 图1
反应的QTOF值计算
Figure1.
HDS quasi-turnover frequency (QTOF) of the reaction over catalysts
图1
反应的QTOF值计算
Figure1.
HDS quasi-turnover frequency (QTOF) of the reaction over catalysts
引用本文:
韩文鹏, 张晔, 李学宽, 唐明兴, 周立公, 吴明红, 葛晖. 配合物的配位基团性质对CoMo/γ-Al2O3催化剂加氢脱硫性能的影响[J]. 燃料化学学报,
2017, 45(11): 1332-1339.

Citation: HAN Wen-peng, ZHANG Ye, LI Xue-kuan, TANG Ming-xing, ZHOU Li-gong, WU Ming-hong, GE Hui. Effect of coordinating groups of chelating agents on the hydrodesulfurization over CoMo/γ-Al2O3 catalysts[J]. Journal of Fuel Chemistry and Technology, 2017, 45(11): 1332-1339.
Citation: HAN Wen-peng, ZHANG Ye, LI Xue-kuan, TANG Ming-xing, ZHOU Li-gong, WU Ming-hong, GE Hui. Effect of coordinating groups of chelating agents on the hydrodesulfurization over CoMo/γ-Al2O3 catalysts[J]. Journal of Fuel Chemistry and Technology, 2017, 45(11): 1332-1339.
配合物的配位基团性质对CoMo/γ-Al2O3催化剂加氢脱硫性能的影响
摘要:
选择四种不同配位基团的双齿配位分子乙二胺(EN)、乙醇胺(EA)、乙二醇(EG)和丙二酸(MA)对CoMo/γ-Al2O3催化剂改性,比较了它们对二苯并噻吩HDS性能的影响。结果表明,其活性顺序为CoMo(EN)> CoMo(EA)> CoMo(EG)≈CoMo(MA)> CoMo,反应以直接脱硫路径为主,随反应温度升高,加氢路径的占比增加,加入配合物后可以促进加氢路径脱硫,CoMo(EN)催化剂具有最高的加氢活性。采用UV-vis、EA、XPS和HRTEM等手段对催化剂进行表征,结果表明,-NH2与Co2+有强络合作用,-COOH主要是静电作用,而-OH与钴离子没有相互作用。配位基团和Co2+的相互作用,与HDS活性直接相关。配合物与Co2+的结合可以有效生成Co-Mo-S活性相,且配合物碳化减弱载体与活性相的相互作用,有利于生成有更高本征活性的II型活性相。
-
关键词:
- 有机配合物
- / 双齿配位分子
- / CoMo/γ-Al2O3
- / 加氢脱硫
- / 二苯并噻吩
English
Effect of coordinating groups of chelating agents on the hydrodesulfurization over CoMo/γ-Al2O3 catalysts
Abstract:
The modified CoMo/γ-Al2O3 catalyst was prepared by addition of ethylene diamine (EN), ethanolamine (EA), ethylene glycol (EG) or malonic acid (MA). The effect of four bidentate molecules with different coordination groups on the dibenzothiophene HDS was compared. And the catalytic activity is determined in the sequence of CoMo (EN) > CoMo (EA) > CoMo (EG)≈CoMo (MA) > CoMo. For all catalysts, the direct desulfurization route is dominated, but with the increase of reaction temperature, the desulfurization by hydrogenation route become more apparent. Chelating agents facilitate the HDS reaction through hydrogenation route. CoMo (EN) catalyst presents the highest hydrogenation ability. The catalysts were characterized by UV-vis, EA, XPS and HRTEM. The results show that NH2 group has a strong complexing interaction with Co2+. COOH group mainly has an electrostatic interaction with cobalt ion. Meanwhile, OH group hardly interacts with Co2+. It is noted that the HDS activity is directly related to the interaction between coordinating groups and Co2+. The combination of coordinating molecules with Co2+ leads to the effective formation of Co-Mo-S active center, and the carbonization of chelating decreases the interaction of the support with active phases, facilitating the formation of type Ⅱ active phases which has a higher intrinsic catalytic activity.
-
Key words:
- organic chelating agent
- / bidentate molecule
- / CoMo/γ-Al2O3
- / hydrodesulfurization
- / dibenzothiophene
-
加氢脱硫(HDS)是石油炼制和煤焦油加工的最重要环节之一,主要目的是脱除油品中的含硫杂质等,减少环境污染和提高油品质量[1]。近年来,环境法规对于油品中的硫含量限制越来越严格,美国对于柴油中的硫含量要求不大于15 μg/g,中国和欧盟则不大于10 μg/g[2]。超深度HDS使得柴油加工难度增加,成本升高,有效的解决方法是开发更高活性的催化剂。
氧化铝负载的Co(Ni)MoS2是工业上目前最常用的HDS催化剂。但由于氧化铝与活性相有强相互作用,易于形成较低活性的Ⅰ型Co(Ni)-Mo-S活性中心,同时助剂离子与载体形成尖晶石后,硫化会形成独立的Co9S8或NiSx相,因此, 表现出较低的HDS活性[3]。为此,近年来使用配合物辅助的方法来改进催化剂的制备。适量的添加配合物如环己二基四乙酸(CyDTA)、乙二胺四乙酸[4](EDTA)、次氮基三乙酸[5](NTA)、柠檬酸[6](CA)、三乙二醇[7](TEG)、聚乙二醇(PEG)、乙二胺[8](EN)或马来酸[9]等到前驱体溶液中,经浸渍且不经焙烧,可以制备高HDS活性的催化剂。研究表明,配合物的作用主要体现在三方面:延缓Co或Ni助剂的硫化,促使Co(Ni)-Mo-S活性相的形成;与Mo阴离子配位形成Mo配合物,减弱载体与金属的相互作用,形成有更高本征活性的Ⅱ型活性相;形成Co-Mo-C活性结构[10]。
对于硫化钼基加氢脱硫催化剂的制备可以选择多种有机配合物,但可选择的配位基团主要有三类:氨基类(-NH2)、羧基类(-COOH)和羟基类(-OH)。虽然已有大量研究涉及了多种有机配合物对HDS性能的影响[11, 12],但对于不同配位基团与金属离子的相互作用以及配位基团对催化结构和反应性能的影响还缺乏深刻认识,制约了配合物加氢脱硫催化剂的开发应用。本研究选择四种骨架结构类似但配位基团不同的双齿配位分子乙二胺、乙醇胺、乙二醇和丙二酸,采用分步浸渍的方法,不经焙烧制备出四种配合物改性的CoMo/γ-Al2O3催化剂,采用紫外可见光谱(UV-vis)、元素分析(EA)、X射线光电子能谱(XPS)和高分辨透射电镜(HRTEM)等,研究了配位基团对催化剂的结构和性质的调控,通过比较不同配合物对二苯并噻吩(DBT)的加氢脱硫性能的影响,揭示了不同配位基团在催化剂制备、硫化和反应过程中的作用,为配合物改性工业加氢脱硫催化剂提供了理论指导。
1 实验部分
1.1 催化剂的制备
以商业γ-Al2O3为载体,破碎筛分到40-60目,120 ℃烘干4 h后备用。将载体与一定浓度的七钼酸铵氨水溶液混合,等体积浸渍8 h后,经110 ℃干燥8 h和450 ℃焙烧4 h,得到Mo/γ-Al2O3。将Co(NO3)2与配位分子(乙二胺、乙醇胺、乙二醇或丙二酸)按物质的量比n(Co):n(配合物)=1:1.5制备成浸渍液,按吸水率等体积浸渍Mo/γ-Al2O3,4 h后,在110 ℃干燥8 h,不经焙烧得到改性催化剂,分别标记为CoMo(EN)、CoMo(EA)、CoMo(EG)和CoMo(MA)。将Co(NO3)2水溶液等体积浸渍Mo/γ-Al2O3,4 h后在110 ℃干燥8 h,450 ℃焙烧4 h,得到对照催化剂,标记为CoMo。催化剂CoO、MoO3的理论负载量分别为3.2%和16.0%(质量分数,按焙烧后计算)。
1.2 催化剂的硫化和二苯并噻吩脱硫活性评价
催化剂硫化和DBT加氢脱硫反应采用微型固定床反应器。所用反应器是长300 mm,内径6 mm的不锈钢管。筛分成40-60目的催化剂大约2 mL(相当于1.10 mmol MoO3和0.42 mmol CoO)装于反应器恒温区。系统压力为4.0 MPa,氢气流量为32 mL/min。硫化时,在氢气气氛下,100 min由室温升至200 ℃,然后以0.08 mL/min泵入1.5% (质量分数)的二甲基二硫醚(DMDS)正壬烷溶液,在200 ℃恒温2 h,以2.5 ℃/min升至350 ℃,在350 ℃恒温4 h,然后降至反应温度。反应评价在280、240、220、200和180 ℃温度点进行,反应原料油为800 μg (S)/g的DBT十氢萘溶液,以0.08 mL/min泵入,取样前使反应稳定12 h。液相产物使用配备FID检测器的Shimadzu GC-2010气相色谱仪分析。DBT脱硫反应为准一级反应[13],其转换频率(QTOF)计算公式为:
式中,r为反应速率,单位:mol/(molMo·min); x为二苯并噻吩转化率;F为反应物的单位摩尔流率,单位:mol/min;m为基于Mo的摩尔数,由于HDS反应在Co-Mo-S活性中心进行,Mo为主催化剂,所以用Mo的摩尔数近似代表活性中心数量。
为计算反应活化能,反应速率常数kHDS计算公式为[14]:
式中,F为二苯并噻吩的进料速率,单位:mol/min;W为催化剂质量,单位:g;x为转化率。
反应活化能(Ea)计算公式为:
式中,A为指前因子,Ea为反应活化能。
1.3 催化剂的表征
紫外-可见光谱(UV-vis):催化剂样品在美国Varian仪器公司的CARY 300型紫外可见分光光度仪上进行测试,扫描200-800 nm,硫酸钡作参考样。
元素分析(EA):采用德国Elementar公司的Vario EL Cube分析仪,测定反应后催化剂上的硫含量和碳含量。
X射线光电子能谱(XPS):采用英国Kratos公司的Axis Ultra Dld光谱仪。以Al Kα靶(1 486.6 eV)作为激发光源,真空度保持在3×10-8 Pa以下,功率150 W,以C 1s(284.6 eV)谱峰作为内标,对荷电效应引起的谱峰移动进行校准。使用XPSPEAK Version 4.1软件进行XPS谱图的拟合,采用灵敏度因子计算催化剂表面离子的相对浓度。
高分辨透射电镜(HRTEM):催化剂样品的形貌观察是在Tecnai G2 F20 S-Twin显微镜上进行,操作电压200 kV,活性相晶粒粒径分布通过随机选择400个以上的颗粒进行统计。(Co)MoS2晶粒的平均长度(L)和平均堆叠层数(N)根据以下公式计算[15]:
式中,Li代表晶粒长度,Ni代表晶粒层数,ni代表相同长度或堆叠层数的晶粒数量。
2 结果与讨论
2.1 配位基团对催化剂加氢脱硫性能的影响
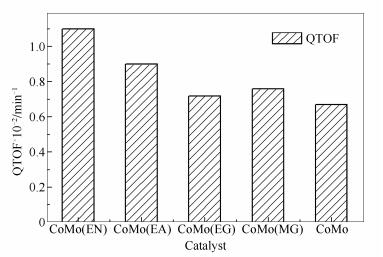
图 1为在200 ℃稳定状态下根据公式(1)计算的不同催化剂的QTOF值。由图 1可知,与CoMo对照催化剂相比,CoMo(EN)、CoMo(EA)、CoMo(EG)和CoMo(MA)的HDS活性分别提高了63.1%、34.1%、7.4%和13.6%。说明配合物的络合能力会直接影响制备的催化剂的反应活性。
图1
反应的QTOF值计算
Figure1.
HDS quasi-turnover frequency (QTOF) of the reaction over catalysts
由于乙二胺中的两个-NH2可以与Co2+形成强的双齿配位,因此,具有很强的络合能力。乙醇胺中的-NH2容易与Co2+配位,但羟基与Co2+作用很弱,导致该配位分子与-NH2的相互作用有所下降。丙二酸的-COOH虽然有一定的络合能力,但由于在酸性条件下不能充分解离,其配位能力受到较大限制,因此,主要与钴离子发生静电相互作用。而乙二醇中两个-OH与Co2+的相互作用很弱。图 2为四种双齿配体分子与Co2+相互作用的示意图。
 图2
配位分子与Co2+相互作用示意图
Figure2.
Scheme of interaction between chelating molecular and Co2+
图2
配位分子与Co2+相互作用示意图
Figure2.
Scheme of interaction between chelating molecular and Co2+
随着配合物与Co2+络合能力的增强,Co2+硫化延迟更加明显,这使得Co助剂更易于在Mo硫化以后配位到MoS2片层的侧边[16]。同时,配合物的碳化可以在氧化铝与活性相直接形成碳层,减弱活性金属与载体γ-Al2O3间的强相互作用,促进Mo的硫化,从而也使得催化剂活性有所提高。而对照催化剂形成的Co-Al尖晶石结构会影响Co的硫化,而且Co的预先硫化还可能形成基本没有脱硫活性的Co9S8。
2.2 配合物对催化剂活化能的影响
由公式(2)和(3)计算反应速率,取180、200和220 ℃温度点的数据作图,根据斜率计算反应的活性能,具体见图 3。
 图3
催化剂的lnkHDS与1/T的关系图以及计算的活化能
Figure3.
Relation of lnkHDS to 1/T and calculated apparent activation energy of catalysts
图3
催化剂的lnkHDS与1/T的关系图以及计算的活化能
Figure3.
Relation of lnkHDS to 1/T and calculated apparent activation energy of catalysts
由图 3可知,与对照的CoMo催化剂相比,加入配位分子的催化剂,反应的活化能不同程度的降低,其顺序为CoMo (EN) < CoMo (EA) < CoMo (EG)≈CoMo (MA) < CoMo。这表明配位分子的配位能力越强,会形成更多的具有更高本征催化活性的活性中心,使得表观活化能降低。由此推断配合物会促进Ⅱ型Co-Mo-S活性相的形成。
2.3 配合物和反应温度对DBT脱硫路径的影响
DBT类化合物主要有两种脱硫路径,即直接脱硫路径(DDS)和加氢路径(HYD)。DDS途径是反应分子中的硫原子被直接脱除;HYD途径则是分子中的芳环先加氢然后再脱硫。研究发现,CoMo催化剂以DDS途径为主[17, 18],产物为联苯(BP),加氢路径产物为环己基苯(CHB)。
图 4为各种催化剂在不同反应温度下的DBT脱硫产物分布图。
 图4
反应温度和配合物对脱硫产物选择性的影响
Figure4.
Effect of reaction temperature and chelating agents on the product selectivity of HDS
图4
反应温度和配合物对脱硫产物选择性的影响
Figure4.
Effect of reaction temperature and chelating agents on the product selectivity of HDS
由图 4可知,随着反应温度的升高,环己基苯(CHB)的选择性逐渐增大,说明提高温度有利于通过HYD途径脱硫。在低温(200和240 ℃)低转化率条件下,基本通过DDS途径脱硫,而当温度接近工业反应温度280 ℃时,HYD途径占比逐渐增大,这说明芳烃加氢的活性能更高,需要在较高温度下进行。本研究显示配位分子的加入有利于在280 ℃下DBT的芳烃加氢,而且配合能力越强,HYD路径的占比越高。因为芳烃加氢能力的提升有利于空间位阻的含硫分子如4-甲基二苯并噻吩和4, 6-二甲基二苯并噻吩的硫脱除,同时芳烃的加氢可以提高十六烷值,改善柴油品质。
2.4 紫外可见光谱表征
图 5为催化剂前体的紫外可见光谱谱图。根据文献报道[19],对于CoMo/γ-Al2O3催化剂,600 nm附近出现的Co2+三重峰代表非活性的CoAl2O4尖晶石结构的形成。由图 5可知,经焙烧的CoMo催化剂前体在600 nm附近出现明显的三重峰,表明Co2+和γ-Al2O3形成强的相互作用。而配合物改性的CoMo (EN)、CoMo (EA)、CoMo (EG)和CoMo (MA)催化剂前体,Co2+吸收峰分别蓝移至480、520和550 nm,说明加入配合物后不形成CoAl2O4尖晶石,这有利于硫化时Co2+迁移到MoS2相形成Co-Mo-S活性结构。而从本质上讲,反应的催化活性与配位分子中的配位基团与Co2+的络合能力强弱直接相关,从Co2+的蓝移程度推断出配位基团配位强弱顺序为:-NH2> -COOH> -OH。
 图5
催化剂前体的UV-vis光谱谱图
Figure5.
UV-vis spectra of precursor of the catalysts
图5
催化剂前体的UV-vis光谱谱图
Figure5.
UV-vis spectra of precursor of the catalysts
2.5 元素分析
催化剂经过二苯并噻吩的HDS反应后进行了硫和碳元素分析,结果见表 1。
 表 1
反应后催化剂S和C含量分析
Table 1.
Analysis of S and C contents of spent catalysts
表 1
反应后催化剂S和C含量分析
Table 1.
Analysis of S and C contents of spent catalysts
Catalyst S contents w/% C contents w/% Degree of sulfidation* /% CoMo (EN) 6.49 2.35 87 CoMo (EA) 6.04 1.97 81 CoMo (EG) 5.78 2.24 77 CoMo (MA) 5.81 2.13 78 CoMo 5.50 2.26 71 *:the calculation of sulfidity is based on transformation all Mo and Co atoms into MoS2 and CoS 由表 1可知,与对照的CoMo催化剂相比,加入配合物的催化剂,硫含量都有不同程度的提高,其顺序为CoMo (EN)>CoMo (EA)> CoMo (EG)≈CoMo (MA),说明配位分子能促进催化剂的硫化,有利于形成活性相,而且配位基团的配位能力越强,硫化程度越高。加入配位分子的催化剂与对照催化剂相比,碳含量没有发生明显变化,说明配位分子在硫化或反应过程虽然会有碳化发生,但并不一定会造成催化剂碳含量的相应增加。
2.6 X射线光电子能谱表征
图 6为反应后催化剂的Mo 3d XPS谱图。据文献报道[20],Mo具有不同的价态,结合能在229.0、231.2和232.7 eV附近的峰归于Mo 3d5/2的Mo4+、Mo5+及Mo6+,在226.2 eV附近的峰归于S 2s。催化剂中未硫化的Mo为+6价,硫化后Mo的价态将降低到+4和+5价,但由于氧化铝与载体的强相互作用,使得Mo不能完全硫化,图 6中Mo的XPS分峰显示,尽管+4和+5价的Mo占绝大多数,但仍有一部分的Mo处于+6价,未能得到硫化还原。
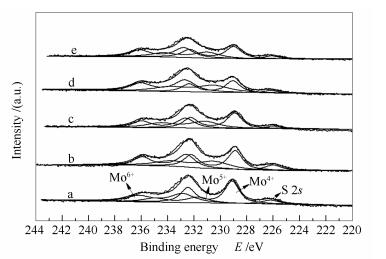 图6
反应后催化剂的Mo 3d XPS谱图
Figure6.
Mo 3d XPS spectra of the spent catalysts after HDS tests
图6
反应后催化剂的Mo 3d XPS谱图
Figure6.
Mo 3d XPS spectra of the spent catalysts after HDS tests
不同价态Mo的相对比例,以及催化剂表面的S/Mo比(以结合能在161 eV附近的S 2p峰计算)的结果见表 2。
表 2
反应后催化剂表面成分分析
Table 2.
Surface components determined by XPS of the spent catalysts
Catalyst Mo 3d5/2 E/eV FWHM E/eV Relative percentage/% S/Mo CoMo (EN) 229.1 1.31 49 1.9 231.7 2.00 19 232.8 2.20 32 CoMo (EA) 228.9 1.34 41 1.8 231.2 2.50 29 232.7 1.63 30 CoMo (EG) 228.9 1.32 38 1.7 230.5 2.88 31 232.6 1.57 31 CoMo (MA) 229.0 1.46 37 1.7 231.1 2.24 28 232.8 1.72 35 CoMo 229.0 1.31 27 1.6 230.5 2.90 34 232.7 1.90 39 由表 2可知,对于CoMo (EN)、CoMo (EA)、CoMo (EG)和CoMo (MA),Mo4+相对含量分别为49%、41%、38%和37%,高于CoMo催化剂的27%,而Mo4+和Mo5+的相对含量之和也高于对照催化剂。说明加入配合物使Mo得到更充分的还原硫化。S/Mo比的结果显示出相同的变化趋势,说明随着配合物基团络合能力增强,催化剂的硫化更加完全,这与表 1中硫元素的分析和硫化度的计算结果一致。
2.7 高分辨透射电镜表征
对反应后的催化剂做了HRTEM表征,具体见图 7。由图 7可知,在高倍电镜下观察到六边形(Co)MoS2片层的侧边呈现出条纹状,条纹的间距大约是0.62 nm,条纹的长度代表了晶粒宽度,条纹的个数代表堆叠层数。图 7显示(Co)MoS2催化剂颗粒的宽度基本处于1-4 nm,不同催化剂的颗粒宽度分布区别不大。催化剂的堆叠层数在1-4层,多数居于2层左右。
 图7
反应后催化剂的高分辨透射电镜照片
Figure7.
HRTEM images of spent catalysts
图7
反应后催化剂的高分辨透射电镜照片
Figure7.
HRTEM images of spent catalysts
表 3列出测量超过400个颗粒后统计的催化剂中(Co)MoS2微晶粒的平均宽度和平均堆叠层数。结果表明,加入配合物的催化剂,活性颗粒的平均宽度高于对照CoMo催化剂,平均堆叠层数也有不同程度的增加。其平均宽度和堆叠层数增加的程度与配位基团与Co金属离子的结合能力基本一致,反映出配体结合能力的强弱会影响活性相与载体的相互作用。
表 3
反应后催化剂(Co)MoS2晶粒的平均宽度和平均堆叠层数
Table 3.
Average slab length and stacking number of (Co)MoS2 grains in spent catalysts
Catalyst Average slab length /nm Average stacking number CoMo(EN) 2.97 1.87 CoMo(EA) 2.89 1.80 CoMo(EG) 2.86 1.67 CoMo(MA) 2.85 1.63 CoMo 2.80 1.58 加入配合物未经焙烧的催化剂,由于配合物与钴离子相互作用,一方面使Co的硫化延迟,使得Co离子可以配位在预先形成的MoS2相的侧边,形成所谓的Co-Mo-S活性中心;另一方面,由于Co在MoS2侧边配位,可以抑制Mo-O-Al键的形成[21],同时配合物的碳化降低活性相与载体的相互作用,使得催化剂更易于形成有高本征活性的Ⅱ型Co-Mo-S活性相。
总的来讲,出于环境和经济方面的考虑,应用配位分子对传统CoMo加氢脱硫催化剂进行改性已经成为制备新型催化剂过程中一个非常重要的方向。从之前的螯合剂出发,EDTA中的羧基和氨基基团(-COOH,-NH2)能够与Co2+作用,延迟Co2+的硫化,形成Co-Mo-S活性相,提高DBT加氢脱硫活性。NTA为典型的羧基配合物,Lelias等[5]研究了NTA对CoMoS2催化剂结构和活性的影响,发现NTA能明显促进常压噻吩和高压苯并噻吩加氢脱硫反应,但对DBT加氢脱硫反应却没有明显促进作用。而对另一种羧基配合物柠檬酸(CA), Rinaldi等[22]和Valencia等[23]研究发现,CA能很好促进DBT加氢脱硫过程。EN为典型的氨基配合物,氨基基团(-NH2)能够与Co2+形成双配位,同样能够提高DBT加氢脱硫活性。另外,乙二醇类物质为典型的羟基配合物,如TEG和PEG,其自身与Co2+作用力较弱,但其中的羟基官能团(-OH)能与γ-Al2O3中羟基和Al3+位点作用减弱活性相与载体的相互作用,从而促进形成Co-Mo-S活性相,提高催化活性[8]。综合考虑,近些年的研究主要是针对三种基团配位分子:氨基、羟基和羧基对催化剂进行改性。但是比较这三类基团对CoMo催化剂作用影响方面的研究报道还很少。因此,本研究尝试采用几种结构简单的双齿配位分子对CoMo催化剂进行改性,经实验和表征结果可知,对CoMo催化剂而言,其活性和选择性与配位分子同Co2+的作用力强弱有关,这对研究配位分子的作用和指导配合物的选择具有重要意义。但在具体制备催化剂过程中,配合物的作用同时受到多方面的因素,其中,pH值就是一个非常重要的因素,酸碱性能够影响配位分子与活性金属离子的配位形式和配合强弱,从而影响催化活性和选择性,因此,未来作者还将研究pH值对配合物改性的加氢脱硫催化剂的影响。
3 结论
以双齿配位分子为有机添加剂,采用分步浸渍法,未经焙烧制备的改性CoMo催化剂,与对照焙烧的CoMo催化剂相比,配位分子的加入能够不同程度提高催化剂对于DBT的HDS活性,其顺序为CoMo (EN)>CoMo (EA)>CoMo (EG)≈CoMo (MA)>CoMo,活性的增加程度与配位基团(-NH2, -COOH, -OH)同Co2+的作用力强弱直接相关。乙二胺和乙醇胺中的-NH2能够与Co2+形成强络合,推迟钴的硫化,减弱活性相与载体的相互作用,有利于形成具有更高本征活性的Ⅱ型Co-Mo-S活性中心,降低反应活化能。丙二酸和乙二醇中的-OH和-COOH与Co2+作用较弱,但其碳化形成中间碳物种减弱了活性组分与载体的相互作用,使得其脱硫活性也有一定程度的提高。同时配合物的加入促进HDS反应通过加氢路径进行,这有利于柴油的超深度脱硫和油品提质。
-
-
[1]
王腾飞, 张晔, 葛晖, 唐明兴, 周立公, 吕占军, 李学宽. 硫代硫酸铵预硫化的Mo/AC催化剂加氢脱硫性能的研究[J]. 燃料化学学报, 2015,43,(2): 202-207. WANG Teng-fei, ZHANG Ye, GE Hui, TANG Ming-xing, ZHOU Li-gong, LÜ Zhan-jun, LI Xue-kuan. Hydrodesulfurization of thiophene over Mo/AC catalyst presulfided by ammonium thiosulfate[J]. J Fuel Chem Technol, 2015, 43(2): 202-207.
-
[2]
范强. 柴油超深度加氢脱硫技术长周期生产国Ⅴ柴油的工业应用[J]. 当代石油石化, 2016,24,(12): 21-29. doi: 10.3969/j.issn.1009-6809.2016.12.005FAN Qiang. Industrial application of diesel ultra deep hydrodesulfurization technology producing V diesel oil[J]. Pet Petrochem Today, 2016, 24(12): 21-29. doi: 10.3969/j.issn.1009-6809.2016.12.005
-
[3]
TOPSOE H. The role of Co-Mo-S type structures in hydrotreating catalysts[J]. Appl Catal A:Gen, 2007, 322: 3-8. doi: 10.1016/j.apcata.2007.01.002
-
[4]
HIROSHIMA K, MOCHIZUKI T, HONMA T. High HDS activity of CoMo/Al2O3 modified by some chelates and their surface fine structures[J]. Appl Sur Sci, 1997, 21(6): 433-436.
-
[5]
LELIAS M A, KOOYMAN P J, MARIEY L. Effect of NTA addition on the structure and activity of the active phase of cobalt-molybdenum sulfide hydrotreating catalysts[J]. J Catal, 2009, 267(1): 179.
-
[6]
ESCOBAR J, BARRERA M C, GUTIERREZ A W. Benzothiophene hydrodesulfurization over NiMo/alumina catalysts modified by citric acid. Effect of addition stage of organic modifier[J]. Fuel Process Technol, 2017, 156: 33-42. doi: 10.1016/j.fuproc.2016.09.028
-
[7]
CIROSPEREZ J, GOMEZ , SERRA M. On the role of triethylene glycol in the preparation of highly active Ni-Mo/Al2O3, hydrodesulfurization catalysts:A spectroscopic study[J]. Appl Catal B:Enriron, 2015, 166-167: 560-567. doi: 10.1016/j.apcatb.2014.11.039
-
[8]
HUI G, LI X, QIN Z. Effects of carbon on the sulfidation and hydrodesulfurization of CoMo hydrating catalysts[J]. Korean J Chem Eng, 2009, 26(2): 576-581. doi: 10.1007/s11814-009-0098-6
-
[9]
BUI N Q, GEANTET C, BERHAULT G. Maleic acid, an efficient additive for the activation of regenerated CoMo/Al2O3, hydrotreating catalysts[J]. J Catal, 2015, 330: 374-386. doi: 10.1016/j.jcat.2015.07.031
-
[10]
GE H, WEN X D, RAMOS M A. Carbonization of ethylenediamine coimpregnated CoMo/Al2O3 catalysts sulfided by organic sulfiding agent[J]. ACS Catal, 2014, 4(8): 2556-2565. doi: 10.1021/cs500477x
-
[11]
CATTANEO R, WEBER T, SHIDO T. A quick EXAFS study of the sulfidation of NiMo/SiO2, hydrotreating catalysts prepared with chelating ligands[J]. J Catal, 2000, 191(1): 225-236. doi: 10.1006/jcat.1999.2784
-
[12]
IWAMOTO R, KAGAMI N, ⅡNO A. Effect of polyethylene glycol addition on hydrodesulfurization activity over CoO-MoO3/Al2O3 catalyst[J]. J Jpn Pet Inst, 2005, 48(4): 237-242. doi: 10.1627/jpi.48.237
-
[13]
VALENCIA D, KLIMOVA T. Kinetic study of NiMo/SBA-15 catalysts prepared with citric acid in hydrodesulfurization of dibenzothiophene[J]. Catal Commun, 2012, 21(9): 77-81.
-
[14]
INFANTES M A, ROMERO P A, SANCHEZ G V. Role of Cs on hydrodesulfurization activity of RuS2 catalysts supported on a mesoporous SBA-15 type material[J]. ACS Catal, 2011, 1(3): 175-186. doi: 10.1021/cs100053e
-
[15]
左东华, 聂红, VrinatM, 石亚华, LacroixM, 李大东. 硫化态NiW/AI2O3催化剂加氢脱硫活性相的研究Ⅰ. XPS和HREM表征[J]. 催化学报, 2004,25,(4): 309-314. ZUO Dong-hua, NIE Hong, VRINAT M, SHI Ya-hua, LACROIX M, LI Da-dong. Study on the hydrodesulfurization active phase in sulfided NiW/Al2O3 catalyst Ⅰ. XPS and HREM characterization[J]. Chin J Chem, 2004, 25(4): 309-314.
-
[16]
RANA M S, RAMIREZ J, GUTIERREZ A A, ANCHEYTA J, CEDENO L, MAITY S K. Support effects in CoMo hydrodesulfurization catalysts prepared with EDTA as a chelating agent[J]. J Catal, 2007, 246(1): 100-108. doi: 10.1016/j.jcat.2006.11.025
-
[17]
白天忠, 刘继华, 柳伟, 宋永一, 孙厚祥, 包洪洲. 柴油加氢脱硫机理的研究进展[J]. 广东化工, 2011,38,(9): 92-93. BAI Tian-zhong, LIU Ji-hua, LIU Wei, SONG Yong-yi, SUN Hou-xiang, BAO Hong-zhou. Study on the mechanism of diesel hydrodesulfurization[J]. Guangdong Chem Ind, 2011, 38(9): 92-93.
-
[18]
徐永强, 赵瑞玉, 商红岩, 赵会吉, 刘晨光. 二苯并噻吩和4-甲基二苯并噻吩在Mo和CoMo/γ-Al2O3催化剂上加氢脱硫的反应机理[J]. 石油学报, 2003,19,(5): 14-21. XU Yong-qiang, ZHAO Rui-yu, SHANG Hong-yan, ZHAO Hui-ji, LIU Chen-guang. Mechanism of hydrodesulfurization of dibenzothiophene and 4-methyldibenzothiophene on Mo/γ-Al2O3 and CoMo/γ-Al2O3[J]. Acta Pet Sin, 2003, 19(5): 14-21.
-
[19]
PAPADOPOULOU C, VAKROS J, MATRALIS H K. Preparation, characterization, and catalytic activity of CoMo/γ-Al2O3 catalysts prepared by equilibrium deposition filtration and conventional impregnation techniques[J]. J Colloid Interface Sci, 2004, 274(1): 159-166. doi: 10.1016/j.jcis.2003.11.041
-
[20]
QIU L, XU G. Peak overlaps and corresponding solutions in the X-ray photoelectron spectroscopic study of hydrodesulfurization catalysts[J]. Appl Surf Sci, 2010, 256(11): 3413-3417. doi: 10.1016/j.apsusc.2009.12.043
-
[21]
BERIT H, HENRIK T. A density functional study of the chemical differences between type Ⅰ and type Ⅱ MoS2-based structures in hydrotreating catalysts[J]. J Phys Chem B, 2005, 109(6): 2245-2253. doi: 10.1021/jp048842y
-
[22]
RINALDI N, KUBOTA T, OKAMOTO Y. Effect of citric acid addition on Co-Mo/B2O3/Al2O3 catalysts prepared by a post-treatment Method[J]. Ind Eng Chem Res, 2009, 48(23): 10414-10424. doi: 10.1021/ie9008343
-
[23]
VALENCIA D, KLIMOVA T. Citric acid loading for MoS2-based catalysts supported on SBA-15. New catalytic materials with high hydrogenolysis ability in hydrodesulfurization[J]. Appl Catal B:Enriron, 2013, 129(2): 137-145.
-
[1]
-

图 1 反应的QTOF值计算
Figure 1 HDS quasi-turnover frequency (QTOF) of the reaction over catalysts
reaction conditions: t =200 ℃, p=4.0 MPa, V(H2)/V(oil) ≈400, LHSV=2.4 h-1

图 2 配位分子与Co2+相互作用示意图
Figure 2 Scheme of interaction between chelating molecular and Co2+

图 3 催化剂的lnkHDS与1/T的关系图以及计算的活化能
Figure 3 Relation of lnkHDS to 1/T and calculated apparent activation energy of catalysts

图 4 反应温度和配合物对脱硫产物选择性的影响
Figure 4 Effect of reaction temperature and chelating agents on the product selectivity of HDS

图 5 催化剂前体的UV-vis光谱谱图
Figure 5 UV-vis spectra of precursor of the catalysts
a: CoMo(EN); b: CoMo(EA); c: CoMo(EG); d: CoMo(MA); e: CoMo

图 6 反应后催化剂的Mo 3d XPS谱图
Figure 6 Mo 3d XPS spectra of the spent catalysts after HDS tests
a: CoMo (EN) spent catalyst; b: CoMo (EA) spent catalyst; c: CoMo (EG) spent catalyst; d: CoMo (MA) spent catalyst; e: CoMo spent catalyst

图 7 反应后催化剂的高分辨透射电镜照片
Figure 7 HRTEM images of spent catalysts
(a): CoMo (EN) spent catalyst; (b): CoMo (EA) spent catalyst; (c): CoMo (EG) spent catalyst; (d): CoMo (MA) spent catalyst; (e): CoMo spent catalyst
表 1 反应后催化剂S和C含量分析
Table 1. Analysis of S and C contents of spent catalysts
Catalyst S contents w/% C contents w/% Degree of sulfidation* /% CoMo (EN) 6.49 2.35 87 CoMo (EA) 6.04 1.97 81 CoMo (EG) 5.78 2.24 77 CoMo (MA) 5.81 2.13 78 CoMo 5.50 2.26 71 *:the calculation of sulfidity is based on transformation all Mo and Co atoms into MoS2 and CoS  下载: 导出CSV
下载: 导出CSV
表 2 反应后催化剂表面成分分析
Table 2. Surface components determined by XPS of the spent catalysts
Catalyst Mo 3d5/2 E/eV FWHM E/eV Relative percentage/% S/Mo CoMo (EN) 229.1 1.31 49 1.9 231.7 2.00 19 232.8 2.20 32 CoMo (EA) 228.9 1.34 41 1.8 231.2 2.50 29 232.7 1.63 30 CoMo (EG) 228.9 1.32 38 1.7 230.5 2.88 31 232.6 1.57 31 CoMo (MA) 229.0 1.46 37 1.7 231.1 2.24 28 232.8 1.72 35 CoMo 229.0 1.31 27 1.6 230.5 2.90 34 232.7 1.90 39
下载: 导出CSV
表 3 反应后催化剂(Co)MoS2晶粒的平均宽度和平均堆叠层数
Table 3. Average slab length and stacking number of (Co)MoS2 grains in spent catalysts
Catalyst Average slab length /nm Average stacking number CoMo(EN) 2.97 1.87 CoMo(EA) 2.89 1.80 CoMo(EG) 2.86 1.67 CoMo(MA) 2.85 1.63 CoMo 2.80 1.58
下载: 导出CSV
-








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 1751
- HTML全文浏览量: 942
 下载:
下载:
