboxylic acid (10 mmol), phenol or pyridine-2-ol (10 mmol), 4-(dimethylamino)pyridine (DMAP, 1 mmol) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiamide hydrochloride (EDC•HCl, 10 mmol) in THF (50 mL) was stirred overnight at 25 ℃. The resulting mixture was filtered and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography [silica gel, V(ethyl ether):V(petroleum ether)=1:2~1:5 as eluent], affording a corresponding aryl ester 1a~1r.
3.4 Aminolysis reaction for synthesis of versatile amides
The general procedure for the aminolysis reaction was as follows: The catalyst (20 mg), aryl ester 1 (0.2 mmol), quaternary ammonium salts 2 (0.4 mmol) and chlorobenzene (2.0 mL) were added into the reaction tube. The reaction mixture was stirred at 120 ℃ for 24 h. After cooling to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography [silica gel, V(ethyl acetate):V(petroleum ether)=1:2~1:5 as the eluent] to afford the desired amide 3.
N, N-Dibutylfuran-2-carboxamide (3aa)[28]: Yield 87% (38.8 mg). 1H NMR (500 MHz, CDCl3) δ: 7.45 (s, 1H), 6.99 (d, J=3.4 Hz, 1H), 6.46 (dd, J=3.3, 1.7 Hz, 1H), 3.61~3.32 (m, 4H), 1.74~1.53 (m, 4H), 1.44~1.28 (m, 4H), 0.93 (t, J=7.4 Hz, 6H); MS (ESI) m/z: 265.25 [M+H+CH3CN]+.
N, N-Dibutylpicolinamide (3ba)[29]: Yield 86% (40.2 mg). 1H NMR (500 MHz, CDCl3) δ: 8.58 (d, J=4.5 Hz, 1H), 7.83~7.73 (m, 1H), 7.56 (d, J=7.8 Hz, 1H), 7.32 (dd, J=7.0, 5.4 Hz, 1H), 3.50 (t, J=8.0 Hz, 2H), 3.33 (t, J=8.0 Hz, 2H), 1.74~1.62 (m, 2H), 1.59~1.45 (m, 2H), 1.45~1.37 (m, 2H), 1.19~1.05 (m, 2H), 0.97 (t, J=7.4 Hz, 3H), 0.77 (t, J=7.4 Hz, 3H); MS (ESI) m/z: 235.32 [M+H]+.
N, N-Dibutylpyrazine-2-carboxamide (3ca)[30]: Yield 58% (27.3 mg). 1H NMR (500 MHz, CDCl3) δ: 8.87 (d, J=1.4 Hz, 1H), 8.61 (d, J=2.5 Hz, 1H), 8.54 (dd, J=2.5, 1.6 Hz, 1H), 3.52 (t, J=7.7 Hz, 2H), 3.35 (t, J=7.8 Hz, 2H), 1.72~1.63 (m, 2H), 1.62~1.51 (m, 2H), 1.46~1.36 (m, 2H), 1.22~1.11 (m, 2H), 0.98 (t, J=7.4 Hz, 3H), 0.81 (t, J=7.4 Hz, 3H).
N, N-Dibutyl-1H-indole-2-carboxamide (3da)[31]: Yield 72% (39.2 mg). 1H NMR (500 MHz, CDCl3) δ: 9.81 (s, 1H), 7.68 (d, J=8.0 Hz, 1H), 7.46 (d, J=8.2 Hz, 1H), 7.28 (dd, J=10.6, 3.4 Hz, 1H), 7.14 (t, J=7.5 Hz, 1H), 6.78 (d, J=1.5 Hz, 1H), 3.65 (d, J=60.9 Hz, 4H), 1.75 (s, 4H), 1.44 (dd, J=14.6, 7.3 Hz, 4H), 1.00 (t, J=6.8 Hz, 6H).
N, N-Dibutylbenzamide (3ea)[32]: Yield 46% (21.4 mg). 1H NMR (500 MHz, CDCl3) δ: 7.43~7.30 (m, 5H), 3.49 (s, 2H), 3.18 (s, 2H), 1.64 (s, 2H), 1.55~1.32 (m, 4H), 1.13 (d, J=5.9 Hz, 2H), 0.98 (s, 3H), 0.78 (s, 3H); MS (ESI) m/z: 234.25 [M+H]+.
N, N-Dibutyl-1-naphthamide (3fa)[33]: Yield 45% (25.5 mg). 1H NMR (500 MHz, CDCl3) δ: 7.89~7.83 (m, 2H), 7.83~7.78 (m, 1H), 7.55~7.49 (m, 2H), 7.49~7.44 (m, 1H), 7.41~7.36 (m, 1H), 3.89~3.73 (m, 1H), 3.52~3.39 (m, 1H), 3.13~2.93 (m, 2H), 1.88~1.69 (m, 2H), 1.58~1.34 (m, 4H), 1.04 (t, J=7.3 Hz, 4H), 1.02~0.98 (m, 2H), 0.65 (t, J=7.4 Hz, 3H); MS (ESI) m/z: 284.25 [M+H]+.
N, N-Dibutyl-4-cyanobenzamide (3ga)[34]: Yield 83% (42.8 mg). 1H NMR (500 MHz, CDCl3) δ: 7.70 (d, J=8.2 Hz, 2H), 7.46 (d, J=8.2 Hz, 2H), 3.56~3.40 (m, 2H), 3.22~3.02 (m, 2H), 1.75~1.59 (m, 3H), 1.53~1.43 (m, 2H), 1.43~1.34 (m, 2H), 1.18~1.08 (m, 2H), 0.98 (t, J=7.3 Hz, 3H), 0.79 (t, J=7.3 Hz, 3H); MS (ESI) m/z: 259.25 [M+H]+.
N, N-Dibutyl-4-nitrobenzamide (3ha)[33]: Yield 81% (45.0 mg). 1H NMR (500 MHz, CDCl3) δ: 8.27 (d, J=8.7 Hz, 2H), 7.53 (d, J=8.7 Hz, 2H), 3.62~3.45 (m, 2H), 3.19~3.07 (m, 2H), 1.73~1.60 (m, 2H), 1.56~1.45 (m, 2H), 1.45~1.37 (m, 2H), 1.19~1.05 (m, 2H), 0.99 (t, J=7.4 Hz, 3H), 0.80 (t, J=7.3 Hz, 3H); MS (ESI) m/z: 279.20 [M+H]+.
4-Bromo-N, N-dibutylbenzamide (3ia)[34]: Yield 65% (40.6 mg). 1H NMR (500 MHz, CDCl3) δ: 7.52 (d, J=8.3 Hz, 1H), 7.23 (d, J=8.3 Hz, 2H), 3.47 (t, J=6.3 Hz, 2H), 3.17 (t, J=6.1 Hz, 2H), 1.76~1.53 (m, 1H), 1.53~1.33 (m, 4H), 1.20~1.05 (m, J=6.8 Hz, 2H), 0.97 (t, J=5.6 Hz, 3H), 0.80 (t, J=6.6 Hz, 3H); MS (ESI) m/z: 312.30 [M+H]+.
N, N-Dibutyl-2-(1H-indol-2-yl)acetamide (3ja): Yellow solid. Yield 69% (39.5 mg). 1H NMR (500 MHz, CDCl3) δ: 8.36 (s, 1H), 7.60 (d, J=7.9 Hz, 1H), 7.35 (d, J=8.1 Hz, 1H), 7.23~7.16 (m, 1H), 7.15~7.09 (m, 1H), 7.07 (d, J=2.2 Hz, 1H), 3.82 (d, J=0.4 Hz, 2H), 3.42~3.32 (m, 2H), 3.32~3.21 (m, 2H), 1.60~1.46 (m, 4H), 1.35~1.26 (m, 4H), 0.95~0.89 (m, 6H); 13C NMR (126 MHz, CDCl3) δ: 171.5, 136.4, 127.2, 122.9, 121.7, 119.1, 118.4, 111.5, 109.0, 48.3, 45.9, 31.3, 29.8, 25.8, 20.3, 20.1, 13.9, 13.7; MS (ESI) m/z: 285.15 [M-H]-. Anal. calcd for C18H26N2O: C 75.48, H 9.15, N 9.78; found C 75.47, H 9.20, N 9.69.
N, N-Diethylpicolinamide (3bf)[35]: Yield 53% (18.9 mg). 1H NMR (500 MHz, CDCl3) δ: 8.61 (d, J=4.0 Hz, 1H), 7.85 (t, J=7.6 Hz, 1H), 7.59 (d, J=7.8 Hz, 1H), 7.42~7.32 (m, 1H), 3.59 (q, J=7.1 Hz, 2H), 3.37 (q, J=7.0 Hz, 2H), 1.29 (t, J=7.2 Hz, 3H), 1.16 (t, J=7.1 Hz, 3H); MS (ESI) m/z: 179.13 [M+H]+.
N, N-Dipropylpicolinamide (3bg)[36]: Yield 54% (22.2 mg). 1H NMR (500 MHz, CDCl3) δ: 8.57 (d, J=4.6 Hz, 1H), 7.77 (td, J=7.7, 1.7 Hz, 1H), 7.55 (d, J=7.8 Hz, 1H), 7.35~7.29 (m, 1H), 3.47 (t, J=7.5 Hz, 2H), 3.30 (t, J=7.7 Hz, 2H), 1.79~1.64 (m, 2H), 1.64~1.50 (m, 2H), 0.98 (t, J=7.4 Hz, 3H), 0.73 (t, J=7.4 Hz, 3H); MS (ESI) m/z: 207.30 [M+H]+.
3.5 Catalysts recycle experiment
After each reaction cycle, the solvent, substrate and products were removed by centrifugation. The separated catalyst was washed thoroughly with 0.1 mol/L NaOH ethanol solution (twice) and distilled water (4 times), and then washed twice with ethanol, followed by centrifugal separation and drying at 80 ℃ for 12 h. The recovered catalyst was used for the next cycle.
3.6 Hot filtration test
The hot filtration test is performed by filtering the model reaction mixture through a preheated Celite pad after the reaction for 2 h. The filtrate was detected by GC to obtain the conversion of 1a and TON. Then the filtered reaction solution continued to react for 2 h under normal conditions, and was detected by GC again.[37]
Supporting Information The GCMS analysis of reactions of 1a with 2a and 1b with 2e, 1H NMR, 13C NMR and MS spectra of products. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
-
-
[1]
(a) Pattabiraman, V. R. ; Bode, J. W. Nature 2011, 480, 471. (b) Allen, C. L. ; Williams, J. M. J. Chem. Soc. Rev. 2011, 40, 3405. (c) Valeur, E. ; Bradley, M. Chem. Soc. Rev. 2009, 38, 606.
-
[2]
Bai, C. H.; Yao, X. F.; Li, Y. W. ACS Catal. 2015, 5, 884. doi: 10.1021/cs501822r
-
[3]
(a) Nekkanti, S. ; Veeramani, K. ; Kumar, N. P. ; Shankaraiah, N. Green chem. 2016, 18, 3439. (b) Wu, K. ; Huang, Z. L. ; Ma, Y. Y. ; Lei, A. W. RSC Adv. 2016, 6, 24349.
-
[4]
Arefi, M.; Saberi, D.; Karimi, M.; Heydari, A. ACS Comb. Sci. 2015, 17, 341. doi: 10.1021/co5001844
-
[5]
Ouyang, K.; Hao, W.; Zhang, W. X.; Xi, Z. Chem. Rev. 2015, 115, 12045. doi: 10.1021/acs.chemrev.5b00386
-
[6]
(a) Bao, Y. S. ; Baiyin, M. ; Agula, B. ; Jia, M. L. ; Bao, Z. J. Org. Chem. 2014, 79, 6715. (b) Bao, Y. S. ; Bao, Z. ; Agula, B. ; Baiyin, M. ; Jia, M. L. J. Org. Chem. 2014, 79, 803.
-
[7]
(a) Li, Y. M. ; Jia, F. ; Li, Z. P. Chem. -Eur. J. 2012, 18, 5150. (b) Porcheddu, A. ; Luca, L. D. Adv. Synth. Catal. 2012, 354, 2949.
-
[8]
Xu, K.; Hu, Y. B.; Zhang, S.; Zha, Z. G.; Wang, Z. Y. Chem.-Eur. J. 2012, 18, 9793. doi: 10.1002/chem.v18.32
-
[9]
Du, B.; Sun, P. Sci. China Chem. 2014, 57, 1176.
-
[10]
Gao, L.; Tang, H.; Wang, Z. Chem. Commun. 2014, 50, 4085. doi: 10.1039/c4cc00621f
-
[11]
(a) Cassar, L. ; Foa, M. ; Gardano, A. J. Organomet. Chem. 1976, 121, C55. (b) Bhardwaj, M. ; Sahi, S. ; Mahajan, H. ; Paul, S. ; Clark, J. H. J. Mol. Catal. A: Chem. 2015, 408, 48. (c) Zhang, J. T. ; Li, D. Y. ; Chen, H. ; Wang, B. J. ; Liu, Z. X. ; Zhang, Y. H. Adv. Synth. Catal. 2016, 358, 792.
-
[12]
(a) Wang, X. ; Zhu, L. Z. ; Chen, S. H. ; Xu, X. H. ; Au, C. T. ; Qiu, R. Org. Lett. 2015, 17, 5228. (b) Lin, C. L. ; Li, D. ; Wang, Y. B. ; Yao, J. Z. ; Zhang, Y. H. Org. Lett. 2015, 17, 1328. (c) Fabrizi, G. ; Goggiamani, A. ; Sferrazza, A. ; Cacchi, S. Angew. Chem. , Int. Ed. 2010, 49, 4067. (d) Thirupathi, N. ; Puri, S. ; Reddy, T. J. ; Sridhar, B. ; Reddya, M. S. Adv. Synth. Catal. 2016, 358, 303. (e) Xu, P. ; Han, F. S. ; Wang, Y. H. Adv. Synth. Catal. 2015, 357, 3441. (f) Gadge, S. T. ; Khedkar, M. V. ; Lanke, S. R. ; Bhanage, B. M. Adv. Synth. Catal. 2012, 354, 2049.
-
[13]
(a) Kim, H. J. ; Kim, J. ; Cho, S. H. ; Chang, S. J. Am. Chem. Soc. 2011, 133, 16382. (b) Xue, Q. C. ; Xie, J. ; Li, H. M. ; Cheng, Y. X. ; Zhu, C. J. Chem. Commun. 2013, 49, 3700. (c) Zhao, D. ; Wang, T. ; Shen, Q. ; Li, J. X. Chem. Commun. 2014, 50, 4302. (d) Wei, W. ; Zhang, C. ; Xu, Y. ; Wan, X. B. Chem. Commun. 2011, 47, 10827. (e) Liu, L. H. ; Yun, L. ; Wang, Z. K. ; Fu, X. F. ; Yan, C. H. Tetrahedron Lett. 2013, 54, 5383. (f) Li, D. J. ; Yang, T. H. ; Su, H. L. ; Yu, W. Adv. Synth. Catal. 2015, 357, 2529. (g) Hao, W. J. ; Du, Y. ; Wang, D. ; Jiang, B. ; Gao, Q. ; Tu, S. J. ; Li, G. G. Org. Lett. 2016, 18, 1884. (h) Zhang, H. ; Dong, D. Q. ; Hao, S. H. ; Wang, Z. L. RSC Adv. 2016, 6, 8465. (i) Tan, B. ; Toda, N. ; Barbas, C. F. Angew. Chem. , Int. Ed. 2012, 51, 12538. (j) Sun, J. W. ; Wang, Y. ; Pan, Y. J. Org. Chem. 2015, 80, 8945. (k) Liu, Z. ; Zhang, J. ; Chen, S. ; Shi, E. ; Xu, Y. ; Wan, X. Angew. Chem. , Int. Ed. 2012, 51, 3231. (l) Uyanik, M. ; Okamoto, H. ; Yasui, T. ; Ishihara, K. Science 2010, 328, 1376.
-
[14]
(a) Hirao, T. ; Yamada, N. ; Ohshiro, Y. ; Agawa, T. J. Organomet. Chem. 1982, 236, 409. (b) Hosomi, A. ; Hoashi, K. ; Kohra, S. ; Tominaga, Y. ; Otaka, K. ; Sakurai, H. J. Chem. Soc. , Chem. Commun. 1987, 570.
-
[15]
(a) Wenkert, E. ; Han, A. -L. ; Jenny, C. -J. J. Chem. Soc. , Chem. Commun. 1988, 975. (b) Reeves, J. T. ; Fandrick, D. R. ; Tan, Z. ; Song, J. J. ; Lee, H. ; Yee, N. K. ; Senanayake, C. H. Org. Lett. 2010, 12, 4388.
-
[16]
Blakey, S. B.; MacMillan, D. W. C. J. Am. Chem. Soc. 2003, 125, 6046. doi: 10.1021/ja034908b
-
[17]
(a) Xie, L. -G. ; Wang, Z. -X. Angew. Chem. , Int. Ed. 2011, 50, 4901. (b) Zhang, X. -Q. ; Wang, Z. -X. J. Org. Chem. 2012, 77, 3658.
-
[18]
Zhang, X. Q.; Wang, Z.-X. Org. Biomol. Chem. 2014, 12, 1448. doi: 10.1039/c3ob41989d
-
[19]
(a) Lei, Y. ; Zhang, R. ; Wu, L. ; Wu, Q. ; Mei, H. ; Li, G. Appl. Organomet. Chem. 2014, 28, 310. (b) Lei, Y. ; Zhang, R. ; Wu, Q. ; Mei, H. ; Xiao, B. ; Li, G. J. Mol. Catal. A: Chem. 2014, 381, 120.
-
[20]
Rozita, Y.; Brydson, R.; Comyn, T. P.; Scott, A. J.; Hammond, C.; Brown, A.; Chauruka, S.; Hassanpour, A.; Young, N. P.; Kirkland, A. I. ChemCatChem 2013, 5, 2695. doi: 10.1002/cctc.v5.9
-
[21]
Halima, T. B.; Zhang, W.; Yalaoui, I.; Hong, X.; Yang, Y.; Houk, K. N.; Newman, S. G. J. Am. Chem. Soc. 2017, 139, 1311. doi: 10.1021/jacs.6b12329
-
[22]
Sun, J. W.; Fu, Y. S.; He, G. Y.; Sun, X. Q.; Wang, X. Catal. Sci. Technol. 2014, 4, 1742. doi: 10.1039/c4cy00048j
-
[23]
Pillo, T.; Zimmermann, R.; Steiner, P.; Hüfner, S. J. Phys.: Condens. Matter 1997, 9, 3987. doi: 10.1088/0953-8984/9/19/018
-
[24]
Murata, S.; Miura, M.; Nomura, M. J. Org. Chem. 1989, 54, 4700. doi: 10.1021/jo00280a049
-
[25]
(a) North, M. Angew. Chem. , Int. Ed. 2004, 43, 4126. (b) Murahashi, S. I. ; Komiya, N. ; Terai, H. ; Nakae, T. J. Am. Chem. Soc. 2003, 125, 15312. (c) Murahashi, S. I. ; Komiya, N. ; Terai, H. Angew. Chem. , Int. Ed. 2005, 44, 6931.
-
[26]
Guo, S. M.; Qian, B.; Xie, Y. J.; Xia, C. G.; Huang, H. M. Org. Lett. 2011, 13, 522. doi: 10.1021/ol1030298
-
[27]
Zhang, D. L.; Bao, Z.; Bao, Y. S. J. Phys. Chem. C 2015, 119, 20426. doi: 10.1021/acs.jpcc.5b04735
-
[28]
Naumov, Y. A.; Dremova, V. P.; Kost, A. N.; Mentus, A. N.; Smirnova, S. N. Tr. Vses. Nauch.-Issled. Inst. Dezinfek. Steriliz. 1970, 2, 24.
-
[29]
Houghton, R. P.; Williams, C. S. Tetrahedron Lett. 1967, 40, 3929.
-
[30]
Kushner, S.; Dalalian, H.; Sanjurjo, J. L.; BachJr, F. L.; Safir, S. R.; SmithJr, V. K.; Williams, J. H. J. Am. Chem. Soc. 1952, 74, 3617. doi: 10.1021/ja01134a045
-
[31]
周啓霆, 嵇汝运, 化学学报, 1962, 28, 236.Chow, C. T.; Chi, J. Y. Acta Chim. Sinica 1962, 28, 236 (in Chinese).
-
[32]
Wu, K.; Huang, Z. L.; Ma, Y. Y.; Lei, A. W. RSC Adv. 2016, 6, 24349. doi: 10.1039/C6RA02153K
-
[33]
Pathak, U.; Bhattacharyya, S.; Pandey, L. K.; Mathur, S.; Jain, R. RSC Adv. 2014, 4, 3900. doi: 10.1039/C3RA45781H
-
[34]
Fang, T.; Gao, X. H.; Tang, R. Y.; Zhang, X. G.; Deng, C. L. Chem. Commun. 2014, 50, 14775. doi: 10.1039/C4CC07378A
-
[35]
Bao, Y. S.; Bao, A.; Bao, Z.; Bai, Y.; Jia, M. J. Org. Chem. 2014, 79, 803. doi: 10.1021/jo4023974
-
[36]
Bhattacharya, A.; Plata, R. E.; Villarreal, V.; Muramulla, S.; Wu, J. Tetrahedron Lett. 2006, 47, 505. doi: 10.1016/j.tetlet.2005.11.063
-
[37]
Reay, A. J.; Fairlamb, I. J. S. Chem. Commun. 2015, 51, 16289. doi: 10.1039/C5CC06980G
-
[1]
-

Figure 2 TEM images of fresh 3 wt% Pd/γ-Al2O3 (a) and used 3 wt% Pd/γ-Al2O3 (b), respectively, and PdNP size distributions of fresh (c) and used catalysts (d)
Table 1. Optimization of reaction conditionsa
Entry Catalyst Solvent Conv./% Select./% TON 1 3wt% Pd/γ-Al2O3 PhCl 88 99 31 2 3wt% Pd/Nd2O3 PhCl 24 76 8 3 3wt% Pd/Sm2O3 PhCl 44 91 15 4 3wt% Pd/ZrO2 PhCl 72 88 25 5 3wt% Pd/SiO2 PhCl 50 92 17 6 5wt% Pd/γ-Al2O3 PhCl 75 91 26 7 1wt% Pd/γ-Al2O3 PhCl 45 91 16 8 3wt% Pd/γ-Al2O3 Ethylbenzene 52 86 18 9 3wt% Pd/γ-Al2O3 Isopropylbenzene 42 89 14 10 3wt% Pd/γ-Al2O3 p-Xylene 31 79 10 11b 3wt% Pd/γ-Al2O3 Water — — — 12 3wt% Pd/γ-Al2O3 V(PhCl):V(water)=2.5:1 73 99 13c 3wt% Pd/γ-Al2O3 PhCl 47 97 16 14d 3wt% Pd/γ-Al2O3 PhCl 76 95 27 15 PdCl2 PhCl 85 99 8.5 16 PdCl2/L-Lysine PhCl 80 99 8 17e Pd(OAc)2 PhCl 60 93 6 a Reaction conditions: 1a (0.2 mmol), 2a (2 equiv.), catalyst (20 mg), air, 120℃, 24 h. Conversion and selectivity were based on 1a, determined by GC. b Water (100 ℃). c The reaction temperature was 100 ℃. d The reaction temperature was 130 ℃. e Catalyst (10 mol%).  下载: 导出CSV
下载: 导出CSV
Table 2. Substrate scope of various perfluorophenyl carboxylates 1 with different quaternary ammonium salts 2a
Entry Ester Quaternary ammonium salt Product Yieldb/% 1 2a 86 2 2a 58 3 2a 72 4 2a 46 5 2a 45 6 2a 83 7 2a 81 8 2a 65 9 2a 69 10 1b 3ba 73 11 1b 3ba 61 12 1b 3ba 31 13 1b 3ba 63 14 1b 53 15 1b 54 16 1b 3bf 46 a Reaction conditions: 1 (0.2 mmol), 2 (2 equiv.), 3 wt% Pd/γ-Al2O3 (20 mg), air, 120 ℃, 24 h. bIsolated yield.
下载: 导出CSV
Table 4. Characterization results of BET and AAS of different Pd/γ-Al2O3 catalysts
Sample SBET/(m2•g-1) Pd loading/wt% γ-Al2O3 101 — 3 wt% Pd/γ-Al2O3 (fresh) 112 2.91
下载: 导出CSV
Table 5. Results of hot filtration test
Time/h Conv.a/% TOF 0 0 0 2 27.21 4.80 aGC data.
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 20
- 文章访问数: 1788
- HTML全文浏览量: 215
 下载:
下载:
