
图 1.
PNIPAM应用示意图
Figure 1.
Schematic representation of the application of PNIPAM

智能型高分子聚(N-异丙基丙烯酰胺)(PNIPAM)由于其对环境温度具有很好的响应性,故又称为温敏性高分子聚合物。PNIPAM拥有一个低临界溶解温度(LCST),在30~35℃之间[1, 2],并且在此范围内PNIPAM产生亲水/疏水间的可逆构象变化[3],进而发生相分离。若加以改性,可以得到拥有可调相转移温度的聚合物,从而进一步加强聚合物材料的温敏性能[4]。PNIPAM分子内含有酰胺基和异丙基两种基团,若温度在LCST以下,可以通过酰胺基与水的氢键使PNIPAM在水中稳定存在,水分子包围在疏水基团周围,此时PNIPAM具有亲水性,其在水溶液中为伸展态;逐渐升温,PNIPAM与水的水合氢键被削弱,分子内的异丙基之间的相互吸引力增强。若温度在LCST以上,氢键彻底断裂,自身内部形成氢键使内部结构坍塌,由伸展态转为球形结构[5~7],体积缩小约90%[8],此时PNIPAM具有疏水性。而当温度再度恢复到LCST以下时,PNIPAM也恢复为亲水性结构,故这种温敏性行为具有可逆性。这种特殊的材料在药物负载运输、基因传递、化学分析和表面浸润性等领域里引起了广泛兴趣。
活性可控自由基聚合(CRP)主要包括原子转移自由基聚合(ATRP)[9]、单电子转移活性自由基聚合(SET-LRP)、可逆加成-断裂链转移(RAFT)聚合[10]及电子转移催化剂再生活化原子转移自由基聚合(ARGET-ATRP)等。线性PNIPAM可通过CRP合成得到,并且制备的PNIPAM分子量分布都维持在一个较窄的范围内。
ATRP是近年发展非常成功的一种CRP方式[11, 12]。2003年,Masci等[13]采用氯丙酸乙酯(ECP)/CuCl/三(2-甲氨基乙基)胺(Me6TREN)作引发催化剂、N, N-二甲基甲酰胺(DMF)/水(体积比1:1)作溶剂,室温下通过ATRP合成了分子量分布较窄的PNIPAM。Stover等[14]对Masci的方法进行改进,采用异丙醇和叔丁醇这些多支化醇类作溶剂,氯丙酸甲酯/CuCl/Me6TREN等比例聚合,由于多支化醇类可以与单体及聚合体的酰胺基产生氢键作用,从而减少它们与催化剂及增长链之间的反应,因此可分别以高达90%和79%转化率得到具有分子量可控性好及分子量多分布窄的PNIPAM。研究发现,这些PNIPAM随分子量的增加其LCST降低,由于齐聚物更易受末端疏水性基团的影响,所以低分子量的PNIPAM其LCST较低、变化较缓、温度较宽;而高分子量的PNIPAM其LCST较高、变化迅速、范围较小。
2011年,Voelcker等[15]以溴代异丁酸乙酯(EBIB)作引发剂,采用CuBr/CuBr2/五甲基二乙烯三胺(PMDETA)作为催化系统,在水溶液中通过ATRP室温下即可快速合成PNIPAM。并且,通过表面引发原子转移自由基聚合(SI-ATRP)可将PNIPAM接枝到多孔硅表面。在实验中,首先在无水甲苯中将引发剂丙基三乙氧基硅烷(BiBAPTES)粘结在多孔硅表面完成硅烷化反应,采用相同的配体与催化剂,室温下成功地在多孔硅表面接枝PNIPAM。
RAFT聚合在合成聚合物过程中极其重要,这是由于除具有ATRP的特点外,其可用的单体非常丰富。2002年,Ganachand等[16]以偶氮二异丁腈(AIBN)作引发剂、二硫代氨基甲酸苄基和枯基酯作链转移剂(CTA),在60℃下通过RAFT聚合合成了可控分子量的PNIPAM。2003年,Olcamoto等[17]以1-苯乙基二硫代苯乙酸酯(PEPD)为CTA、AIBN作引发剂,以甲醇/甲苯混合液(体积比1:1)为溶剂,在路易斯酸氟甲烷磺酸钇[Y(OTf)3]催化下,通过RAFT聚合成功合成了可控分子量的全同立构的PNIPAM。2004年,Zhu等[18]以硫代苯甲酸苄基酯(EBTB)为CTA、AIBN为引发剂,按照NIPAM:EBIB:AIBN=200:2:1的比例在60℃下通过RAFT聚合成功合成了末端为二硫酯的可控分子量的PNIPAM。同年,Tenhu等[19]分别以4-氰基-4-(硫代苯甲酰硫基)戊酸(CPADB)及硫代苯甲酸枯基酯为CTA,利用RAFT聚合制备了末端分别为羧基和苯丙基的两种PNIPAM衍生物。这两种衍生物通过共价作用接枝到纳米金表面形成单层保护簇(MPC),通过量热法研究发现,两种MPC的PNIPAM链都呈现两个相转变温度,其吸热峰在温度较低处尖而窄,在温度较高处较宽。随着MPC浓度增加,第一个吸热峰都只有轻微的变化,但第二个吸热峰极大地向低温转变且两种MPC的转变程度差别较大,协同性较低。他们提出了一个合理的假设,将MPC细分为内外两部分,内部的PNIPAM紧贴纳米金,高密度、低含水,呈现第一个相转变温度;外部的PNIPAM为自由卷曲态,低密度、高含水,呈现的第二个相转变温度受MPC浓度及PNIPAM末端基团的影响,故末端疏水的枯基-PNIPAM要比亲水的CPA-PNIPAM相转变温度高。
此外,SET-LRP也被用来合成PNIPAM。2011年,Turan等[20]以2-溴异丁酰溴/CuBr/联吡啶作引发催化剂、巯基乙醇作CTA,在DMF中通过SET-LRP制备PNIPAM。先将巯基乙醇和2, 2′-联吡啶溶于DMF,进行5次冷冻-抽气-解冻后加入CuBr,于22℃反应24h完成CuBr的歧化反应,得到纳米铜Cu(0);然后加入NIPAM和2-溴异丁酰溴,于90℃进行聚合。结果表明,通过SET-LRP制备的PNIPAM具有低分子量和低聚合度,且随着分子量的降低,其LCST略微升高。
2007年,Matyjaszewski等[21]提出ARGET ATRP。Shivapooja等[22]通过此法在硅片上成功接枝了可控的高分子量、高纯度的PNIPAM聚合物刷。在无水甲苯中通过室温下长时间反应,将2-溴-2-甲基丙酸(3-三甲氧基硅基)丙酯粘结在干净的硅片表面作为ATRP引发剂,并采用PMDETA作配体、Cu(II)Br/维生素C作催化剂,在水/甲醇(体积比1:1)混合液中,将高分子量的PNIPAM聚合物刷接枝到硅片表面。这种方法简单、快速、方便,只需要很少量的二价铜催化剂(少于0.1‰),同时此合成过程避免了在反应结束后除去过渡金属,从而避免了聚合物纯化时造成的损失。
界面上PNIPAM修饰的制备方法常包括“接枝到”和“接枝自”。其中“接枝到”主要依赖于功能化PNIPAM的末端基团与各种载体表面互补基团的共价作用[19, 23, 24]。这种方法简单易操作,被广泛使用[25],研究者可以在接枝到各种载体表面之前,按需要提前将PNIPAM功能化。而“接枝自”依据表面引发的聚合反应[22, 25~30]或者与载体表面存在的双链共聚[31]可以在各种载体表面形成均一可控厚度的聚合物壳层。
PNIPAM由于其独特的性质而被广泛应用于药物传递、智能化纳米金、智能化表面材料、共聚物、传感分析及表面浸润性等领域(图 1)。
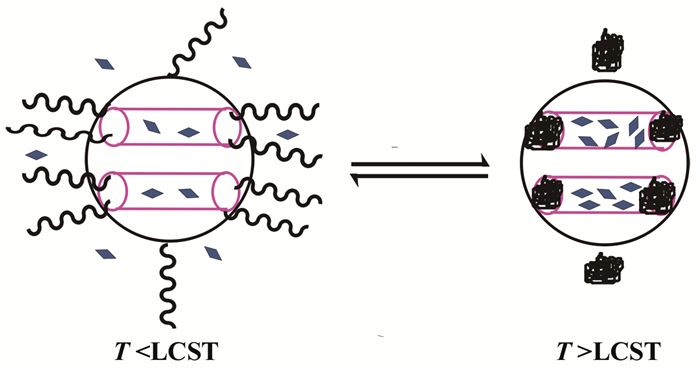
2008年,You等[32]通过RAFT聚合制备PNIPAM,再通过己胺的氨解作用将末端的三硫代酯还原为巯基,然后与二吡啶基二硫醚作用结合为末端为二硫吡啶的PNIPAM-S-S-Py聚合物。在实验中,他们通过正硅酸乙酯与巯基丙基三甲氧基硅烷作用,制备了孔壁修饰巯基链的微小介孔二氧化硅纳米颗粒(MSN-SH),并与PNIPAM-S-S-Py作用最终制得MSN-PNIPAM温敏性纳米颗粒。MSN-PNIPAM于室温下可以负载荧光素,若温度在LCST以上,PNIPAM塌陷为球形,回缩至孔壁,堵住微孔孔洞,阻止荧光素外泄;若温度在LCST以下,PNIPAM自由膨胀,打开孔洞,荧光素释放。这种方法可以用于药物的释放或基因的传递,简单示意如图 2所示。
2013年,Mohapatra等[31]将PNIPAM微凝胶与壳聚糖修饰的化学还原氧化石墨烯(CRGO)结合,制备了可以负载抗癌药物阿霉素(DOX)的具有温敏性的氧化石墨烯纳米凝胶(CGN)。他们首先将氧化石墨烯与聚苯乙烯磺酸钠(PSS)、肼共同作用制备了PSS覆盖的CRGO片,然后利用氯乙酸将表面的羟基转换成羧基,得到CRGO-COOH,并通过1-乙基-(3-二甲氨基丙基)碳二亚胺(EDC)/N-羟基琥珀酰亚胺(NHS)活化羧基,将带正电荷、亲水的、能够增加纳米颗粒的分散性而防止颗粒聚集的壳聚糖接枝到石墨烯表面,从而制备了壳聚糖修饰的CRGO。再将EDC、NHS活化的丙烯酸与上述壳聚糖修饰的CRGO反应得到丙烯酸化的壳聚糖-CRGO复合物。然后此复合物与NIPAM、二丙烯酸聚乙二醇酯共聚制备了可以负载抗癌药物DOX的温敏性水凝胶CGN。此CGN纳米平台在近红外光照射下可以产生可控的药物释放,同时在37℃或42℃时对细胞无毒。荧光图像显示,将负载药物的水凝胶(DOX-CGN)与癌细胞在37℃共同孵育时,药物可进入细胞质;在42℃孵育时,药物可深入细胞核,且一旦近红外光照射可观察到药物迅速释放。这是由于红外激光辐射下,由于CRGO的光热效应,光被吸收并转化成热,此热能使PNIPAM急剧收缩,使凝胶快速释放药物。与单纯的DOX相比,凝胶释放的药物可以快速深入细胞核,从而达到更好的治疗效果。一旦停止辐照,温度降低,PNIPAM回复原状,凝胶停止释放药物。该方法拓展了PNIPAM在药物控制释放领域的发展前景。
2015年,Stroeve等[30]以具有生物相容性的超顺磁Fe3O4为核、介孔SiO2(MSNs)为壳,制备了一种新型的核壳介孔SiO2纳米粒子(CS-MSNs)。与You等[32]制备的MSN-PNIPAM相比,此纳米材料的孔径更大,可以负载的药物选择性更多。在纳米材料表面接枝PNIPAM聚合物刷后可作为控制药物释放的关卡。在实验中,首先合成CS-MSNs,经三甲苯水热反应将孔扩张后得到CS-MSNs-EP,利用氨丙基三乙氧基硅烷在纳米颗粒表面修饰上氨基,再通过酰溴进一步修饰上Br基团,然后以PMDETA为配体、CuBr为催化剂,在异丙醇和水的混合液中通过ATRP成功接枝PNIPAM得到CS-MSNs-EP-PNIPAM。在37℃时,CS-MSNs-EP-PNIPAM与溶菌酶(Lys)共同孵育可成功负载Lys得到CS-MSNs-EP-PNIPAM-Lys。在室温下,PNIPAM为伸展态,可堵塞孔洞,阻止孔内Lys释放;而在生理温度时,PNIPAM塌陷,暴露孔洞,释放负载的Lys。在两种革兰氏阳性菌的存在下,研究了不同温度时CS-MSNs-EP-PNIPAM-Lys选择性释放Lys的行为。结果表明,25℃时,0.50mg/mL的纳米粒子对细菌无毒;而在37℃时,由于Lys的释放大大抑制了细菌的生长。这种纳米载体还将有望用于负载类似尺寸的蛋白质和其他药物,这对PNIPAM用于药物输送具有重要意义。
功能化纳米金由于其可以用于生物学、电化学、催化体系及可视材料,因而得到了广泛应用。表面接枝温敏性的聚合物PNIPAM,使智能化的纳米金能够对温度有非常灵敏的反应,因而被广泛用于生物化学传感领域。
PNIPAM修饰的智能化纳米金的制备一般有两种方法,即“接枝到”和“接枝自”。其中“接枝到”主要依赖于功能化PNIPAM的末端基团与微载体表面互补基团的共价作用[23~25]。而“接枝自”依据表面引发的聚合反应,可以在微载体表面形成均一可控厚度的聚合物壳层[25~28]。
2003年,Tenhu等[33]通过三种方法制备了PNIPAM-AuNPs复合物。其中,一步合成法是将一端为枯基的PNIPAM与氯金酸直接作用;二步合成法是利用水合肼的肼解作用将枯基直接还原为巯基,然后再与氯金酸作用;三步合成法则是先采用偶氮二氰基戊酸作引发剂,通过传统自由基聚合制备PNIPAM-COOH,然后通过酰胺键接枝末端为氨基的巯基乙胺制备PNIPAM-SH,再与氯金酸作用制备PNIPAM-AuNPs复合物。研究发现,通过以上三种方法制备的复合物其表面的PNIPAM壳层比溶液中自由卷曲态的PNIPAM的尺寸略大,这说明在纳米金表面的PNIPAM形态是呈伸展态的。
2007年,Tenhu等[34]通过RAFT聚合制备了临近PNIPAM的末端连接二硫酯键的聚甲基丙烯酸共聚物(PMAA)-b-PNIPAM,再在三乙基氢化硼锂的存在下,在无水四氢呋喃中与氯金酸直接作用,由于三乙基氢化硼锂可将二硫酯键还原为巯基,进而制备了PMAA-b-PNIPAM包被的纳米金。PMAA对pH有很灵敏的刺激响应性,使得此智能化纳米金对温度及pH都有很好的刺激响应性行为。
Yusa等[35]发现盐浓度对PNIPAM功能化的纳米金的温敏性有很大影响。他们通过RAFT聚合合成了窄分子量分布的PNIPAM,先利用乙醇胺的氨解作用将PNIPAM末端的二硫酯还原为巯基,再通过巯基作用将PNIPAM接枝到纳米金表面。研究发现,50mmol·L-1氯化钠盐溶液中的PNIPAM其LCST要比水溶液中PNIPAM的LCST低。当将温度从25℃升到40℃时,50mmol·L-1盐溶液中的纳米金从粉红变为灰蓝色;当温度从40℃再降为25℃时,溶液由灰蓝重新变为粉红色。随着温度的循环,PNIPAM构象发生可逆变化,溶液也体现出相应的可逆的颜色变化,但水溶液中的纳米金复合物却并无相应的颜色变化。研究进一步发现,25℃下当盐离子的浓度小于300mmol·L-1时,溶液为粉红,随着盐浓度增加溶液吸光度并无变化,说明在此盐浓度范围内纳米金可以稳定存在;而40℃时,随着盐浓度升高,溶液开始转为灰蓝,溶液吸光度也开始升高,当盐离子浓度达到50mmol·L-1时,溶液吸光度趋于稳定,纳米金聚集达到稳定水平。可见盐离子浓度对PNIPAM修饰的智能化纳米金的温敏性有很大影响。
2009年,Tenhu等[36]通过RAFT聚合合成了一系列不同分子量的NIPAM齐聚物,然后通过NIPAM齐聚物与氯金酸的一锅法反应将NIPAM齐聚物接枝到纳米金表面。研究发现,自由态的NIPAM齐聚物其LCST随分子量的升高而降低;但当PNIPAM接枝在纳米金表面时,随着分子量的升高其LCST反而升高。这是因为纳米金表面的PNIPAM由于分子量低故而链短且接枝密度高,致使链伸展得更开,更多的氢键作用发生在链间而不是链与水之间,进而加强了链间作用,使接枝在纳米金表面的PNIPAM的LCST降低。同时研究发现,金核的大小对于NIPAM齐聚物的相转移温度影响较大,但当分子量较大时,金核的影响不再明显。
2007年,Singh等[23]报道了一种空心PNIPAM微凝胶的合成方法。他们将胶体金与末端为氨基的PNIPAM-NH2共同孵育,使PNIPAM吸附在纳米金表面,然后在氮气中将吸附有PNIPAM的纳米金置于70℃保温,PNIPAM坍塌附着在纳米金颗粒表面形成疏水层,然后加入适量的NIPAM、N, N-亚甲基二丙烯酰胺和表面活性剂十二烷基硫酸钠水溶液反应1h,而后注入过硫酸铵(APS)引发反应形成PNIPAM微凝胶。若在加APS之前加入共聚单体丙烯酸(AAC)或者4-丙烯酸胺-荧光素(AFA),可引发形成PNIPAM-co-AAC共聚微凝胶或显示荧光的PNIPAM-co-AFA共聚微凝胶。在微凝胶形成后,加入氰化钾溶液可刻蚀金核形成[Au(CN)4]-,从而得到空心的PNIPAM微凝胶。研究发现,随着微凝胶壳层厚度的增大,Au的刻蚀速率变慢,但是在相同的单体浓度下,PNIPAM-co-AAC共聚微凝胶的刻蚀速率还是较PNIPAM微凝胶的刻蚀速率要快。这是因为AAC的羧基和PNIPAM-co-AAC更大的自由体积促使氰根离子更容易穿过壳层接近纳米金核。此类PNIPAM空心微凝胶可用于封装小分子和蛋白质,进而在药物输送和基因负载等方面大有前途。
2013年Mirkin等[37]将末端修饰巯基的单链DNA(ssDNA)与末端巯基化的PNIPAM共组装在纳米金表面,当温度在LCST以下时,PNIPAM在金表面呈伸展态,且其链长度高于DNA,可将DNA掩盖;当温度在LCST以上时,PNIPAM坍塌缩短,从而暴露DNA,若溶液中存在与其互补的ssDNA,纳米金上的DNA通过碱基互补配对形成双链(dsDNA),从而使得相邻纳米金间的距离缩小,呈现出纳米金的颜色转变。这个动态智能系统可进一步发展用于生物分子、蛋白质、多肽等领域的检测。
2010年,Caykara等[29]通过引发剂固定反应将2-溴丙酰溴固定在覆盖羟基的亲水硅片表面,再利用SI-ATRP技术,成功地在硅片表面自组装上分布均匀的PNIPAM聚合物刷(如图 3)。不同于水溶液中PNIPAM聚合物的相转移温度在一个较窄范围内,这种接枝在材料表面的干燥的聚合物刷其相转移温度范围较宽,且分为两个阶段。第一个相转变温度在27℃,是由靠近硅片表面的聚合物刷内部区域的PNIPAM塌陷造成的,此区域的PNIPAM密度较高,使得PNIPAM链间链内的作用都较强,故而在较低温度时即可先引发此区域PNIPAM的构象变化。第二阶段相转变温度在38℃,是由远离硅表面的聚合物刷外部区域的PNIPAM塌陷造成的,此区域链稀疏密度较低,链间的作用较弱,故而相转变温度也较高。这种通过表面引发自由基聚合制备的PNIPAM聚合物刷可用作可调型智能化表面材料。

2001年,Maeda等[38]将PNIPAM与DNA共聚,发展了一种新型的特殊序列寡核苷酸的沉淀分离体系。他们将甲基丙烯酸N-羟琥珀酸亚胺酯与氨基修饰的寡核苷酸(dT)8通过酰胺化反应制备乙烯基修饰的寡核苷酸(dT)8,然后与NIPAM单体在APS及四甲基乙二胺的共同作用下完成共聚制得PNIPAM-(dT)8共聚物。在盐浓度高于1.5mol·L-1时此共聚物可高选择性地与(dA)8形成三重结构,通过加热,使PNIPAM发生构象变化,在此温度下离心,即可成功将(dA)8沉淀分离出来;同时研究发现,寡核苷酸(dA)s链越长,则越容易沉淀出来。因此,PNIPAM-DNA共聚物可用于特殊序列寡核苷酸的分离体系,亦可用于多聚腺苷酸RNA的分离。
2002年,Maeda等[39]以同样的方法制备了PNIPAM-DNA共聚物,然后在高于LCST温度将PNIPAM脱水成核,成功制备了DNA包被的PNIPAM胶体,并研究其与单链靶向DNA杂交时胶体颗粒的稳定性改变情况。结果表明,杂交后最外端有突出的单链靶向DNA使胶体最为稳定,突出长度越长胶体越稳定;杂交后内端有突出的单链靶向DNA次之,同样突出长度越长胶体越稳定;完全互补配对的单链靶向DNA使胶体最不稳定,这是由于dsDNA比ssDNA上负电荷明显增多,使溶液中金属离子与其亲和力增强,故而胶体稳定性降低引发聚集。此外,胶体稳定性的改变伴随着可视的浊度变化。由于碱基缺失往往易引发癌症,因此相应的DNA包被的PNIPAM胶体可用于癌症方面的染色体诊断。
2011年Takarada等[40]报道了通过RAFT聚合制备可控分子链长度的PNIPAM-DNA双嵌段共聚物。他采用CPA-RAFT试剂,合成末端为二硫酯键的PNIPAM,然后利用NaBH4还原二硫酯键为巯基,再与依据Takarada等[41]的方法制备的5’端修饰顺丁烯二酰亚胺的DNA反应,最终制得PNIPAM-DNA共聚物。在温度高于LCST时使PNIPAM坍塌发生构象变化从而制备PNIPAM-DNA胶体颗粒。当溶液中存在完全互补配对的靶向DNA时,胶体会以非交联方式聚集。此类胶体具有以下特性:(1)由于静电和熵排斥使此微胶颗粒可在高离子强度溶液中保持稳定;(2)当溶液存在完全互补的靶向DNA时,胶体稳定性迅速降低且以非交联方式聚集;(3)当靶向DNA末端为错配碱基时,胶体稳定性明显增强。
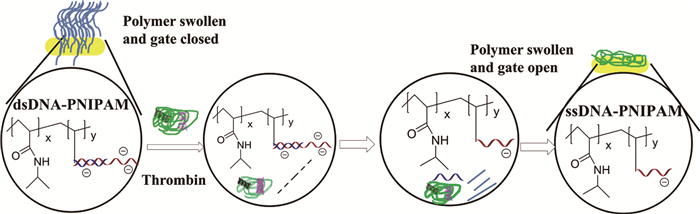
2015年,Yamaguchi等[42]通过在多孔基质孔隙接枝PNIPAM与包含凝血酶适配体的ssDNA的共聚物制备了分子识别门控膜。共聚的DNA在起始部位与凝血酶适配体形成双链结构,dsDNA带有较多的负电荷,DNA链间较强的静电作用使聚合物膨胀,孔隙呈关闭态;当溶液中存在凝血酶时,凝血酶与适配体强有力的识别使dsDNA解链成ssDNA,DNA间的静电斥力降低,从而使得接枝的PNIPAM收缩,孔隙释放,如图 4所示。通过改变接枝的适配体可使分子识别膜用于多种分子的识别,所以这种分子识别膜作为传感和药物输送系统在环境和药物领域极具发展潜力。
2013年Chang等[43]先通过NIPAM与交联剂亚甲基二丙烯酰胺(BIS)在引发剂的作用下制备了PNIPAM微凝胶,然后与Au3+共同作用制备了包含光致发光金纳米点的微凝胶(Au ND-PNIPAM微凝胶),此微凝胶可以通过离心洗涤纯化,与只能通过透析及胶体分离纯化的金纳米点相比,操作简单。Hg2+可以与金纳米点形成Au-Hg结构并且可以引发微凝胶聚集,致使520nm处的荧光减弱,引发荧光猝灭,并且在一定的Hg2+浓度区间内,此信号响应强度与汞离子浓度呈线性关系,故而此微凝胶适用于Hg2+的检测。且此微凝胶更耐盐,在500mmol·L-1 NaCl盐溶液中都可高效、高选择性地检测Hg2+,因而此微凝胶可以用于成分较复杂的环境中和生物样品中Hg2+的检测。
Maeda等[44]研究发现,接枝dsDNA的PNIPAM比接枝ssDNA的PNIPAM聚集程度更大,这是由于dsDNA比ssDNA有更强的构象限制,减少了DNA构象变异,弱化了DNA间的熵斥力,使PNIPAM更易趋向聚集,这种聚集现象是由熵效应控制的。Yamaguchi等[45]发现,ssDNA-PNIPAM比dsDNA-PNIPAM聚集程度更大,这是由于dsDNA比ssDNA的负电荷多,DNA间的静电斥力更强,阻止dsDNA-PNIPAM聚集,这种聚集现象是由热焓效应控制的。在此基础上,他们[46]通过将嵌入剂插入dsDNA中,可以转换PNIPAM-DNA聚集的这两种控制因素。并且,通过将嵌入剂插入dsDNA中可以转换这两种不同的聚集现象控制因素。实验中发现,dsDNA-PNIPAM随着温度升高只有些微聚集,这是由于dsDNA上的负电荷产生的静电斥力阻止其聚集。但是当带正电荷的嵌入剂二氨基吖啶(DAA)插入DNA中后,随着温度升高发生明显聚集,且插入的DAA越多,聚集越明显,这可能是由于DAA带正电荷,屏蔽了DNA上的负电荷,弱化了静电斥力;同时DAA的插入限制了DNA的构象变异,增强了构象限制,弱化了构象的熵斥力,且随着温度升高熵效应增强,由热焓控制因素转为熵控制因素。DAA的加入引起的聚集效应明显弱于dsDNA,这是由于DAA对dsDNA的绑定亲和力更强。通过将大量的嵌入剂或高绑定亲和力的嵌入剂插入DNA中,极大地影响了PNIPAM-DNA的聚集现象的控制因素,同时,这种被放大的聚集现象使PNIPAM这种材料可用于嵌入剂的检测。
2015年,Lu等[47]报道了鸟嘌呤(G)-四联体和温度引发的可逆的双重刺激响应PNIPAM水凝胶。接枝了富含G的核苷酸的PNIPAM在有钾离子存在下,可使链内部的G-四联体聚集连接PNIPAM链,从而形成水凝胶,在除去钾离子后,水凝胶又恢复到溶液状态;同时,若将水凝胶于40℃加热可形成固体,而于20℃冷却后又可恢复到凝胶态,故而形成了一个双重刺激响应的PNIPAM水凝胶,在液态-凝胶-固态循环转变。他们将血红素绑定到G-四联体交联形成的水凝胶中,将其作为一种接触反应基质,这种纳米材料可以释放辣根过氧化酶,可催化双氧水氧化苯胺形成导电性的聚苯胺。聚苯胺沉淀堆积,卷入凝胶的孔内,形成了导电性PNIPAM-聚苯胺混合水凝胶,且具有光滑无孔的刚性表面。将绑定血红素的G-四联体交联的PNIPAM水凝胶作为催化水凝胶用于导电聚苯胺的沉淀极具创新性,同时也阐述了导电水凝胶在电子学应用方面的新概念,并且,通过简单的湿印或涂布工艺可制备用于电子电路的导电聚合物。
PNIPAM在LCST附近具有亲水/疏水及链伸展/卷曲的体积相转变性质。由于这些特点,PNIPAM已被应用到表面浸润性领域,如抗污(自清洁)[48, 49]、分离[50, 51]、微流[52, 53]等方面。PNIPAM表面在LCST以下呈亲水性,会减少对有机污染物的吸附,在LCST以上为疏水性,相对易吸附污染物,可通过改性获得具有自清洁的PNIPAM共聚物,用于制备自清洁膜。Ye等[48]制备了表面具抗污能力的PNIPAM-聚甲基丙烯酸乙二醇酯(PEGMA)共聚物刷。其亲水的聚乙二醇侧链(该侧链在LCST上下均能保持伸展且遮蔽了共聚物主链)的加入,有效解决了PNIPAM在LCST以上时的疏水性问题。该共聚物刷(PEGMA摩尔分数在5%~10%)显示出良好的表面自清洁性能,经简单的变温水清洗,污染物残留可低至0.2%。将PNIPAM用于制备可再生性萃取水凝胶,可分离不同分子量的溶质,适用生物大分子的浓缩和提纯。PNIPAM还可用于制备温敏性水凝胶膜[51],通过直接制备或将PNIPAM接枝于高分子膜表面来得到智能型膜。PNIPAM还可用于制备温敏性接枝膜[50],它克服了水凝胶膜的机械强度差的缺陷,可对酶、蛋白质等进行分离。关于在微流控方面的应用,2010年李志明[53]研究了温敏PNIPAM整体柱超控微流体,通过在玻璃芯片上构建集成化的温敏整体柱塞,研究了它在微流控及样品预富集分离方面的特性及应用。
本文主要介绍了PNIPAM的制备及其在药物传递、智能化纳米金、智能化表面材料、PNIPAM-DNA共聚物、传感分析及表面浸润性等领域的应用。在制备PNIPAM时应注重开发简单、快速、经济的制备方法,通过不同方法对PNIPAM进行改性,或与其他单体共聚,可拓宽温敏性聚合物材料的研究领域。但是,目前其应用方面的研究还不够全面,例如在生物医学方面的应用研究仍有待加强。相信不久的将来PNIPAM会得到更深入的研究和更广泛的应用。
翟茂林, 哈鸿飞.高分子通报, 1999, (2):37~76. http://edu.wanfangdata.com.cn/Periodical/Detail/kxjsygc200709041
周啸, 何其维.高分子学报, 1992, 1(1):74~80.
祝黔江, 陶朱.高分子学报, 1997, 1(5):585~588. http://www.oalib.com/paper/4712960
A Laschewsky, E D Rekaï, E Wischerhoff. Macromol. Chem. Phys., 2001, 202(2):276~286. doi: 10.1002/(ISSN)1521-3935
J K Dong, J Y Heo, K S Kim et al. Macromol. Rapid Commun., 2003, 24(8):517~521. doi: 10.1002/marc.200390076
N Yamada, T Okano, H Sakai et al. Makromol. Rapid Commun., 1990, 11(11):571~576. doi: 10.1002/marc.1990.030111109
V Mittal, N B Matsko, A Butté et al. Eur. Polym. J., 2007, 43(12):4868~4881. doi: 10.1016/j.eurpolymj.2007.10.012
H G Schild. Prog. Polym. Sci., 1992, 17(2):163~249. doi: 10.1016/0079-6700(92)90023-R
M Kamigaito, T Ando, M Sawamoto et al. Chem. Rev., 2001, 101(12):3689~3746. doi: 10.1021/cr9901182
E Rizzardo, J Chiefari, B Y K Chong et al. Macromol. Symp., 1999, 143(1):291~307. doi: 10.1002/masy.v143.1
K Matyjaszewski, J Xia. Chem. Rev., 2001, 101(9):2921~2990. doi: 10.1021/cr940534g
T Pintauer, K Matyjaszewski. Atom transfer radical polymerization (ATRP) and addition (ATRA) and applications//Encyclopedia of radicals in chemistry, biology and materials, Wiley: New York, 2012.
M Giancarlo, G Laura, C Vittorio et al. Macromol. Rapid Commun., 2004, 25(4):559~564. doi: 10.1002/(ISSN)1521-3927
Y Xia, X Yin, N A D Burke et al. Macromolecules, 2005, 38(14):2275~2283.
R B Vasani, S J Mcinnes, M A Cole et al. Langmuir, 2011, 27(12):7843~7853. doi: 10.1021/la200551g
F Ganachaud, M J Monteiro, R G Gilbert. Macromol. Symp., 2002, 150(1):275~281. https://www.iupac.org/publications/pac/79/8/1463/references/index.html
B Ray, Y Isobe, K Morioka et al. Macromolecules, 2003, 36(3):543~545. doi: 10.1021/ma0257595
M Q Zhu, L Q Wang, G J Exarhos et al. J. Am. Chem. Soc., 2004, 126(9):2656~2657. doi: 10.1021/ja038544z
J Shan, J Chen, M Nuopponen et al. Langmuir, 2004, 20(11):4671~4676. doi: 10.1021/la0363938
E Turan, A Zengin, T Caykara. J. Polym. Sci. A, 2011, 49(23):5116~5123. doi: 10.1002/pola.v49.23
K Matyjaszewski, H Dong, W Jakubowski et al. Langmuir, 2007, 23(8):4528~4531. doi: 10.1021/la063402e
P Shivapooja, L K Ista, H E Canavan et al. Biointerphases, 2012, 7(1-4):1~9.
N Singh, L A Lyon. Chem. Mater., 2007, 19(19):719~726.
F J Xu, K G Neoh, E T Kang. Prog. Polym. Sci., 2009, 34(8):719~761. doi: 10.1016/j.progpolymsci.2009.04.005
W Huang, G L Baker, M L Bruening. Angew. Chem., 2001, 113(8):1558~1560. doi: 10.1002/(ISSN)1521-3757
S Peleshanko, V V Tsukruk. Prog. Polym. Sci., 2008, 33(5):523~580. doi: 10.1016/j.progpolymsci.2008.01.003
P Liu, J Tian, W Liu et al. Polym. J., 2003, 35(4):379~383. doi: 10.1295/polymj.35.379
D R Walt, T K Mandal, M S Fleming. USP: 6, 720, 007.
E Turan, S Demirci, T Caykara. Thin Solid Films, 2010, 518(21):5950~5954. doi: 10.1016/j.tsf.2010.05.103
E Yu, I Galiana, R Martínezmáñez et al. Colloids Surf. B, 2015, 135652~660.
C Y Wang, U S Garapati, S Ravi et al. Nanomed. Nanotechnol., 2013, 9(7):903~911. doi: 10.1016/j.nano.2013.01.003
Y Z You, K K Kalebaila, S L Brock et al. Chem. Mater., 2008, 20(10):3354~3359. doi: 10.1021/cm703363w
J Shan, M Nuopponen, H Jiang et al. Macromolecules, 2003, 36(12):4526~4533. doi: 10.1021/ma034265k
M Nuopponen, H Tenhu. Langmuir, 2007, 23(10):5352~5357. doi: 10.1021/la063240m
S Yusa, K Fukuda, T Yamamoto et al. Langmuir, 2007, 23(26):12842~12848. doi: 10.1021/la702741q
J Shan, Y Zhao, N Granqvist et al. Macromolecules, 2009, 42(7):2696~2701. doi: 10.1021/ma802482e
K Zhang, X Zhu, F Jia et al. J. Am. Chem. Soc., 2013, 135(38):14102~14105. doi: 10.1021/ja408465t
T Mori, D Umeno, M Maeda et al. Biotechnol. Bioeng., 2001, 72(3):261~268. doi: 10.1002/(ISSN)1097-0290
T Mori, M Maeda. Polym. J., 2002, 34(8):624~628. doi: 10.1295/polymj.34.624
K Isoda, N Kanayama, D Miyamoto et al. React. Funct. Polym., 2011, 71(3):367~371. doi: 10.1016/j.reactfunctpolym.2010.11.020
N Kanayama, H Shibata, A Kimura et al. Biomacromolecules, 2009, 10(4):805~813. doi: 10.1021/bm801301b
Y Sugawara, T Tamaki, T Yamaghchi. Polymer, 2015, 62:86~93. doi: 10.1016/j.polymer.2015.02.027
L Y Chen, C M Ou, W Y Chen et al. Appl. Mater. Interf., 2013, 5(10):4383~4388. doi: 10.1021/am400628p
H Mori, Y Maeda, S Kubota et al. Polym. J., 2002, 34(34):687~691.
Y Sugawara, T Tamaki, H Ohashi et al. Soft Matter, 2013, 9(12):3331~3340. doi: 10.1039/c3sm27230c
Y Sugawara, T Tamaki, T Yamaguchi et al. Polym. Chem., 2014, 5(16):4612~4616. doi: 10.1039/C4PY00600C
C Lu, W Guo, X Qi et al. Chem. Sci., 2015, 6(11):6659~6664. doi: 10.1039/C5SC02203G
Y Ye, J Huang, X Wang. ACS Appl. Mater. Interf., 2015, 7(40), 22128~22136. doi: 10.1021/acsami.5b07336
S Yu, Z Chen, J Liu et al. J. Membr. Sci., 2012, 392(2), 181~191.
Y J Choi, T Yamaguchi, S I Nakao. Ind. Eng. Chem. Res., 2000, 39(7), 2491~2495. doi: 10.1021/ie9907627
H Feil, H B You, J Feijen et al. J. Membr. Sci., 1991, 64(3), 283~294. doi: 10.1016/0376-7388(91)80099-R
Z Li, Q He, D Ma et al. Anal. Chim. Acta, 2010, 665(2), 107~112. doi: 10.1016/j.aca.2010.03.024
李志明. 浙江大学博士学位论文, 2010.


图 3 硅片表面引发NIPAM的ATRP示意图
Figure 3 The diagram of the ATRP of NIPAM on the silicon surface

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

 下载:
下载: