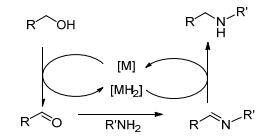
 图 图式1
金属催化醇胺化反应的氢转移策略
Figure 图式1.
Hydrogen transfer strategy for metal-catalyzed amination of alcohols
图 图式1
金属催化醇胺化反应的氢转移策略
Figure 图式1.
Hydrogen transfer strategy for metal-catalyzed amination of alcohols
引用本文:
吴慧, 武俊成, 杜正银. 后过渡金属催化醇对胺的N-烷基化反应研究进展[J]. 有机化学,
2017, 37(5): 1127-1138.
doi:
10.6023/cjoc201610034

Citation: Wu Hui, Wu Juncheng, Du Zhengyin. Progress in Late Transition Metal-Catalyzed N-Alkylation of Amines with Alcohols[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1127-1138. doi: 10.6023/cjoc201610034
Citation: Wu Hui, Wu Juncheng, Du Zhengyin. Progress in Late Transition Metal-Catalyzed N-Alkylation of Amines with Alcohols[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1127-1138. doi: 10.6023/cjoc201610034
后过渡金属催化醇对胺的N-烷基化反应研究进展
摘要:
胺的N-烷基化反应是一类重要的有机合成反应,其合成产物烷基化胺更是涉及到包括化工、医疗、医药以及国防等重要领域.近年来随着化学技术,尤其是绿色催化合成技术的发展,使得该类反应已成为当前有机反应的研究热点之一.以醇作为胺的N-烷基化试剂,水是唯一的副产物,因此具有绿色环保、简单可靠的特点.但是绝大多数该类反应都需要过渡金属催化剂.从脂肪胺、芳香胺、杂环芳香胺等含氮化合物出发,综述了近年来金属催化的以醇为烷基化试剂的胺的N-烷基化反应的研究进展,主要介绍了各均相、多相金属催化反应体系,通过比较指出了每一方法的优缺点,并对今后氮烷基化反应的发展方向进行了展望.
English
Progress in Late Transition Metal-Catalyzed N-Alkylation of Amines with Alcohols
Abstract:
N-Alkylation of amines is a class of important organic reaction. The products of this reaction are related to the important technological fields including chemical, medical, pharmaceutical and defense. In recent years, the development and need of green catalytic synthesis technology make such C-N bond formation reaction via N-alkylation become one of the current research focuses of organic chemistry. Because water is the only by-product, the N-alkylation of amines with alcohols as alkylating agents is green, environmental, simple and reliable. However, the most of such reactions require a late transition metal catalyst. In this paper the progress in late transition metal-catalyzed N-alkylation reaction with alcohols as alkylating agents starting from various aliphatic, aromatic and heterocyclic aromatic amines in recent years is reviewed. Various homogeneous and heterogeneous catalytic systems as well as the substrate application scope of each method involved in this reaction are mainly introduced. The expectation and development direction about this N-alkylation in future are suggested.
-
Key words:
- amine
- / alcohol
- / N-alkylation
- / late transition metal
- / green chemistry
-
氮的烷基化反应是一类重要的有机合成反应, 其合成产物涉及到包括化工、医疗、医药及国防等重要科技领域.近年来随着化学技术, 尤其是催化合成技术的发展, 该类反应已成为有机化学的研究重点之一.目前, 氮的烷基化反应大多是从含氮化合物出发, 以卤代烃、醛、胺等物质作为烷基化试剂, 在一定的条件下经缩合、氧化、还原、交叉偶联等反应得到烷基化胺.关于这一C—N键构建方面的研究, 早期有著名的Ullmann反应[1]和Goldberg反应[2].由于当时条件所限, 这些方法往往存在许多不足: (1) 反应中使用有机卤化物, 副产对环境有害的卤化氢和大量酸性废水; (2) 没有较好的催化体系, 反应往往需要200 ℃及以上高温; (3) 需要加入很多强碱作为缚酸剂, 产生大量无机固体废物; (4) 原子利用率低, 产物选择性差.
上世纪八十年代随着贵金属钯催化体系在卤代芳烃的胺化反应中的成功应用, Hartwig[3~7]、Buchwald[8~11]等研究小组对该催化体系进行不断的改进和完善, 建立了重要的C—N键构建方法——Buchwald-Hartwig胺化反应法.这一方法的发现极大的推进了该反应的发展, 但是由于反应中仍然需要脱除卤化氢并采用了高毒性的含磷配体, 使得该反应在其他领域的应用大大受到限制.
随着现代社会的发展, 绿色化学的崛起, 在保证高产率、高选择性的前提下, 更加高效、廉价、低毒性的催化体系成为当下该领域的追求.经过多年的研究发展, 醇已经被证实在反应中具有和卤代烷烃、卤代芳烃相同的性质, 成为新一代的烷基化试剂, 并且以醇作为烷基化试剂与芳胺反应合成取代芳胺, 具有原料廉价易得, 原子利用率高, 副产物只有水等诸多优点.因此以醇作为烷基化试剂的绿色有机合成方法, 越来越受到研究者青睐.根据催化反应体系的不同可将醇胺化反应大致分为以下三类: (1) 金属催化, 分均相金属催化和多相金属催化反应; (2) 有机小分子催化, 即采用有机化合物、离子液体等其他小分子化合物来催化该类反应; (3) 无催化剂, 即采用无机金属盐或碱类化合物来促进该类反应.
过渡金属催化醇胺化反应的机理因醇的结构和催化体系的不同而不同, 主要有以下三种机理: (1) 直接胺化或碳正离子机理, 指在质子酸作用下醇脱水形成碳正离子, 其后发生胺化反应; (2) 氢转移胺化[12], 又称“借氢策略”, 即在过渡金属催化下醇脱氢转变成醛, 醛与胺缩合成亚胺, 亚胺再被加氢还原成饱和胺, 如Scheme 1所示, 反应不需外加氢源; (3) 配位-亲核胺化, 类似Tsuji-Trost反应, 指烯丙醇先与过渡金属形成烯丙基配位络合物, 进而发生亲核胺化生成产物, 如Scheme 2所示钯催化烯丙醇的胺化反应机理[13]; (4) 氧化-氢转移胺化, 即醇先被氧化成醛, 醛与胺反应生成亚胺, 亚胺在金属催化下与醇发生氢转移反应生成产物仲胺, 如Scheme 3所示的需氧氧化-氢转移胺化机理.脂肪醇在过渡金属催化下大多经历氢转移机理发生胺化反应.
图 图式1
金属催化醇胺化反应的氢转移策略
Figure 图式1.
Hydrogen transfer strategy for metal-catalyzed amination of alcohols
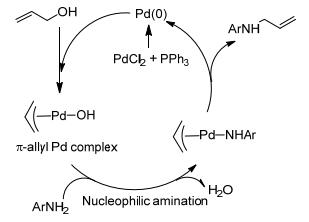 图 图式2
钯催化烯丙醇的胺化反应机理
Figure 图式2.
Mechanism of palladium-catalyzed amination of allylic alcohols
图 图式2
钯催化烯丙醇的胺化反应机理
Figure 图式2.
Mechanism of palladium-catalyzed amination of allylic alcohols
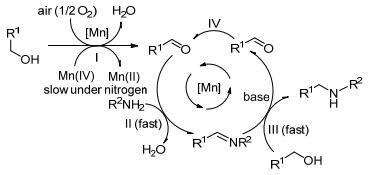 图 图式3
MnO2催化合成单取代胺的反应机理
Figure 图式3.
Mechanism of MnO2-catalyzed synthesis of monosubstituted amines
图 图式3
MnO2催化合成单取代胺的反应机理
Figure 图式3.
Mechanism of MnO2-catalyzed synthesis of monosubstituted amines
本文从脂肪胺、芳香胺、杂环芳香胺等含氮化合物出发, 以金属在周期表中所在的族为分类线索, 综述了近10年来金属催化的以醇为烷基化试剂的胺的N-烷基化反应的研究进展, 主要介绍了各均相、多相金属催化反应体系, 通过比较指出了每一方法的优缺点, 并对今后氮烷基化反应的发展方向进行了展望.
1 VIIB族金属催化的胺与醇的反应
2011年, Xu课题组[14]以苯磺酰胺和苄醇的反应为模板, 进行了一系列金属氧化物催化剂的筛选, 发现毒性较低的MnO2是该反应的良好催化剂.作者对反应条件进行筛选后, 最终表明胺醇物料比为3:4, 以10~20 mol% MnO2催化, K2CO3为碱, 在无溶剂条件下主要得到单取代产物.反应底物不仅适用于磺酰胺, 芳香胺也有很好的反应结果.机理研究表明这是一个锰催化需氧氧化-氢转移的串联反应过程(Scheme 3).
2 Ⅷ族金属催化的胺与醇的反应
2.1 铁、钌催化醇胺化反应
最早的Ru催化体系用于胺与醇的反应是在1981年由Grigg[15]和Watanabe[16]分别报道的. 1984年, Watanabe等[17]进一步采用醇作为胺的烷基化试剂, 分别对Ru催化剂的种类及用量, 胺与醇的结构和取代基的适用性进行了详细地研究, 确定了以RuCl2为催化剂, PPh3为配体, 胺醇物料比为1:2的催化体系, 催化芳香胺和伯醇、仲醇(如2-丙醇、2-丁醇)反应生成单取代产物, 但是仲醇与胺反应的单取代产物产率较低.这是较早研究后过渡金属催化的以氢转移机理进行的醇胺化反应的几例报道之一.
在2007年, Williams课题组[18]以[Ru(p-cymene)-Cl2]2为催化剂催化苯乙醇和叔丁胺的反应(Eq. 1), 结果发现在膦配体dppf、K2CO3、3 分子筛存在下反应较好.该反应体系不足之处是催化体系相对复杂, 反应时间也长达24 h, 反应底物种类不多, 收率范围为69%~100%, 不是很稳定, 变化比较大.
2014年, zdemir课题组[19]以苯并咪唑类化合物L1~L6为配体, 与[Ru(p-cymene)Cl2]2反应制得“半夹心型”钌(Ⅱ)配合物1~6.随后该课题组以苯胺和苄醇的反应为模板反应开始了催化条件的筛选, 发现以1为催化剂, KOBu-t为碱, 在150 ℃下反应15 h, 单取代产物产率高达98% (Scheme 4).模板反应不变, 对配体进行筛选时, 他们发现催化剂1~6都可以在适当的碱条件下催化模板反应得到产率超过95%的单取代N-烷基化产物.这比前述Williams课题组单纯以[Ru(p-cymene)-Cl2]2为催化剂要好得多.但遗憾的是该体系没有尝试去催化脂肪醇与脂肪胺的反应.
 图 图式4
Ru(Ⅱ)催化剂的制备
Figure 图式4.
Preparation of Ru(Ⅱ) catalysts
图 图式4
Ru(Ⅱ)催化剂的制备
Figure 图式4.
Preparation of Ru(Ⅱ) catalysts
多相催化体系较均相催化体系相比具有贵金属负载量少, 催化剂可回收利用的优点. 2009年, Mizuno课题组[20]以苯胺与苄醇的反应为模板, 较系统地研究了RuO2, Ru(OH)x, RuCl3•nH2O, RuCl2(PPh3)3, [RuCl2(p-cymene)]2, Ru(acac)3, Ru3(CO)12等钌化合物、络合物作为均相催化剂和RuClx/Al2O3, Ru/C, Ru/HAP(羟基磷灰石), Ru(OH)x/Al2O3等不同载体负载的钌化合物作为多相催化剂的催化效果, 发现一个压强Ar保护下, 以Ru的含量为5 mol%的Ru(OH)x/Al2O3为催化剂时反应产物选择性好、收率高.当反应完成后, 把催化剂过滤出来, 用NaOH (0.1 mol/L)溶液和水进行洗涤, 然后烘干再催化同样的反应, 同样的条件下, 产物产率基本没有变化, 说明催化剂循环利用效率很高.研究表明不需要添加任何助催化剂、碱和配体, 该催化体系可催化各种芳香族胺和杂环芳胺与芳香醇反应转化成相应的仲胺.该方法的不足之处是不能很好地催化脂肪胺与醇的烷基化反应, 如苯甲醇与苯胺反应产率为95%, 而苯甲醇与叔丁胺反应产率为74%.机理研究[21~26]证实该反应经历过程是转氢过程, 作者在一个大气压氧气条件下, Ru(OH)x/Al2O3催化苯甲醇与苯胺的反应仅产生亚胺, 并没有N-苄基苯胺形成.为了进一步验证氢化亚胺的氢是否来自苯甲醇, 作者进一步进行了实验验证, 在醇的存在下, 亚胺才能转化为胺(Scheme 5).类似的, 该课题组[27]还将Ru(OH)x负载在TiO2上作为醇胺化反应的多相催化剂, 发现以Ru为2 mol%的载入量的Ru(OH)x/TiO2, 该催化体系不仅可以催化几种杂环芳香醇和杂环芳香胺的反应, 而且产率较高.如图所示, 该催化体系催化苄醇和邻苯二胺快速合成咪唑(Eq. 2).反应完成后, 可以通过过滤回收Ru(OH)x/TiO2催化剂, 并且回收的催化剂重新使用也表现出高催化性能.
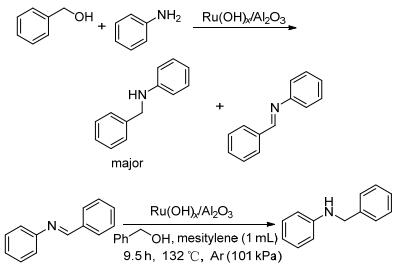 图 图式5
Ru(OH)x/Al2O3催化剂的应用
Figure 图式5.
Application of Ru(OH)x/Al2O3 catalyst
图 图式5
Ru(OH)x/Al2O3催化剂的应用
Figure 图式5.
Application of Ru(OH)x/Al2O3 catalyst
以磁性铁氧化物作为催化剂载体可使多相贵金属催化剂更易回收、减少损失. 2011年, Ramon等[28]在对Fe3O4负载的Co(OH)2, Ni(OH)2, Cu(OH)2, Ru(OH)2, Pd(OH)2, Pd(Ⅱ/0)/Cu等一系列催化剂筛选后, 指出Ru(OH)2/Fe3O4能够很好地催化醇与胺、磺酰胺、亚磺酰胺的氮烷基化反应(Scheme 6), 其大部分底物的单取代产物的产率高达99%.当反应完成, 用磁铁将催化剂捕获, 用甲苯洗涤, 用于反应循环.当催化剂重复使用10个循环, 反应产率还是很好, 均高于90%.
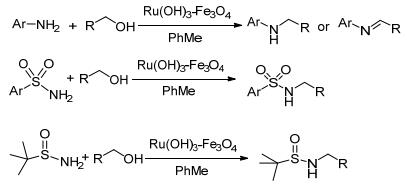 图 图式6
Ru(OH)2/Fe3O4催化剂的应用
Figure 图式6.
Application of Ru(OH)2/Fe3O4 catalyst
图 图式6
Ru(OH)2/Fe3O4催化剂的应用
Figure 图式6.
Application of Ru(OH)2/Fe3O4 catalyst
2014年, Vos课题组[29]以2 wt%的三价钌Ru/HAP为催化剂, 钙羟基磷灰石(HAP)作为载体, 高效实现了醇对胺的N-烷基化反应.反应过程中不需要添加碱, 而且催化剂在反应之前也不需要碱处理.研究表明该方法具有非常广泛的底物适用性:芳伯胺、仲胺、环状脂肪胺、长链脂肪胺以及苄醇、长链脂肪醇都有很好的反应活性, 比如正辛胺和正辛醇的反应(Eq. 3).比较而言, 脂肪胺与脂肪醇的反应选择性较芳胺和苄醇的反应选择性差, 收率和芳胺和苄醇的反应收率相比也稍低.这是目前报道的醇胺化反应催化效果最好的体系之一.
尽管贵金属催化剂催化活性很高, 但由于其稀有性和高成本促使化学家努力寻找廉价金属替代物. 2011年, Saito课题组[30]以FeX3为催化剂, 以卤代烷作烷基化试剂时经常用到的氨基酸L7~L15作配体(图 1), 催化苯胺与苄醇的反应.通过一系列精细筛选, 最后确定以FeBr3为催化剂、焦谷氨酸(L7)为配体, 1, 2, 4-三甲基苯(TMB)作溶剂, 1, 2, 3, 4, 5-五甲基环戊二烯(Cp*H)作为添加剂(Eq. 4).该方法尽管可以以较高的产率得到单取代产物, 但是反应温度高, 反应时间也较长.
 图 1
氨基酸衍生物L7~L15的结构
Figure 1.
Structures of amino acid derivatives L7~L15 FeBr3
图 1
氨基酸衍生物L7~L15的结构
Figure 1.
Structures of amino acid derivatives L7~L15 FeBr3
2.2 铑、铱催化醇胺化反应
徐清等[31]以Ru、Rh、Ir的简单化合物和络合物为催化剂研究了磺酰胺与苄醇的交叉偶联反应, 并通过大量对比实验对反应机理做了很详细的研究.作者认为, 由于胺、酰胺通常能使催化剂失活, 这是金属催化N-烷基化反应中醇的厌氧脱氢活化为醛和氢化金属的瓶颈.相对于需要大量的金属配合物或者添加配体来活化催化剂的厌氧醇氢自动转移反应, 金属催化的醇需氧氧化, 在热力学上更可行、更有效、更有利于醛的生成. RhCl3、RuO2等许多更简单、无配体的金属催化剂在有氧条件下也可以高效地完成催化N-烷基化反应, 产物收率高达99%, 具体反应机理类似图 3的描述, 即醇在金属催化下先被氧气氧化成醛, 其后再发生缩合和氢转移反应生成产物.该方法的突出优点是不需要惰性气体保护, 也不用添加配体, 更不用提前制备金属络合物, 操作简单, 产物收率高.
对于金属催化的N-烷基化反应, 发现并制备新型、高效催化剂和配体是一个重要的研究内容. 2009年Kempe课题组[32]首先通过1 equiv.的N-P化合物7和0.5 equiv.的[IrCl(cod)2] (cod=1, 5-cyclooctadiene), 以四氢呋喃为溶剂, 在室温下合成新型铱金属络合物8, 随后将其应用到2-氨基吡啶与苄醇的反应中, 其优点在于反应温度低, 70 ℃下, 2-氨基吡啶与苄醇以1:1, 0.05 mol% [IrCl(cod)2], 0.1 mol%Py2NPiPr2 (7), 0.2 mL二甘醇二甲醚作溶剂, 添加KOBu-t作为碱的情况下, 产物收率高达98% (Scheme 7).邻位和对位取代的苄醇可以很好地和芳胺反应, 产率在71%~93%, 取代苄醇、脂肪醇(如甲醇、正丁醇)可以和2-氨基吡啶、2, 6-二氨基吡啶、2-氨基-6-苄基吡啶胺进行反应, 当邻位取代基位阻比较大时, 反应需要负载量0.2 mol% Ir增加到0.6 mol% Ir, 以及延长反应时间.该反应催化体系简单高效, 是合成N-苄基吡啶-2-胺类化合物的有效方法.
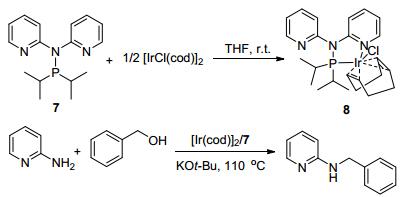 图 图式7
铱络合物8的制备及应用
Figure 图式7.
Preparation and application of Ir complex 8
图 图式7
铱络合物8的制备及应用
Figure 图式7.
Preparation and application of Ir complex 8
尽管铱络化物8是2-氨基吡啶和苄醇反应的有效催化剂, 但是却不能催化苯胺与苄醇的反应, 只有将铱的加入量增加到0.6 mol%(原来0.05 mol%), 才能实现原料的完全转化和90%以上的产物产量.作者认为氨基吡啶的存在可能改变了催化剂的性能.随后他们用核磁共振谱对反应进行跟踪, 发现配合物8确实会在碱的存在下与2-氨基吡啶形成一个新的化合物9.课题组以PyHNPR2 (R=i-Pr)为原料, 经两步反应, 最终以80%的产率得到络合物9 (R=i-Pr).用同样的方法, 以PyHNPR2(R=Ph), 得到络合物10 (R=Ph).实验发现以0.05~0.4 mol%的9为催化剂, 在70 ℃的较低温度下即可催化芳胺和苄醇的N-烷基化反应, 单取代产物的收率高达98% (Scheme 8).
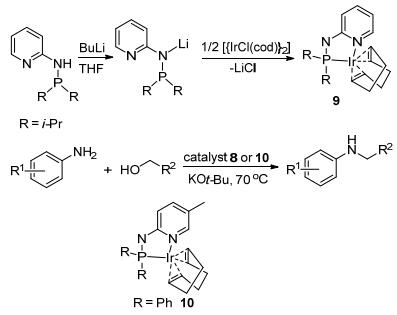 图 图式8
络合物9和10的制备和应用
Figure 图式8.
Preparation and application of complexes 9 and 10
图 图式8
络合物9和10的制备和应用
Figure 图式8.
Preparation and application of complexes 9 and 10
我们知道水分子是氢的良好受体, 且水是最便宜, 安全和环保的绿色溶剂, 耐水性和水溶性催化剂的开发已经成为一个活跃的研究领域[33, 34]. 2010年, Fujita课题组[35]以[Cp*Ir(NH3)3][I]2为催化剂, 在水相中140 ℃下成功实现了氨水与醇的N-烷基化反应, 该催化剂克服了难溶于水和空气中不稳定的困难, 催化氨水与伯醇和仲醇合成多种叔胺和仲胺, 且催化剂可以循环利用并保持高活性.同年, 作者还以[Cp*Ir(NH3)3][I]2为催化剂, 水为溶剂, 在回流温度下催化芳胺与苄醇的反应[36], 发现其在纯水相中的催化活性非常高, 不仅能使芳伯胺与各种苄醇反应以83%~93%的收率获得单取代产物, 而且可以催化多种仲胺、长链脂肪胺与长链烷醇的反应; 苄胺和端二醇在上述条件下还可以一步制备取代的环胺(Scheme 9).该反应机理包括三个基本步骤(脱氢、亚胺的形成和转移氢化), 也是典型的氢转移机理.上述结果说明水对该催化剂催化的醇胺化氢转移反应没有影响.这是目前报道的在纯水相中进行的最高效的醇胺化反应, 是名副其实的烷基化胺的绿色合成方法.
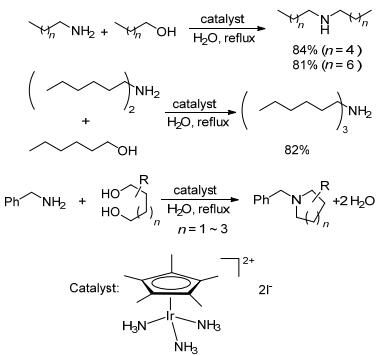 图 图式9
[Cp*Ir(NH3)3]I2的应用
Figure 图式9.
Application of [Cp*Ir(NH3)3]I2
图 图式9
[Cp*Ir(NH3)3]I2的应用
Figure 图式9.
Application of [Cp*Ir(NH3)3]I2
2.3 镍、钯催化醇胺化反应
我们知道钯催化的交叉偶联反应是构建C—C键的有效方法之一, 纳米钯颗粒在C—C键的偶联反应中也表现出极高的活性[37~39].那么, 纳米钯能否应用于醇胺化催化反应中呢? García课题组[40]以PdCl2为原料, 经甲基化、热解还原、零价钯生长三步制备了纳米钯颗粒, 并将其应用到甲醇和十六烷胺的模版反应中, 发现在1.72 MPa氢气存在下二者在密闭体系中100 ℃反应72 h可以100%的收率得到十六烷胺的异丙醇单取代产物(Scheme 10).该催化体系催化十六烷胺和仲醇(异丙醇)、叔醇(叔丁醇)合成相应的单取代产物, 催化多种脂肪胺和脂肪醇合成单取代产物, 但这一催化体系的缺点是只有脂肪胺和脂肪醇(甲醇、异丙醇、叔丁醇)参与反应, 没有涉及芳香醇和芳香胺, 随着氢气的压力由1.72MPa降至1.38 MPa, 十六烷胺的异丙醇单取代产物的产率由100%降至60%.
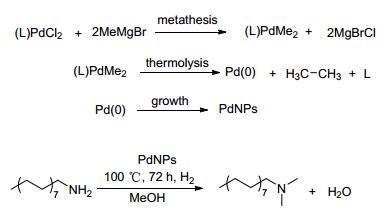 图 图式10
Pd NPs的制备与应用
Figure 图式10.
Preparation and application of Pd NPs
图 图式10
Pd NPs的制备与应用
Figure 图式10.
Preparation and application of Pd NPs
在醇胺化反应中, 脂肪族伯醇、仲醇及仲胺因反应活性低, 易氧化而成为研究的一个难点. 2013年, Seayad研究小组[41]以PdCl2为催化剂, 通过筛选大量的单膦、双膦配体, 最终以1, 2-双二苯基膦乙烷(dppe)为配体、LiOH为添加剂, 在100~150 ℃下实现了多种伯胺、环仲胺与伯醇的烷基化反应, 也可使易氧化的脂肪族伯醇、仲醇与仲胺发生N-烷基化反应生成相应的单取代产物, 收率在73%~98%间(Scheme 11).该方法的优点是直接以氯化钯作为催化剂, 不用制备复杂的钯络合物, 避免了无水无氧操作; 缺点是反应时间较长, 普遍在24 h以上, 反应温度也较高.
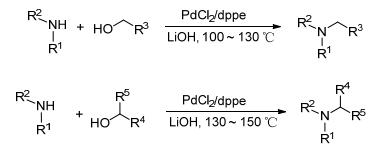 图 图式11
氯化钯催化的醇胺化反应
Figure 图式11.
PdCl2-catalyzed amination of alcohols
图 图式11
氯化钯催化的醇胺化反应
Figure 图式11.
PdCl2-catalyzed amination of alcohols
以烯丙醇作为烷基化试剂与胺发生交叉偶联反应是近年来发展的一种制备烯丙基胺的简单、高效的方法, 采用非对称结构的烯丙基仲醇作为烷基化试剂还可以构建手性烯丙基碳. 2014年, Beller课题组[42]用Pd(dba)2催化外消旋2-环己烯-1-醇与苯胺的对映选择性胺化, 对配体、添加剂、溶剂、反应温度等进行了一系列的筛选和优化, 发现以手性磷酸10为手性诱导试剂, 以亚磷酰胺L16为配体, 在四氢呋喃溶剂中25 ℃下反应48~ 72 h, 以接近100%的定量产率得到对映选择性产物, e.r.值为96:4~85:15 (Eq. 5).催化体系中手性磷酸还能促进活性烯丙基钯中间体的形成.以反应活性较差的链烷基烯丙基醇代替2-环己烯-1-醇也能取得很好的反应结果.与传统的N-烯丙基化方法相比, 这种烯丙醇的Tsuji-Trost反应更具有实用性, 有利于医药片段的准确定向合成.
近来, 我们课题组[43]以烯丙醇为烷基化试剂对芳胺进行了水相N-烷基化反应研究(Scheme 12).条件优化结果表明, 以2 mol% PdCl2为催化剂, 5 mol%三苯基膦为配体, 以二氧六环和水的混合物(体积比2:1) 作为溶剂, 添加10 mol%的三氟乙酸, 当芳胺与烯丙醇的物质的量比为1:3, 在回流温度下反应结果最好, 产物收率最高90%.当苯胺邻位无取代基时, 单烯丙基取代产物和双烯丙基取代产物的物质的量比在2:1~4:1间; 当苯胺邻位有一个或两个取代基时, 单取代产物的选择性几乎为100%;苯胺上取代基的电子效应对反应影响不大, 位阻效应则较明显.反应过程中烯丙基伯醇、仲醇会发生重排, 主要生成相应的空间位阻较小的烯丙基产物; 烯丙基叔醇如2-甲基丁-3-烯-2-醇则生成100%的重排产物.互为异构体的3-丁烯-2-醇和2-丁烯-1-醇的胺化反应结果说明, 该烯丙基化反应经历了(π-烯丙基)钯络合物中间体后发生亲核胺化, 即配位-亲核胺化机理(图 2).这是首例由氯化钯催化的水相中的烯丙醇胺化反应, 底物适用范围广, 反应条件温和, 操作简单.
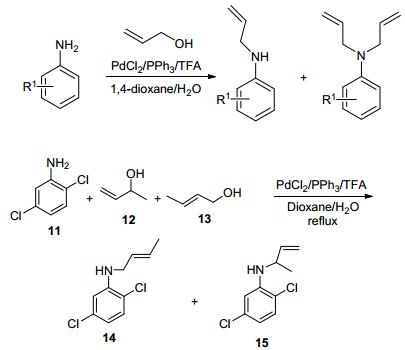 图 图式12
PdCl2催化芳香胺的烯丙基化
Figure 图式12.
PdCl2-catalyzed allylation of aromatic amines
图 图式12
PdCl2催化芳香胺的烯丙基化
Figure 图式12.
PdCl2-catalyzed allylation of aromatic amines
2011年, Shi等[44]用Fe2O3负载的钯(Ⅱ)催化苯胺与苄醇的反应, 生成N-苄基苯胺、亚胺、N, N-二苄基苯胺三种产物, 总收率为100%, 三者的比例是47:1:2 (Eq. 6).将该催化剂应用到取代苯胺和取代苄醇的反应中发现, 可以通过反应时间和反应物的位阻大小来控制产物, 位阻小的反应物反应时间短则得到单取代产物, 延长反应时间则有双取代产物生成; 对于位阻大的反应物, 延长反应时间并没有双取代产物生成.该方法的优点是不需要添加碱, 缺点是反应温度较高.在机理验证过程中, 课题组设计了一个N-苄基苯胺与对甲基苯胺和苄醇的竞争反应.从结果可以看出, 对甲基苯胺首先会和苄醇反应, 产物是亚胺和单取代产物, 没有得到双取代的叔胺产物, 说明N-苄基苯胺没有参与反应.所以可以通过简单地改变反应时间来合成单烷基化产物或二烷基化产物(Eq. 7).
对于醇胺化反应的多相催化体系, 除了上述铁氧化物、HAP外, 其他金属氧化物、铝镁水滑石(HT)、碳材料也可作载体. Sabater课题组[45]将贵金属Pd(Ⅱ)、Pt(Ⅱ)、Au(Ⅲ)负载到MgO、HAP、HT、活性炭上, 并将其应用到苯胺与苄醇的反应中.如表 1, 负载催化剂中发现Pd(0)/MgO活性最高, Pd的载入量仅为0.8 %, 在氮气保护下, 胺的转化率最高达到99%, 亚胺产率为13%, 单取代产物的产率为80% (Eq. 8).该催化方法可以催化脂肪醇(长链醇、丙烯醇类、二醇)、芳香醇和苯胺、二胺生成相应的亚胺、单取代产物、环二胺.
 表 1
负载钯催化剂的筛选
Table 1.
Screening of supported palladium catalysts
表 1
负载钯催化剂的筛选
Table 1.
Screening of supported palladium catalysts
Entry Catalyst Yield/% of amine Yield/% of imine 1 Pd/MgO (0.8%) 79 13 2 Pd/C (5%) 28 23 3 Pd/HT (0.55%) 49 11 4 Pd/HAP (0.55%) 26 39 5 Au/MgO (1.0%) 38 49 6 Pt/MgO (1.0%) 61 21 表 1 负载钯催化剂的筛选
Table 1. Screening of supported palladium catalysts2013年, Shiraishi等[46]根据载有贵金属粒子的半导体二氧化钛在室温惰性气体氛围下, 可由光激发促进醇脱氢产生醛为依据, 尝试将纳米钯负载到二氧化钛上催化胺与醇的烷基化反应.作者在催化剂筛选过程中发现该反应的效率强烈地依赖于钯颗粒的大小, 当钯的载入量为0.3 wt%且钯的颗粒粒径为2~2.5 nm时, Pd/TiO2显示出极高的催化活性.该催化剂在λ>300 nm的光照下, 氮气保护, 室温反应3~16 h, 可使苯甲醇、脂肪醇与脂肪胺、芳香胺反应的单取代产物收率范围为82%~98%.底物拓展表明乙醇、丁醇等活性较低的链烷醇都可以和多种苯胺类化合物反应得到相应的单取代产物.这是目前报道的贵金属负载量最少的醇胺化反应的多相催化体系.
2014年, Pera-Titus研究小组[47]以氧化锰(Ⅲ/Ⅳ)八面体分子筛(K-OMS-2) 为载体, 将高锰酸钾溶液、硫酸钾溶液、浓硝酸混合, 110 ℃下回流24 h, 将得到的红褐色固体洗涤、过滤, 在120 ℃干燥12 h.随后通过浸渍的方法将纳米Pd负载上去就得到所要的催化剂.将该负载催化剂应用到苯胺和苄醇的反应中, 160 ℃, 3 h, 单取代产物收率96%, 原料苯胺转化率达99%, 遗憾的是:该催化体系只是一个催化剂的详细表征研究, 并没有应用到其他底物拓展(Eq. 9)
2013年, Shimizu研究小组[48]通过离子交换法制得NiO/CaSiO3, 最后通氢气将NiO/CaSiO3还原为Ni(0)/ CaSiO3.在相对温和的条件下, 用该催化剂催化醇和NH3 (0.4 MPa)定向合成伯胺(Eq. 10).底物扩展实验结果表明, 各种脂肪醇都可以发生反应, 使伯胺生成仲胺, 仲胺生成叔胺, 产物收率为70%~89%.此催化剂不仅可回收重新使用, 第二次使用时产率是95%, 催化活性没有降低.该方法的创新之处是用廉价的Ni负载催化剂代替了贵金属, 而催化效果完全可以和贵金属相媲美.
Mashima课题组[49]用Ni(COD)2 (COD为1, 5-环辛二烯)催化烯丙醇类化合物与二苯胺、苯胺的反应, 催化体系中添加四正丁基醋酸铵和分子筛可有效地提高反应产率.在催化机理的研究中, 作者捕获到了烯丙基镍配合物, 说明该反应的机理为Scheme 2所示的配位-亲核胺化.通过对四氯化钛催化烯丙醇类化合物与苯胺类化合物的N-烷基化反应的理论计算, 进一步验证了烯丙基金属配合物中间体的存在, 为人们提供了更可信的机理[50].
3 IB族金属催化胺与醇的反应
2011年, Ramon研究小组[51]以苯胺和苄醇为模版反应对多种后过渡金属化合物、络合物的催化性能进行了详细考察, 确定了以Cu(OAc)2为催化剂, KOBu-t为碱, 130 ℃下, 二氧六环为溶剂的催化体系, 并催化合成了多种单苄基取代的苯胺, 收率最高可达99%.随后该课题组将催化体系中的碱稍做调整, 完成了2-氨基吡啶、苯甲酰胺、磺酰胺和磷酰胺等的醇胺化反应, 并以较高的产率得到单取代产物(Scheme 13).该方法所用催化剂廉价易得, 用量少, 底物适用范围宽, 是一种绿色、经济、可行的醇胺化反应方法.
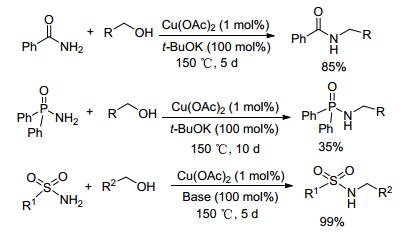 图 图式13
Cu(OAc)2的应用
Figure 图式13.
Application of Cu(OAc)2
图 图式13
Cu(OAc)2的应用
Figure 图式13.
Application of Cu(OAc)2
将银负载在氧化物载体上也能催化苄醇和芳香胺的N-烷基化反应. Shimizu等[52]对Fe、Ni、Co、Zn、Cu、Pt、Ag等后过渡金属和Al2O3、CeO2、MgO、SiO2、SiO2-Al2O3等不同氧化物载体制备的负载催化剂进行活性测试, 筛选出最好的催化剂为平均粒径大小为0.84 nm的Ag/Al2O3, Ag的负载量为5 wt%, 同时需添加5 mol%的水合三氯化铁.该体系不仅可以催化芳基伯醇的胺化反应, 芳基仲醇与芳香胺也能生成单取代的胺(Eq. 11).反应机理研究表明Ag在催化过程中所形成的Ag—H键和其他贵金属(Au、Pd、Pt)相比更能促进中间体亚胺的还原.作者认为这个反应可以进行的四个重要因素是: (1) 金属和氢可以形成一个弱的键; (2) 银有比较小的粒径; (3) 载体既有酸性位又有碱性位; (4) 添加剂具有路易斯酸性.这对今后设计醇胺化反应的多相催化剂具有很好的借鉴意义.
同样以Ag/Al2O3为催化剂, 该催化剂是通过AgNO3和叔丁醇铝化合物共沉淀法制得Ag/Al2O3, 并不是用商业的Al2O3来合成. Jaenicke课题组[53]将银的负载量降为2.4 wt%, 以Cs2CO3为碱, 二甲苯为溶剂, 在120 ℃和氮气保护下, 可高效催化芳胺与苄醇的N-烷基化反应.长链脂肪醇如正丁醇、正戊醇等与芳胺反应也能得到相应的N-烷基化芳胺, 收率为80%~99%.但甲醇、乙醇等低碳醇不反应.当环仲胺六氢吡啶、四氢吡咯与苄醇在上述优化条件下反应时, N-苄基胺的收率大大减少, 主产物成为苯甲酰胺类化合物(Scheme 14).根据以上研究结果Vos等[54]考虑能否将载体进一步改进以提高银的催化性能.为此, 他们首先通过共沉淀法制备了Al2O3-Ga2O3混合氧化物载体, 接着通过浸渍法负载银, 然后在600 ℃煅烧得到Ag/Al2O3-Ga2O3.以苯胺与苄醇的反应为模板, 将Ag/Al2O3-Ga2O3与浸渍法制备的Ag/γ-Al2O3和原位溶胶-凝胶法制备的Ag/γ-Al2O3的催化性能进行对比, 发现Ag/Al2O3-Ga2O3明显表现出更高的催化活性, 3 mol%催化剂, 0.5 mmol NaH, 1 mL甲苯, 110 ℃, 26 h, 胺转化率达到84%, 单取代产率占到96%.载体的X射线衍射(XRD)分析发现, 和纯的氧化镓相比, 这种混合氧化物载体拥有更高的比表面积, 可使更多的Ag原子暴露在载体表面用于催化反应.该催化剂可回收, 并且在温和条件下对醇胺化反应显示活性.
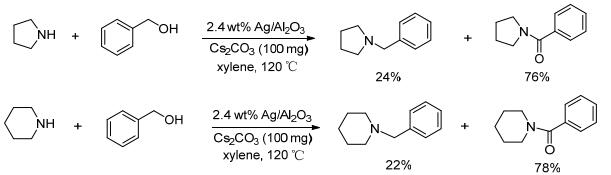 图 图式14
Ag/Al2O3的应用
Figure 图式14.
Application of Ag/Al2O3
图 图式14
Ag/Al2O3的应用
Figure 图式14.
Application of Ag/Al2O3
二氧化钛由于其较好的稳定性以及在光敏催化领域的广泛应用, 成为多相催化体系中载体的不二之选. 2010年曹勇课题组[55]把1.8 nm金纳米粒子负载到TiO2、Al2O3、MgO、ZnO、HAP、CeO2、SiO2等载体上, 以苯胺与苄醇的反应为模板进行催化性能测试, 最后确定以TiO2为载体, 0.5 mol%的Au催化剂, 在120 ℃, N2环境下, 以甲苯为溶剂, 可得到97%~99%的N-苄基苯胺.该催化剂可用于脂肪胺、芳香胺与苄醇的反应, 还可与部分脂肪醇反应生成相应的烷基化胺.不加纳米金, 单独对TiO2的催化作用显示TiO2没有催化活性.降低纳米金的负载量, 提高反应温度, 延长反应时间也能达到类似的催化效果.例如在无溶剂条件下, 以0.0083 mol% Au含量的Au/TiO2为催化剂, 苯胺和苄醇的量由原来的1.5 mmol增加到100 mmol, 在180 ℃, N2下反应96 h, 可得到96%的单取代产物(Scheme 15).这说明该体系催化性能稳定, 可以进行较大规模反应.该方法的优点是不需要任何添加剂或助催化剂, 缺点是需要在无氧条件下进行.
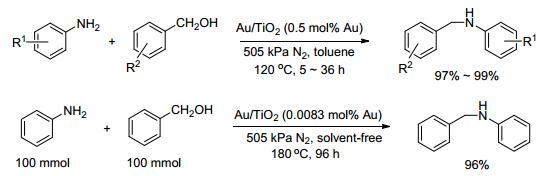 图 图式15
Au/TiO2的应用
Figure 图式15.
Application of Au/TiO2
图 图式15
Au/TiO2的应用
Figure 图式15.
Application of Au/TiO2
一价金络合物对醇胺化反应也具有很好的催化效果. Cheng等[56]以10 mol%三苯基膦一氯化金(Ⅰ)作为催化剂, 加入10 mol%三氟甲烷磺酸银, 在100 ℃二氧六环溶剂中反应48~96 h, 可使取代苯胺和取代苄醇反应, 单取代仲胺产物的收率在58%~92%.该方法也适用于部分脂肪醇的反应, 但是与苄醇相比, 脂肪醇反应的产率明显降低.
4 复合金属催化胺与醇的反应
两种过渡金属化合物或过渡金属与主族金属复合物也能作为醇胺化反应的有效催化剂. 2009年, Likhar研究小组[57]合成Cu-Al水滑石, 以此为催化剂实现了醇和胺的N-烷基化反应.研究小组详细考察了在水滑石制备过程中, Cu/Al的物质的量比对催化剂活性的影响.结果发现当二者的比例为2.5时, 催化剂拥有最好的催化苄醇与芳胺反应的活性.反应在160 ℃, 敞开体系, 无溶剂条件下反应9 h, 一系列取代苄胺、取代苯胺和取代苄醇的反应都可以取得很好的结果, 仲胺产物的收率高达98%.值得注意的是, 脂肪族仲醇由于活性低、位阻大而很少能参与醇胺化反应, 但是在Cu-Al水滑石的催化下二苯基甲醇和苄胺的反应良好, 得到85%的产物(Eq. 12).作者还通过XRD研究了新鲜的CuAl-HT催化剂和循环五次回收催化剂的XRD图案, 表明新鲜催化剂和回收催化剂的XRD图案之间没有差别, 说明催化剂可多次回收利用.
多相催化体系除了前面提到的负载型催化体系外, 混合金属氧化物也成为一个研究热点. 2011年, Shi课题组[58]尝试以市售的金属氧化物作催化剂催化苯胺与苄醇的反应, 在尝试了Ag2O、MoO3后并没有得到想要的反应结果, 于是又尝试了Ag2MoO4, 发现反应产率有所提高, 但选择性较差.随后课题组通过水热法制备了Ag6Mo10O33.以Ag6Mo10O33作为催化剂用于醇胺化反应, 确定了最优反应条件:催化剂用量为40 mg, 醇和胺的物质的量比为5:1, 以KOBu-t为碱, 在160 ℃下反应12 h.该催化体系不仅适用于胺、酰胺和磺酰胺类化合物与醇的N-烷基化反应, 还可以实现酮的烷基化, 收率普遍在90%以上(Scheme 16).
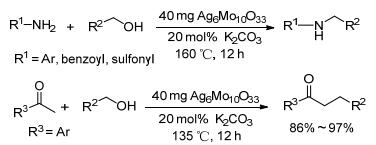 图 图式16
Ag6Mo10O33的应用
Figure 图式16.
Application of Ag6Mo10O33
图 图式16
Ag6Mo10O33的应用
Figure 图式16.
Application of Ag6Mo10O33
2011年余正坤等[59]将Pt-Sn混合金属, H2PtCl6•6H2O的盐酸溶液和SnCl2•2H2O的水溶液在N2(纯度99.995%)气氛下混合以形成Pt-Sn络合物, 然后用浸渍法负载在高比表面积的γ-Al2O3上, 制成双金属负载催化剂.该催化剂在使用前需要在氢气环境下加热到400 ℃活化.课题组在Pt的载入量为0.5 wt%保持不变的情况下, 以苯胺与苄醇的反应为模板反应, 筛选并确定了Pt/Sn比为1:3时, 催化活性最高, 并以此催化了多种芳香胺与芳香醇的反应, 单取代产物产率高达99% (Eq. 13).作者单独对Pt/γ-Al2O3进行了单独实验, 单取代产率只有14%, 更能说明Pt-Sn/γ-Al2O3催化剂的催化活性高.催化剂再循环用于模板反应时, 循环三次催化活性保持相同, 在第四次和第五次循环中, 催化活性逐渐降低, 但仍然可以通过延长反应时间达到所需产物的100%转化率和高产率.
次年, Li研究小组[60]将不同质量比的氯化镍、硝酸铜盐负载到γ-Al2O3上, 再以氢气还原, 得到零价混合金属负载催化剂Ni-Cu/γ-Al2O3.以苯胺与苄醇的反应为模板反应, 考察了混合金属中Ni、Cu质量比对催化性能的影响, 确定Ni-Cu质量比为4.5:1.0, 负载量分别为45 wt% Ni和10 wt% Cu, 同时添加25 mmol% CaCl2时催化活性最好.以此Ni-Cu/γ-Al2O3作为催化剂可合成多种N-苄基苯胺类化合物, 收率最高可达99% (Eq. 14).该催化剂经过两个循环仍具有稳定的催化活性.该方法所用催化剂由两种廉价金属构成, 避免了贵金属的使用, 降低了催化剂的用量, 有良好的应用前景.
2014年Nandan等[61]尝试以瑞尼镍(Raney nickel, R-Ni)为催化剂研究醇胺化反应.他们首先将镍-铝合金(1:1) 悬浮在20%的氢氧化钠溶液中, 随后再将混合物加入40%的氢氧化钠溶液中, 搅拌1 h, 调节溶液的pH值到7, 经离心过滤即得到Raney Ni.以此为催化剂, 二甲苯为溶剂, 在130 ℃下反应30 h, 各种取代苯胺与多种醇都能发生N-烷基化反应, 产物收率高达90%.实验表明R-Ni回收后循环使用两次而催化活性没有明显降低.为了研究反应物的结构对反应活性的影响, 作者还分别设计了脂肪醇和芳香醇、脂肪胺和芳香胺之间的竞争反应, 发现在此条件下脂肪醇比芳基甲醇的反应活性高, 而芳香胺与脂肪胺的反应性相差不大(Scheme 17).利用该方法可使仲胺发生烷基化反应生成叔胺, 如作者合成了药物分子吡贝地尔(piribedil)和比拉明(pyrilami-ne) (Scheme 18).
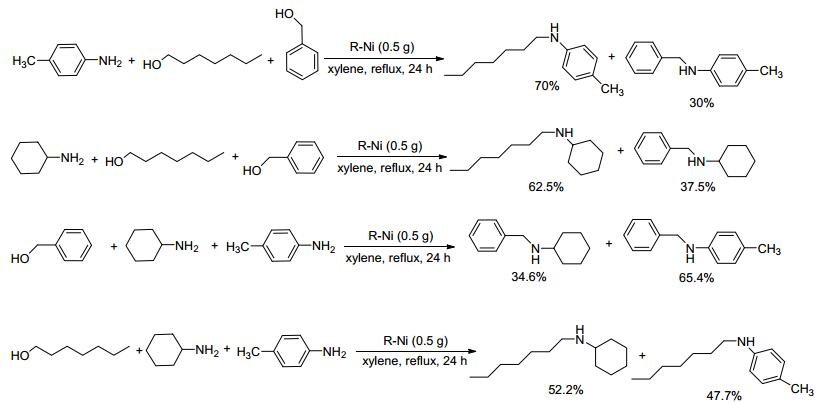 图 图式17
Raney nickel (R-Ni)的应用
Figure 图式17.
Application of Raney nickel (R-Ni)
图 图式17
Raney nickel (R-Ni)的应用
Figure 图式17.
Application of Raney nickel (R-Ni)
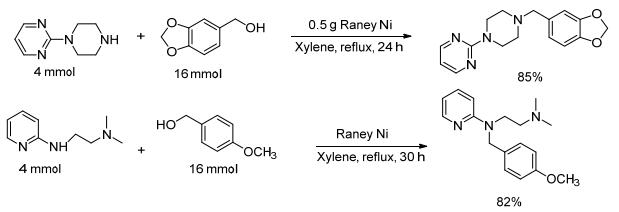 图 图式18
吡贝地尔和比拉明的合成
Figure 图式18.
Synthesis of piribedil and pyrilamine
图 图式18
吡贝地尔和比拉明的合成
Figure 图式18.
Synthesis of piribedil and pyrilamine
5 总结及展望
后过渡金属催化醇与胺的N-烷基化反应是一类重要的、原子利用率高的C—N键的形成反应, 产物仲胺、叔胺在生物、化工、医药、国防等领域有广泛的用途.近年来对该反应的研究已取得了丰硕的成果, 但也存在如下问题: (1) 贵金属催化剂因催化活性高研究较多, 廉价金属催化剂研究较少; (2) 脂肪醇尤其是含有1~4个碳原子的低碳醇的烷基化反应因活性较低, 相关研究较少; (3) 脂肪胺尤其是脂环仲胺因活性低、位阻大而不易发生该反应; (4) 反应的选择性较差, 部分反应除了单烷基化产物外, 还有双烷基化产物、季铵盐及芳烃生成; (5) 反应效率不高, 部分反应即使以单烷基化产物为主, 仍然需要过量2~3倍的醇作为烷基化试剂.
为了解决上述问题, 进一步提高醇胺化反应的效率, 今后对该反应的研究应重点从催化体系和反应体系两个方面考虑.一个高效的催化体系应具有广泛的底物适用范围、很高的目标产物收率、选择性和化学计量的醇.良好的反应体系包括温和的反应温度、环境友好的反应介质、不使用或尽量使用简单易得的添加剂.因此, 今后醇胺化反应的研究方向将会是: (1) 发展具有广泛适用性的、高效的均相廉价金属催化体系; (2) 开发高效非均相后过渡金属催化体系, 特别是纳米金属颗粒负载型催化剂, 筛选具有协同氢转移作用的高效、新型载体; (3) 开发高效贵金属-廉价金属双金属催化剂体系和双廉价金属催化体系, 有效降低贵金属的使用量; (4) 发展可用于水相反应的高效催化体系, 减少有机溶剂的使用对人体和环境的危害.相信随着现代科学和技术的发展, 醇胺化反应的研究将会取得更好的结果, 也必将在有机中间体、农药医药、天然产物分子的合成中成为重要的N-烷基化反应的方法.
-
-
[1]
Ullmann, F.; Bielecki, J. Ber. Dtsch. Chem. Ges. 1901, 34, 2174. doi: 10.1002/(ISSN)1099-0682
-
[2]
Goldberg, I. Ber. Dtsch. Chem. Ges. 1906, 39, 1691. doi: 10.1002/(ISSN)1099-0682
-
[3]
Hartwig, J. F.; Richards, S.; Baranano, D.; Paul, F. J. Am. Chem. Soc. 1996, 118, 3626. doi: 10.1021/ja954121o
-
[4]
Driver, M. S.; Hartwig, J. F. J. Am. Chem. Soc. 1997, 119, 8232. doi: 10.1021/ja971057x
-
[5]
Mann, G.; Hartwig, J. F.; Driver, M. S.; Fernandez-Rivas, C. J. Am. Chem. Soc. 1998, 120, 827. doi: 10.1021/ja973524g
-
[6]
Hartwig, J. F.; Kawatsura, M.; Hauck, S. I..; Shaughnessy, K. H.; Alcazar-Roman, L. M. J. Org. Chem. 1999, 64, 5575. doi: 10.1021/jo990408i
-
[7]
Takemiya, A.; Hartwig, J. F. J. Am. Chem. Soc. 2006, 128, 14800. doi: 10.1021/ja064782t
-
[8]
Strieter, E. R.; Blackmond, D. G.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 13978. doi: 10.1021/ja037932y
-
[9]
Fors, B. P.; Buchwald, S. L. J. Am. Chem. Soc. 2010, 132, 15914. doi: 10.1021/ja108074t
-
[10]
Breitler, S.; Oldenhuis, N. J.; Fors, B. P.; Buchwald, S. L. Org. Lett. 2011, 13, 3262. doi: 10.1021/ol201210t
-
[11]
Ueda, S.; Ali, S.; Fors, B. P.; Buchwald, S. L. J. Org. Chem. 2012, 77, 2543. doi: 10.1021/jo202537e
-
[12]
Ueda, S.; Ali, S.; Fors, B. P.; Buchwald, S. L. Tetrahedron Lett. 2010, 51, 325. doi: 10.1016/j.tetlet.2009.11.009
-
[13]
Zhao, Y.; Foo, S. W.; Saito, S. Angew. Chem., Int. Ed. 2011, 50, 3006. doi: 10.1002/anie.201006660
-
[14]
Yu, X.; Liu, C.; Jiang, L.; Xu, Q. Org. Lett. 2011, 13, 6184. doi: 10.1021/ol202582c
-
[15]
Grigg, R.; Mitchell, T. R. B.; Sutthivaiyakit, S.; Tongpenyai, N. J. Chem. Soc., Chem. Commun. 1981, 611.
-
[16]
Watanabe, Y.; Tsuji, Y.; Ohsugi, Y. Tetrahedron Lett. 1981, 22, 2667. doi: 10.1016/S0040-4039(01)92965-X
-
[17]
Watanabe, Y.; Tsuji, Y.; Ige, H.; Ohsugi, Y.; Ohta, T. J. Org. Chem. 1984, 49, 3359. doi: 10.1021/jo00192a021
-
[18]
Hamid, M. H. S. A.; Williams, J. M. J. Chem. Commun. 2007, 725.
-
[19]
Demir, S.; Coskun, F.; Özdemir, I. J. Organomet. Chem. 2014, 755, 134. doi: 10.1016/j.jorganchem.2014.01.007
-
[20]
Kim, J. W.; Yamaguchi, K.; Mizuno, N. J. Catal. 2009, 263, 205. doi: 10.1016/j.jcat.2009.01.020
-
[21]
Fujita, K.-I.; Enoki, Y.; Yamaguchi, R. Tetrahedron 2008, 64, 1943. doi: 10.1016/j.tet.2007.11.083
-
[22]
Blank, B.; Madalska, M.; Kempe, R. Adv. Synth. Catal. 2008, 350, 749. doi: 10.1002/(ISSN)1615-4169
-
[23]
Yamaguchi, K.; Koike, T.; Kotani, M.; Matsushita, M.; Shinachi, S.; Mizuno, N. Chem. Eur. J. 2005, 11, 6574. doi: 10.1002/(ISSN)1521-3765
-
[24]
Yamaguchi, K.; Koike, T.; Kim, J. W.; Ogasawara, Y.; Mizuno, N. Chem. Eur. J. 2008, 14, 11480. doi: 10.1002/chem.200801655
-
[25]
Hamid, M. H. S. A.; Slatford, P. A.; Williams, J. M. J. Adv. Synth. Catal. 2007, 349, 1555. doi: 10.1002/(ISSN)1615-4169
-
[26]
Hollmann, D.; Tillack, A.; Michalik, D.; Jackstell, R.; Beller, M. Chem. Asian J. 2007, 2, 403. doi: 10.1002/(ISSN)1861-471X
-
[27]
Kim, J. W.; He, J.; Yamaguchi, K.; Mizuno, N. Chem. Lett. 2009, 38, 920. doi: 10.1246/cl.2009.920
-
[28]
Cano, R.; Ramon, D. J.; Yus, M. J. Org. Chem. 2011, 76, 5547. doi: 10.1021/jo200559h
-
[29]
Peeters, A.; Claes, L.; Geukens, I.; Stassen, I.; Vos, D. D. Appl. Catal. A: Gen. 2014, 469, 191. doi: 10.1016/j.apcata.2013.09.051
-
[30]
Zhao, Y.; Foo, S. W.; Saito, S. Angew. Chem., Int. Ed. 2011, 50, 3006. doi: 10.1002/anie.201006660
-
[31]
Liu, C.; Liao, S.; Li, Q.; Feng, S.; Sun, Q.; Yu, X.; Xu, Q. J. Org. Chem. 2011, 76, 5759. doi: 10.1021/jo200862p
-
[32]
Kempe, R.; Blank, B.; Michlik, S. Chem. Eur. J. 2009, 15, 3790. doi: 10.1002/chem.200802318
-
[33]
Saidi, O.; Blacker, A. J.; Lamb, G. W.; Marsden, S. P.; Taylor, J. E.; Williams, J. M. J. Org. Process. Res. Dev. 2010, 14, 1046. doi: 10.1021/op100024j
-
[34]
Saidi, O.; Blacker, A. J.; Farah, M. M.; Marsdenb, S. P.; Williams, J. M. J. Chem. Commun. 2010, 46, 1541. doi: 10.1039/b923083a
-
[35]
Kawahara, R.; Fujita, K.-I.; Yamaguchi, R. J. Am. Chem. Soc. 2010, 132, 15108. doi: 10.1021/ja107274w
-
[36]
Kawahara, R.; Fujita, K.-I.; Yamaguchi, R. Adv. Synth. Catal. 2011, 353, 1161. doi: 10.1002/adsc.201000962
-
[37]
"Xu, G.; Zhang, Y.; Wang, K; Fu, Y.; Du, Z. J. Chem. Res. 2015, 39, 399.
-
[38]
Du, Z.; Zhou, W.; Bai, L.; Wang, F.; Wang, J.-X. Synlett 2011, 369.
-
[39]
Du, Z.; Zhou, W.; Wang, F. Tetrahedron 2011, 67, 4914. doi: 10.1016/j.tet.2011.04.093
-
[40]
Reyes-Rios, G.; García, J. J. Inorg. Chim. Acta 2012, 392, 317. doi: 10.1016/j.ica.2012.03.041
-
[41]
Dang, T. T.; Ramalingam, B.; Shan, S. P.; Seayad, A. M. ACS Catal. 2013, 3, 2536. doi: 10.1021/cs400799n
-
[42]
Banerjee, D.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2014, 53, 1. doi: 10.1002/anie.v53.1
-
[43]
Du, Z.; Yan, Y.; Fu, Y.; Wang, K. Asian J. Org. Chem. 2016, 5, 812. doi: 10.1002/ajoc.v5.6
-
[44]
Zhang, Y.; Qi, X.; Cui, X.; Shi, F.; Deng, Y. Tetrahedron Lett. 2011, 52, 1334. doi: 10.1016/j.tetlet.2011.01.059
-
[45]
Corma, A.; Rodenas, T.; Sabater, M. J. Chem. Eur. J. 2010, 16, 254. doi: 10.1002/chem.v16:1
-
[46]
Shiraishi, Y.; Fujiwara, K.; Sugano, Y.; Ichikawa, S.; Hirai, T. ACS Catal. 2013, 3, 312. doi: 10.1021/cs300756f
-
[47]
Ousmane, M.; Perrussel, G.; Yan, Z.; Clacens, J. M.; Campo, F. D.; Pera-Titus, M. J. Catal. 2014, 309, 439. doi: 10.1016/j.jcat.2013.10.003
-
[48]
Shimizu, K. I.; Kanno, S.; Kon, K.; Siddiki, S. M. A. H.; Tanaka, H.; Sakata, Y. Catal. Today 2014, 232, 134. doi: 10.1016/j.cattod.2013.09.002
-
[49]
Kita, Y.; Sakaguchi, H.; Hoshimoto, Y.; Nakauchi, D.; Nakahara, Y.; Carpentier, J.-F.; Ogoshi, S.; Mashima, K. Chem. Eur. J. 2015, 21, 14571. doi: 10.1002/chem.201502329
-
[50]
Sun, Z.; Wang, Q.; Xu, Y.; Wang, Z. RSC Adv. 2015, 5, 84284. doi: 10.1039/C5RA18503C
-
[51]
Martínez-Asencio, A.; Ramon, D. J.; Yus, M. Tetrahedron 2011, 67, 3140. doi: 10.1016/j.tet.2011.02.075
-
[52]
Shimizu, K.; Nishimura, M.; Satsuma, A. ChemCatChem 2009, 1, 497. doi: 10.1002/cctc.v1:4
-
[53]
Liu, H.; Chuah, G.-K.; Jaenicke, S. J. Catal. 2012, 292, 130. doi: 10.1016/j.jcat.2012.05.007
-
[54]
Geukensa, I.; Vermoortelea, F.; Meledinab, M.; Turnerb, S.; Tendeloob, G. V.; Vos, D. E. D. Appl. Catal. A: Gen. 2014, 469, 373. doi: 10.1016/j.apcata.2013.09.044
-
[55]
He, L.; Lou, X.-B.; Ni, J.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Chem. Eur. J. 2010, 16, 13965. doi: 10.1002/chem.v16.47
-
[56]
Yang, H.; Mao, R.; Luo, C.; Lu, C.; Cheng, G. Tetrahedron 2014, 70, 8829. doi: 10.1016/j.tet.2014.10.007
-
[57]
Likhar, P. R.; Arundhathi, R.; Kantam, M. L.; Prathima, P. S. Eur. J. Org. Chem. 2009, 5383.
-
[58]
Cui, X.; Zhang, Y.; Shi, F.; Deng, Y. Chem. Eur. J. 2011, 17, 1021. doi: 10.1002/chem.v17.3
-
[59]
He, W.; Wang, L.; Sun, C.; Wu, K.; He, S.; Chen, J.; Wu, P.; Yu, Z. Chem. Eur. J. 2011, 17, 13308. doi: 10.1002/chem.201101725
-
[60]
Sun, J.; Jin, X.; Zhang, F.; Hu, W. Liu, J.; Li, R. Catal. Commun. 2012, 24, 30. doi: 10.1016/j.catcom.2012.03.010
-
[61]
Mehta, A.; Thaker, A.; Londhe, V.; Nandan, S. R. Appl. Catal. A: Gen. 2014, 478, 241. doi: 10.1016/j.apcata.2014.04.009
-
[1]
-

图式1 金属催化醇胺化反应的氢转移策略
Scheme 1 Hydrogen transfer strategy for metal-catalyzed amination of alcohols

图式2 钯催化烯丙醇的胺化反应机理
Scheme 2 Mechanism of palladium-catalyzed amination of allylic alcohols

图式3 MnO2催化合成单取代胺的反应机理
Scheme 3 Mechanism of MnO2-catalyzed synthesis of monosubstituted amines
表 1 负载钯催化剂的筛选
Table 1. Screening of supported palladium catalysts
Entry Catalyst Yield/% of amine Yield/% of imine 1 Pd/MgO (0.8%) 79 13 2 Pd/C (5%) 28 23 3 Pd/HT (0.55%) 49 11 4 Pd/HAP (0.55%) 26 39 5 Au/MgO (1.0%) 38 49 6 Pt/MgO (1.0%) 61 21  下载: 导出CSV
下载: 导出CSV
-




















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 79
- 文章访问数: 4176
- HTML全文浏览量: 1132
 下载:
下载:
