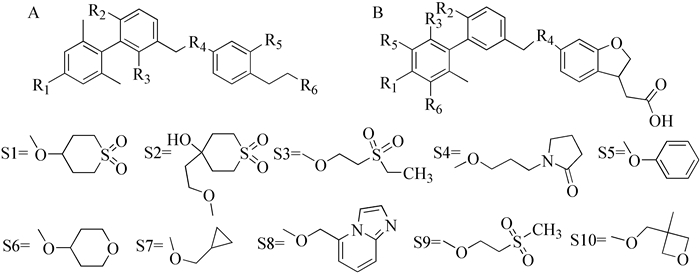
图式 1.
分子母核及取代基结构
Scheme 1.
Molecular cores and substituent structures

GPR40(G-protein-coupled receptor 40)是在G蛋白偶联受体家族中属于A类视紫红质样受体,是一种具有7个跨膜α螺旋结构的G蛋白偶联受体,它可以被中、长链脂肪酸所激活,故又被称为游离脂肪酸受体1(Free fat acid receptor 1,FFAR1)。GPR40蛋白主要分布在胰腺组织中,主要与Gq家族G蛋白α亚基(Gαq)偶联。GPR40激活时,可增强胰岛β细胞的胰岛素分泌。此外,激活的GPR40还可以通过Gs信号途径引起细胞内Ca2+水平的增加以葡萄糖依赖的方式促进胰岛素释放[1]。除胰腺β细胞外,GPR40在胃肠道的肠内分泌细胞中也有表达,Gq和Gs的协同激活是肠内分泌细胞中刺激增量释放的主要途径,可刺激内分泌素的分泌,从而降低血糖[2]。实验表明,GPR40基因敲除小鼠在高脂肪酸供给条件下胆囊收缩素(cholecystokinin)的分泌情况与正常喂养条件下无明显差异,说明GPR40也参与脂肪的摄取及饮食调控。GPR40还广泛存在于脑组织中,特别是黑质和延髓中,GPR40受体的激活可促进神经元细胞突触的增长和神经前体细胞增殖[3]。
GPR40激动剂主要有苯丙酸衍生物、噻唑烷二酮类、4-苯乙炔-二氢肉桂酸类、羧酸的联苯嘧啶巯基/氨基取代物以及酰基间苯三酚类等。其中苯丙酸衍生物研究得最多,如Takeda公司研发的TAK-875是临床研究最快的GPR40小分子激动剂,曾进入三期临床,为GPR40激动剂的研究与开发奠定了很好的基础[4]。
本文首次采用比较分子力场分析法(CoMFA)和比较分子相似性指数分析法(CoMSIA),针对40个苯丙酸类GPR40激动剂分子建立三维定量构效关系(3D-QSAR)模型,分析苯丙酸类激动剂分子结构与生物活性之间的定量关系,同时根据CoMFA和CoMSIA模型提出优化此类激动剂活性的药物设计思路,为设计更加有效的GPR40激动剂提供理论依据。
从文献[5~7]中获得共40个GPR40受体激动剂小分子(分子母核见图式 1),文献所提供活性值为Ki值,根据研究所需,需将这些活性值转换为pKi值(-logKi),见表 1。分子构建及结构优化等过程的运算参数除标明外均设为默认。通过SYBYL-X 2.0软件构建小分子三维结构,并加氢处理。结构优化时,首先以最陡下降法(Steepest descent method)来寻找分子低能构象,参数设置如下,收敛RMS参数:0.05kcal·mol-1·Å-1,最大迭代步数(Max Iterations):6000;随后使用共轭梯度法进一步优化分子构象,参数设置如下,收敛RMS参数:0.001kcal·mol-1·Å-1,最大迭代步数(Max Iterations):1000;最后利用Gasteiger-Hückel法计算分子电荷,即得能量最低的药物分子构象。
 下载:
导出CSV
下载:
导出CSV
| 母核 | 化合物 | R1 | R2 | R3 | R4 | R5 | R6 | pKi |
| A | 1 | S2 | - | - | NH | F | COOH | 7.721 |
| 2 | S1 | - | - | NH | F | COOH | 7.745 | |
| 3 | S3 | - | - | NH | F | COOH | 7.699 | |
| 4 | S2 | - | - | O | F | COOH | 7.721 | |
| 5* | OCH2CH2OCH2CH3 | - | - | NH | F | COOH | 7.745 | |
| 6 | S3 | - | - | O | F | COOH | 7.824 | |
| 7 | S4 | - | - | O | F | COOH | 7.628 | |
| 8 | S6 | - | - | O | F | COOH | 7.699 | |
| 9 | OCH2CH2OCH2CH3 | - | - | O | F | COOH | 7.921 | |
| 10* | S1 | - | - | O | F | COOH | 7.796 | |
| 11 | OCH2CH2OCH2CH3 | - | - | O | - | COOH | 7.638 | |
| 12 | S7 | - | - | O | - | COOH | 7.796 | |
| 13 | - | OCH3 | - | O | - | COOH | 7.328 | |
| 14 | - | - | CH3 | O | - | COOH | 7.237 | |
| 15* | OCH3 | - | - | O | - | COOH | 7.602 | |
| 16 | - | - | - | O | - | - | 7.327 | |
| 17 | - | - | - | O | F | - | 7.495 | |
| 18* | - | - | CH3 | O | F | COOH | 7.004 | |
| B | 19 | S9 | - | CH3 | O | - | Cl | 7.721 |
| 20 | S9 | - | CH3 | O | - | F | 7.745 | |
| 21 | S1 | - | CH3 | O | - | F | 7.509 | |
| 22 | S9 | - | CH3 | O | CH3 | CH3 | 7.481 | |
| 23 | S1 | - | CH3 | O | CH3 | CH3 | 7.398 | |
| 24 | S8 | - | CH3 | O | - | - | 7.842 | |
| 25* | S9 | - | CH3 | O | Cl | Cl | 7.921 | |
| 26 | S4 | - | CH3 | O | - | - | 7.509 | |
| 27 | S9 | - | CH3 | O | - | - | 7.420 | |
| 28 | S2 | - | CH3 | O | - | - | 7.620 | |
| 29 | S1 | - | CH3 | O | - | - | 7.444 | |
| 30* | S3 | - | CH3 | O | - | - | 7.423 | |
| 31 | S10 | - | CH3 | O | - | - | 7.602 | |
| 32 | OCH2CH2OCH2CH3 | - | CH3 | O | - | - | 7.638 | |
| 33 | OCH2CH2OCH2CH3 | - | CH3 | NH | - | - | 7.367 | |
| 34 | OCH2CH2OCH2CH3 | - | CH3 | O | - | CH3 | 7.495 | |
| 35* | - | OCH3 | CH3 | O | - | - | 6.886 | |
| 36 | CH3 | - | CH3 | O | - | - | 7.444 | |
| 37 | CH3 | OCH3 | - | O | - | - | 7.244 | |
| 38 | - | S5 | CH3 | O | - | - | 7.387 | |
| 39 | S9 | - | CH3 | O | - | - | 7.540 | |
| 40* | S5 | - | - | O | - | - | 7.229 | |
| *为测试集分子(8个小分子) | ||||||||
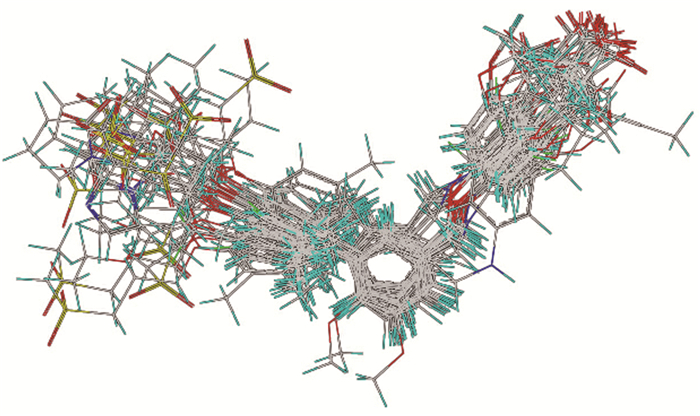
分子叠合效果是构建3D-QSAR模型的关键,实验中需依据力场、药效基团位置等因素来进行叠合,使分子叠合程度达到最高。叠合使用Align Database程序进行,使40个化合物分子的共同部分叠合在同一刚性骨架上。由于3D-QSAR模型假定每一个活性分子以同样的活性方式与受体的同一位点结合,因此选择活性最高的化合物9(pKi 7.921)作为叠合模板以使叠合效果最佳。叠合结果自动生成一个新的数据库。
CoMFA模型的构建是将具有相同结构母环的分子在空间中叠合,使其空间取向尽量一致,然后用一个探针粒子在分子周围的空间中游走,计算探针粒子与分子之间的相互作用,并记录下空间不同坐标中相互作用的能量值,从而探测和计算化合物分子周围的立体场(Steric,S)和静电场(Electrostaic,E)[8]。CoMSIA方法改变了探针粒子与药物分子相互作用能量的计算公式,可探测和计算化合物分子周围的立体场、静电场、疏水场(Hydrophobic,H)、氢键受体场(Acceptor,A)和氢键供体场(Donor,D)。
本文从40个活性分子中随机选择32个分子组成训练集,其余8个分子组成测试集,针对训练集采用偏最小二乘法(PLS)计算得到CoMFA模型和CoMSIA模型,结合pKi活性数据及其相关场描述变化进行分析。采用留一法(Leave-One-Out,LOO)来验证模型可靠性,经过计算可得最佳主成分数和交叉验证系数q2,调整柱滤值(Column filtering value,σ)使q2最大。一般来讲,当q2大于0.5时,认为模型具有良好的统计学意义[9]。之后采用非交叉验证法(Non-validation)建立QSAR模型预测生物活性数据,并得到标准偏差SE、非交叉验证系数r2和显著系数F,以此来评估模型的预测能力。以上实验均在SYBYL-X 2.0软件中完成。
以活性最高的化合物9为叠合模板,将40个化合物分子的公共骨架叠加,结果如图 1所示。所有分子的公共骨架均叠合在一起,并且化合物分子中相似基团的叠合取向也趋于一致,说明叠合效果良好。
采用PLS法对训练集的32个化合物分子建立CoMFA模型,最佳主成分数为3、柱滤值为3.5kJ/mol时,q2最大,为0.527,说明该模型具有良好的统计学意义。显著系数F为85.191,说明所得数据具有较高的显著性。非交叉系数r2为0.901,一般地,当r2大于0.6时,认为该模型具有良好的预测能力[10]。标准偏差SE为0.059,化合物分子pKi实验值与预测值的偏差均小于1个数量级(表 2、3),pKi实验值与预测值之间的线性关系见图 2(a)。另外,CoMFA模型中立体场和静电场的贡献率分别为69.5%和30.5%,说明GPR40激动剂分子周围的立体场对其活性发挥主要作用。
下载:
导出CSV
| 3D-QSAR | ONC | q2 | SE | r2 | F | Field Contribution | ||||
| S | E | H | D | A | ||||||
| CoMFA | 3 | 0.527 | 0.059 | 0.901 | 85.191 | 0.695 | 0.305 | |||
| CoMSIA | 3 | 0.500 | 0.070 | 0.860 | 57.187 | 0.213 | 0.207 | 0.255 | 0.152 | 0.172 |
| ONC:最佳主成分数 | ||||||||||
下载:
导出CSV
| Numbers | Actual pKi | CoMFA | CoMSIA | |||
| Predicted pKi | Residual | Predicted pKi | Residual | |||
| 5 | 7.745 | 7.531 | 0.214 | 7.577 | 0.168 | |
| 10 | 7.796 | 7.525 | 0.271 | 7.619 | 0.177 | |
| 15 | 7.602 | 7.457 | 0.145 | 7.473 | 0.129 | |
| 18 | 7.004 | 7.595 | -0.591 | 7.560 | -0.556 | |
| 25 | 7.921 | 7.484 | 0.437 | 7.463 | 0.458 | |
| 30 | 7.432 | 7.741 | -0.309 | 7.675 | -0.243 | |
| 35 | 6.886 | 7.563 | -0.677 | 7.539 | -0.653 | |
| 40 | 7.229 | 7.497 | -0.268 | 7.525 | -0.296 | |
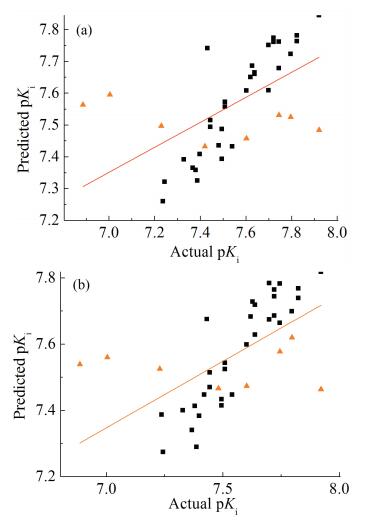
■训练集分子 ▲测试集分子 —全部分子回归线
■Training set molecules ▲Test set molecules — All molecular regression lines
采用PLS法对训练集的32个化合物分子建立CoMSIA模型,最佳主成分数为3、柱滤值为5.8kJ/mol时,q2最大,为0.500,说明该模型具有良好的统计学意义。显著系数F为57.187,说明所得数据具有较好的显著性。非交叉系数r2为0.860,说明该模型具有良好的预测能力。标准偏差SE为0.070,化合物分子pKi实验值与预测值的偏差均小于1个数量级(表 2、3),pKi实验值与预测值之间的线性关系见图 2(b)。图 2反映了训练集及测试集分子生物活性的预测值与理论值间的差异及各点离散程度,图中各点基本分布在直线两侧,表明模型具有良好的稳定性及内部和外部预测能力。另外,CoMSIA模型中立体场(S)、静电场(E)、疏水场(H)、氢键供体场(D)和氢键受体场(A)的贡献率分别为21.3%、20.7%、25.5%、15.5%和17.2%,CoMSIA模型提供了更多场信息,五个场相互作用共同影响了分子活性。
总体数据表明,本研究所建立的CoMFA模型和CoMSIA模型均具有较好的稳定性和预测能力。在药物设计时,应综合考虑两模型提供的信息。
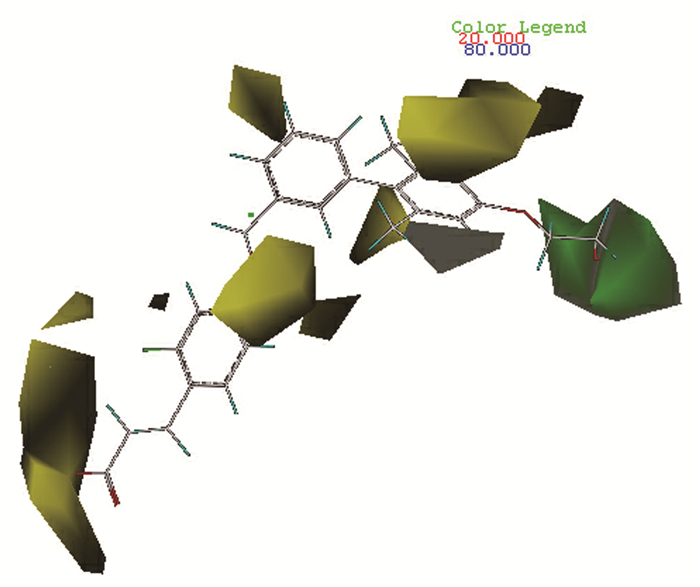
本文以活性最高的化合物9作为参考分子进行分析,其CoMFA模型等势图如图 3所示。其中绿色区域代表增大基团有利于增加化合物活性,黄色区域代表增大基团会降低药物活性,因此可在R1取代基位置增加支链来增大体积,也可用体积较大的取代基如苯基、吡啶基等替代R1,另外,可在R6位右侧减小取代基的体积,比如可用-F、-Cl、-Br、-CH3、-OH等替代R6右侧基团,有较大可能可提高化合物分子的活性。
相比于CoMFA模型,CoMSIA模型提供的信息更为全面,场贡献方面给出了立体场(S)、静电场(E)、疏水场(H)、氢键供体场(D)和氢键受体场(A)五个场的信息。
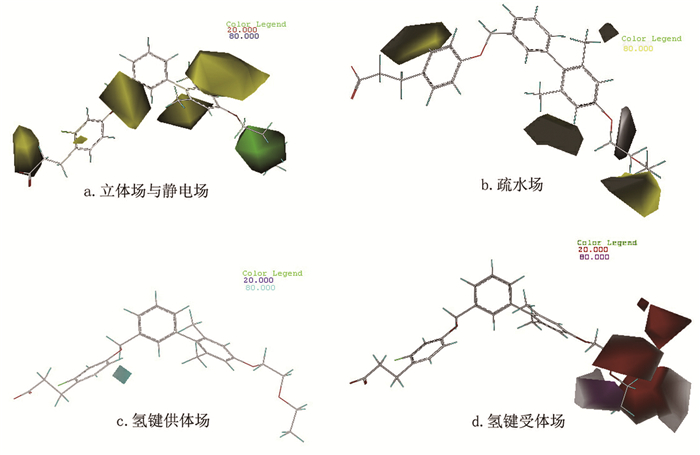
同CoMFA模型,依然选择活性最高的化合物9作为参考分子进行分析。图 4(a)为立体场与静电场贡献图,颜色代表同CoMFA模型,可知R1左侧苯环上取代基以及R4、R6位置不宜引入较大基团,否则会降低分子活性。可以考虑去掉苯环上甲基,引入-F、-Cl、-Br、-OH等体积较小的基团以提高活性。图 4(b)为疏水场贡献图,其中黄色区域表示增加疏水性基团有利于增加活性,白色区域表示增加亲水基团有利于增加活性。可知在R1尾端、R5位置取代疏水性基团(如烷烃、芳香烃和酯基等)可增加分子活性;而在R1中间位置引入羟基、羧基和氨基等亲水基团可增加分子活性。图 4(c)和图 4(d)分别为氢键供体场和氢键受体场,其中蓝绿色区域表示增加氢键供体基团有利于增加活性,紫红色区域代表增加氢键受体有利于增加活性,红色区域则表示过多的氢键受体会降低活性。因此,在R1末端甲基处可适当引入氢键受体基团,R1其他位置则要尽量减少氢键受体基团的引入,理论上可得到活性更好的GPR40受体活性分子。
本文基于三维定量构效关系分子设计方法对GPR40受体激动剂进行了研究,采用偏最小二乘法统计学意义良好的CoMFA和CoMSIA模型,两模型训练集的交叉验证系数q2为0.5~0.6,非交叉验证系数r2为0.8~1.0,两个模型对活性数据pKi的预测结果与实验值基本吻合,说明本实验所构建的两个3D-QSAR模型均具有良好的稳定性和预测能力。结合3D-QSAR等势图给出的立体场、静电场、疏水场、氢键供体场和氢键受体场贡献信息,分析了GPR40受体激动剂分子与受体间的相互作用情况,根据CoMFA等势图可知,在R1取代基位置增加支链来增大体积,或用体积较大的取代基替代R1,也可在R6位右侧减小取代基的体积,能够提高药物分子活性;根据CoMSIA等势图可知,R1左侧苯环上取代基以及R4、R6位置不宜引入较大基团,在R1末端甲基处可适当引入氢键受体基团,R1其他位置则要尽量减少氢键受体基团的引入,理论上可得到活性更好的GPR40激动剂。CoMFA和CoMSIA模型共同提示的药物设计信息基本相符,为进一步设计结构新颖、活性高的GPR40激动剂提供了理论依据。
D D Feng, Z Q Luo, S G Roh et al. Endocrinology 2006; 147(2):674~682. doi: 10.1210/en.2005-0225
A P Liou, X Lu, Y Sei et al. Gastroenterology, 2011, 140(3):903~912. doi: 10.1053/j.gastro.2010.10.012
D Ma, B Tao, S Warashina et al. Neurosci. Res., 2007, 58(4):394~401. https://www.ncbi.nlm.nih.gov/pubmed/17583366
A Srivastava, J Yano, Y Hirozane et al. Nature, 2014, 513(7516):124~127. https://www.ncbi.nlm.nih.gov/pubmed/25043059
S Mikami, S Kitamura, N Negoro et al. J. Med. Chem., 2012, 55(8):3756~3776. doi: 10.1021/jm2016123
N Negoro, S Sasaki, S Mikami et al. J. Med. Chem., 2012, 55(8):3960~3974. doi: 10.1021/jm300170m
N Negoro, S Sasaki, M Ito et al. J. Med. Chem., 2012, 55(4):1538~1552.
曹洪玉, 吴艳华, 任聪等.化学通报, 2018, 81(6):548~554. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20180116002&flag=1
于柯楠, 吴玲, 曹洪玉.广东化工, 2016, 43(05):15~18. doi: 10.3969/j.issn.1007-1865.2016.05.007
李国栋, 彭发, 陈兰美等.化学研究与应用, 2015, 27(2):113~121. doi: 10.3969/j.issn.1004-1656.2015.02.002

图 2 3D-QSAR模型中pKi实验值与预测值的线性关系:CoMFA模型(a);CoMSIA模型(b)
Figure 2 Linear relationship between actual pKi and predicted pKi of 3D-QSAR Models: CoMFA model (a); CoMSIA model (b)
■训练集分子 ▲测试集分子 —全部分子回归线
■Training set molecules ▲Test set molecules — All molecular regression lines
表 1 GPR40受体活性小分子结构及pKi值
Table 1. The molecular structures and pKi value of GPR40 receptors
| 母核 | 化合物 | R1 | R2 | R3 | R4 | R5 | R6 | pKi |
| A | 1 | S2 | - | - | NH | F | COOH | 7.721 |
| 2 | S1 | - | - | NH | F | COOH | 7.745 | |
| 3 | S3 | - | - | NH | F | COOH | 7.699 | |
| 4 | S2 | - | - | O | F | COOH | 7.721 | |
| 5* | OCH2CH2OCH2CH3 | - | - | NH | F | COOH | 7.745 | |
| 6 | S3 | - | - | O | F | COOH | 7.824 | |
| 7 | S4 | - | - | O | F | COOH | 7.628 | |
| 8 | S6 | - | - | O | F | COOH | 7.699 | |
| 9 | OCH2CH2OCH2CH3 | - | - | O | F | COOH | 7.921 | |
| 10* | S1 | - | - | O | F | COOH | 7.796 | |
| 11 | OCH2CH2OCH2CH3 | - | - | O | - | COOH | 7.638 | |
| 12 | S7 | - | - | O | - | COOH | 7.796 | |
| 13 | - | OCH3 | - | O | - | COOH | 7.328 | |
| 14 | - | - | CH3 | O | - | COOH | 7.237 | |
| 15* | OCH3 | - | - | O | - | COOH | 7.602 | |
| 16 | - | - | - | O | - | - | 7.327 | |
| 17 | - | - | - | O | F | - | 7.495 | |
| 18* | - | - | CH3 | O | F | COOH | 7.004 | |
| B | 19 | S9 | - | CH3 | O | - | Cl | 7.721 |
| 20 | S9 | - | CH3 | O | - | F | 7.745 | |
| 21 | S1 | - | CH3 | O | - | F | 7.509 | |
| 22 | S9 | - | CH3 | O | CH3 | CH3 | 7.481 | |
| 23 | S1 | - | CH3 | O | CH3 | CH3 | 7.398 | |
| 24 | S8 | - | CH3 | O | - | - | 7.842 | |
| 25* | S9 | - | CH3 | O | Cl | Cl | 7.921 | |
| 26 | S4 | - | CH3 | O | - | - | 7.509 | |
| 27 | S9 | - | CH3 | O | - | - | 7.420 | |
| 28 | S2 | - | CH3 | O | - | - | 7.620 | |
| 29 | S1 | - | CH3 | O | - | - | 7.444 | |
| 30* | S3 | - | CH3 | O | - | - | 7.423 | |
| 31 | S10 | - | CH3 | O | - | - | 7.602 | |
| 32 | OCH2CH2OCH2CH3 | - | CH3 | O | - | - | 7.638 | |
| 33 | OCH2CH2OCH2CH3 | - | CH3 | NH | - | - | 7.367 | |
| 34 | OCH2CH2OCH2CH3 | - | CH3 | O | - | CH3 | 7.495 | |
| 35* | - | OCH3 | CH3 | O | - | - | 6.886 | |
| 36 | CH3 | - | CH3 | O | - | - | 7.444 | |
| 37 | CH3 | OCH3 | - | O | - | - | 7.244 | |
| 38 | - | S5 | CH3 | O | - | - | 7.387 | |
| 39 | S9 | - | CH3 | O | - | - | 7.540 | |
| 40* | S5 | - | - | O | - | - | 7.229 | |
| *为测试集分子(8个小分子) | ||||||||
 下载: 导出CSV
下载: 导出CSV
表 2 训练集分子的两模型数据参数
Table 2. Statistical parameters of two models for training set molecules
| 3D-QSAR | ONC | q2 | SE | r2 | F | Field Contribution | ||||
| S | E | H | D | A | ||||||
| CoMFA | 3 | 0.527 | 0.059 | 0.901 | 85.191 | 0.695 | 0.305 | |||
| CoMSIA | 3 | 0.500 | 0.070 | 0.860 | 57.187 | 0.213 | 0.207 | 0.255 | 0.152 | 0.172 |
| ONC:最佳主成分数 | ||||||||||
下载: 导出CSV
表 3 两模型测试集分子pKi实验值与预测值比较
Table 3. Comparison of actual and predicted pKi value of the test sets molecules for two models
| Numbers | Actual pKi | CoMFA | CoMSIA | |||
| Predicted pKi | Residual | Predicted pKi | Residual | |||
| 5 | 7.745 | 7.531 | 0.214 | 7.577 | 0.168 | |
| 10 | 7.796 | 7.525 | 0.271 | 7.619 | 0.177 | |
| 15 | 7.602 | 7.457 | 0.145 | 7.473 | 0.129 | |
| 18 | 7.004 | 7.595 | -0.591 | 7.560 | -0.556 | |
| 25 | 7.921 | 7.484 | 0.437 | 7.463 | 0.458 | |
| 30 | 7.432 | 7.741 | -0.309 | 7.675 | -0.243 | |
| 35 | 6.886 | 7.563 | -0.677 | 7.539 | -0.653 | |
| 40 | 7.229 | 7.497 | -0.268 | 7.525 | -0.296 | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: