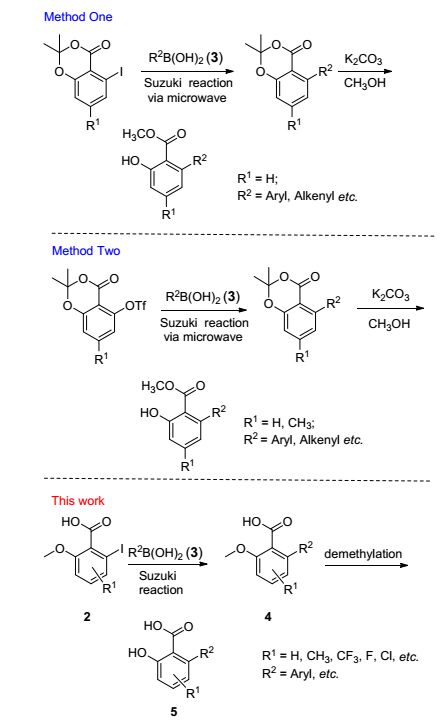
 Figure Scheme1.
Three practical approaches for the synthesis of the 6-substituted salicylates
Figure Scheme1.
Three practical approaches for the synthesis of the 6-substituted salicylates
Citation:
Qu Renyu, Chen Nian, Liu Yuchao, Chen Qiong, Yang Guangfu. An Efficient Synthesis of Functionalized 6-Arylsubstituted Salicylates via Microwave Irradiation[J]. Chinese Journal of Organic Chemistry,
2017, 37(5): 1266-1272.
doi:
10.6023/cjoc201612049

微波辅助下高效合成功能性6-芳基水杨酸类衍生物
摘要:
功能化的6-芳基水杨酸片段广泛地存在于各种天然产物以及活性分子中.在基于杂草抗性的分子设计中,6-芳基取代的水杨酸衍生物作为反抗性乙酰羟酸合成酶抑制剂的关键药效团发挥着至关重要的作用.在前期工作中,探索了两种策略可以合成6-芳基取代水杨酸片段,但不能有效地实现在6-芳基水杨酸片段母核结构中的底物多样化.因此,通过微波辅助的Suzuki偶联反应,成功合成了多取代的6-芳基水杨酸片段.这一方法具有反应时间短、反应收率高等优点,并且实现了底物多样化,为后期合成具有反抗性乙酰羟酸合成酶抑制剂奠定了基础.
-
关键词:
- 功能化6-芳基水杨酸
- / Suzuki交叉偶联反应
- / 微波辐射
- / 电子效应
English
An Efficient Synthesis of Functionalized 6-Arylsubstituted Salicylates via Microwave Irradiation
Abstract:
Functionalized 6-arylsalicylate substructures occur in a variety of pharmacologically relevant natural products and bioactive compounds. Especially 6-arylsubstituted salicylates, as a key pharmacophore of anti-resistant acetohydroxyacid synthase (AHAS) inhibitors have played a lead role in combatting the weed-resistance issues. Previously, we have explored two new methods to synthesize position-6 aryl substituted salicylic acid fragment. However, these two methods failed to introduce various substituents into salicylic acid. Here an efficient method for the synthesis of 6-substituted salicylates is described via a microwave-promoted Suzuki cross-coupling. Due to the obvious advantages of this method, such as a wide range of substrates, smooth and rapid reaction and moderate to excellent yields, this protocol could be utilized to synthesize more anti-resistant AHAS inhibitors.
-
As a common structure motif existing in both natural products and synthetic pharmaceutical drugs, 6-arylsubsti-tuted salicylates have received increasing attention over the past decade due to the synthetic challenges and broad biological activities. [1]
In recent years, we focus our efforts on the discovery of novel inhibitors targeting acetohydroxyacid synthase (AHAS) to combat the resistance issues that could cause sharply reduction of sensitivity toward the AHAS inhibiting herbicide. [2] 6-Bulkysubstituted salicylates as a key pharmacophore have played a lead role in designing anti-resistant AHAS inhibitors. [3] Thus, it is of great importance to develop an efficient methodology to construct substituted 6-arylsalicylates containing library for screening novel inhibitors. Two traditional methods have been developed for the synthesis of functionalized 6-arylsali-cylates, namely transition-metal-catalyzed cyclization reactions and [3+3] cyclizations developed by Chan et al. [4] However, because of rigorous reaction conditions, narrow range of substrates and poor yields, these two methods were insufficient to enrich the structure type of anti-resistant AHAS inhibitors.
Based on this, we have reported two highly efficient methods (Scheme 1) to synthesize 6-arylsubstituted salicy-lates under microwave irradiation. [5] Although these two works greatly enriched the substrate scope in 6-position aryl group of salicylates, introducing substituent group (such as halogen group, electron-withdrawing group) in skeletal structure remained challenging. In order to systematic study the structure-activity relationship (between AHAS inhibitor and anti-resistant property) and discover more potential AHAS inhibitors contained 6-aryl salicylates, we report a new efficient method of the synthesis of the substituted 6-aryl salicylates by using a microwave [6] assisted methodology.
Figure Scheme1.
Three practical approaches for the synthesis of the 6-substituted salicylates
1 Results and discussion
The aryl iodide (2) was prepared based on the ortho directing group assistance strategy. [7] To optimize the conditions for the palladium catalyzed Suzuki cross-coupling reaction, the reaction between aryl iodide (2a) and phenyl boronic acids (3a) as a model was selected. As shown in Table 1, microwave irradiation can dramatically accelerate these reactions and increase the reaction yields. In addition, the reactant concentration, the stoichiometric ratio of the catalyst and base, and the temperature were proved to be important parameters that could significantly affect the reaction yield. It was found that when the reaction temperature increased from 100 to 110 ℃ in the seal microwave tube, the yield increased sharply from 35% to 95% (Table 1, Entries 11 and 7). However, further increasing the temperature to 120 ℃ led to a slight decrease of the yield to 89%. Followed by screening different base such as NaHCO3, NaOH, K2CO3 (Table 1, Entries 1~3), the results showed that K2CO3 (4 equiv.) works well for the reaction. We also noted that these actions could not take place without Pd catalysis. Reducing the palladium loading was unfavorable to the reaction and a much lower yield was observed (Table 1, Entry 9). Thus, the optimum reaction condition should be 1.5 mol% Pd(PPh3)4, 4 equiv. of K2CO3, 110 ℃ and protected by N2. And then, compared to the microwave irradiation, conventional heating methods in the seal tube have longer reaction time and lower reaction yields (Table 1, Entry 12).
 Table 1.
Optimization of the reaction conditionsa
Table 1.
Optimization of the reaction conditionsa

Entry Temp./℃ Pd(PPh3)4/mol% Base (equiv.) Time/min Yieldb/% 1 110c 3 NaHCO3(2) 15 30 2 110 3 NaOH(2) 15 20 3 110 3 K2CO3(2) 15 45 4 110 3 K2CO3(4) 15 69 5 110 2 K2CO3(4) 10 83 6 110 1.5 K2CO3(4) 10 94 7 110 1.5 K2CO3(4) 8 95 8 110 1.0 K2CO3(4) 10 75 9 110 0.5 K2CO3(4) 15 53 10 120 1.5 K2CO3(4) 15 89 11 100 1.5 K2CO3(4) 15 35 12c 110 1.5 K2CO3(4) 15 57 aUnless otherwise noted, all the reactions were preceded in the seal microwave tube and carried out at 1 mmol of scale in the solution of dimethoxyethane (DME) (3 mL) and H2O (0.6 mL) protected by N2. b Isolated yields.c Conventional heating reaction was preceded in the seal tube, DME (10 mL) and H2O (2 mL) was used. Table 1. Optimization of the reaction conditionsaWith the best condition in hand, our attention turned to the evaluation of the scope of this reaction (Table 2). We firstly investigated scope of substrates by employing a variety of aryl boronic acids substituted with electron-donating and electron-withdrawing groups. As shown in Table 2, significant structural variations in the aryl boronic acid components were well tolerated and afforded the corresponding functionalized 6-substituted salicylates 4. Both electron-rich and electron-poor aryl boronic acids could be successfully utilized in this transformation. For instance, methyl, halogen, and trifluoromethyl derivatives showed no significant effects on the transformation and afforded good yields of products in this reaction (Table 2, Entries 1~4). In addition, the large steric hindrance group like 4'-tert-butyl also gave the products in good yield (Table 2, Entry 5).
Table 2.
The reaction with aryl iodide (2) and various arylboronic acids (3)a

Entry R1(2) R2 (3) Product Time/min Yieldb/% 1 5-F (2a) H (3a) 4a 8 95 2 5-F (2a) 4'-F (3b) 4b 7.5 92 3 5-F (2a) 4'-CH3(3c) 4c 8 93 4 5-F (2a) 4'-CF3(3d) 4d 12 90 5 5-F (2a) 4'-t-Bu (3e) 4e 8 95 6 5-Cl (2b) H (3a) 4f 7 93 7 5-CF3 (2c) H (3a) 4g 11 89 8 H (2d) H (3a) 4h 6 96 9 5-CH3 (2e) H (3a) 4l 7 91 10 4-Cl (2f) H (3a) 4j 13 90 aReaction conditions: aryl iodide (2) (1.0 mmol), arylboronic acids (3) (1.0 mmol), Pd(PPh3)4 (1.5 mol%), and K2CO3 (4.0 mmol, 4 equiv.) in DME (3 mL) and H2O (0.6 mL) at 110 ℃under N2protected. bIsolated yield. Table 2. The reaction with aryl iodide (2) and various arylboronic acids (3)aEncouraged by our preliminary findings, we also investigated the substrate scope for skeletal structure of salicylates. It was found that salicylate precursor bearing electron-donating groups like methyl (2e) reacted with borophenylic acid (3a) afforded the expected product (4l) in 91% yields (Table 2, Entries 9). Moreover, the strong electron-withdrawing trifluoromethyl group introduced at skeletal structure of salicylate (2c) was tolerated under the reaction conditions, which could be a complement to the previous method. In addition, salicylates precursor bearing halogen group groups like F, Cl also gave the products in good yields (Table 2, Entries 1~6). And the substituents in 4-position of salicylate precursor did not retard the reaction (Table 2, Entry 10). When we explored the scope of substituents in 6-position salicylate precursor, regrettably, this reaction did not happen, which could be caused by steric hindrance.
Finally, we removed the methyl group of salicylate precursor (Table 2, Entries 1~10) successfully to obtain the target compounds substituted 6-arylsalicylates. For almost all the substrates with different substituents, demethylation proceeded smoothly and afforded moderate to excellent yields of products 5a~5j (Table 3, Entries 1~10).
Table 3.
The synthesis of substituted 6-arylsalicylates through demethylation reactiona

Entry R1 R2 Product Yieldb/% 1 5-F H 5a 92 2 5-F 4'-F 5b 90 3 5-F 4'-CH3 5c 92 4 5-F 4'-CF3 5d 85 5 5-F 4'-t-Bu 5e 87 6 5-Cl H 5f 90 7 5-CF3 H 5g 87 8 H H 5h 93 9 5-CH3 H 5l 93 10 4-Cl H 5j 83 aUnless otherwise noted, all the reactions were carried out 1 mmol scale in the solution of dry dichloromethane (DCM) with 2 equiv. of BBr3 in -78 ℃. bIsolated yield. Table 3. The synthesis of substituted 6-arylsalicylates through demethylation reactiona2 Conclusions
In summary, we have developed a convenient and efficient method for the synthesis of functionalized 6-bulky-substituted salicylates 5a~5j using microwave-assisted Suzuki cross-coupling reactions. The protocol is not only applicable to unsubstituted salicylates (5h), but also to various substituted salicylates (5a~5g, 5l~5j). At the same time, the reactions possess a wide range of substrates and produce a variety of densely functionalized 6-aryl-salicylates 5 in good to excellent yields within a very short time. These important intermediates will provide a solid basis for the discovery of salicylate-containing drugs and agrichemicals.
3 Experimental
3.1 General
Unless otherwise noted, materials were purchased from commercial suppliers and used without further purification. All the solvents were treated by deoxidization. Flash columnchromatography (FC) was performed using 200~300 mesh silicagel.
1H NMR spectra were recorded on a 400/500 (400/500 MHz) spectrophotometers. Chemical shifts are reported from the solvent resonance as the internal standard (DMSO δ 2.50). 13C NMR spectra were recorded on a 500 (125 MHz) spectrophotometers (DMSO δ 39.5) with complete proton decoupling. High-resolution mass spectra (HRMS) were obtained on an Agilent 6224 TOF LC/MS (Agilent Technologies, Santa Clara, CA, USA). Melting points were taken on a Büchi B-545 melting point apparatus (Büchi Labortechnik, Flawil, Switzerland) and were uncorrected. Microwave irradiation reactions were carried out on a Smith synthesizer instrument.
3.2 General procedure for the synthesis of substituted 2-iodo-6-methoxybenzoic acid (2a~2f)
In a 250 mL glass tube, substrate 1 (20 mmol), Pd(OAc)2 (220 mg, 1 mmol), iodobenzene diacetate (6.44 g, 20 mmol), I2 (5.08 g, 20 mmol) were dissolved in N, N-dime-thylformamide (DMF) (40 mL) under atmospheric air.. The tube was sealed with a cap and the reaction mixture was stirred at 100 ℃ for 24 h. The reaction mixture was cooled to room temperature, diluted with ethyl acetate (80 mL) and then washed with 1 mol/L HCl (40 mL×4). The organic phase was washed with saturated NaCl, dried over Na2SO4 and concentrated in a rotary evaporator. The residue was purified by column chromatography on silica gel [V(n-hexane):V(EtO2Ac)=4:1] to give the iodination products 2a~2f.
4-Fluoro-2-iodo-6-methoxybenzoic acid (2a): Yield 79%, white solid. m.p. 191~192 ℃ (recrystallization from EtOAc/n-hexane); 1H NMR (500 MHz, DMSO-d6) δ: 12.88 (s, 1H), 8.04 (d, J=7.0 Hz, 1H), 7.15 (d, J=10.5 Hz, 1H), 3.84 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 165.0, 163.8 (d, JC-F=247 Hz), 160.5 (d, JC-F=10 Hz), 140.6 (d, JC-F=5 Hz), 119.5 (d, JC-F=3 Hz), 101.5 (d, JC-F=29 Hz), 69.6 (d, JC-F=27 Hz), 56.48. HRMS calcd for C8H6FIO3Na [M+Na]+ 318.92433, found 318.92453.
4-Chloro-2-iodo-6-methoxybenzoic acid (2b): Yield 73%, white solid. m.p. 177~178 ℃ (recrystallization from EtOAc/n-hexane); 1H NMR (500 MHz, DMSO-d6) δ: 13.01 (s, 1H), 8.08 (s, 1H), 7.37 (s, 1H), 3.85 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 165.1, 158.9, 141.5, 141.0, 121.9, 114.1, 86.5, 56.4; HRMS calcd for C8H6ClIO3Na [M+Na]+ 334.89478, found 334.89493.
2-Iodo-6-methoxy-4-(trifluoromethyl)benzoic acid (2c): Yield 65%, white solid. m.p. 135~136 ℃ (recrystallizetion from EtOAc/n-hexane); 1H NMR (500 MHz, DMSO-d6) δ: 13.62 (s, 1H), 7.73 (s, 1H), 7.42 (s, 1H), 3.89 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 167.3, 156.0, 135.2, 131.4 (q, JC-F=32 Hz), 126.6, 122.7 (q, JC-F=273 Hz), 108.3, 93.1, 56.6.HRMS calcd for C9H7F3IO3 [M+H]+ 346.93920, found 346.93936.
2-Iodo-6-methoxybenzoic acid (2d): Yield 83%, white solid. m.p. 146~147 ℃ (recrystallization from EtOAc/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.82 (s, 1H), 7.83 (d, J=2.4 Hz, 1H), 7.77 (dd, J=8.8, 2.4 Hz, 1H), 6.95 (d, J=8.8 Hz, 1H), 3.78 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 165.9, 157.9, 141.1, 138.4, 123.7, 115.3, 82.2, 55.9; HRMS calcd for C8H7IO3Na [M+Na]+ 300.93376, found 300.93354
2-Iodo-6-methoxy-4-methylbenzoic acid (2e): Yield 80%, white solid. m.p. 144~145 ℃ (recrystallization from EtOAc/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.66 (s, 1H), 7.97 (s, 1H), 7.13 (s, 1H), 3.79 (s, 3H), 2.39 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 165.5, 158.6, 146.4, 140.2, 120.4, 114.5, 88.7, 55.9, 27.8; HRMS calcd for C9H9IO3Na [M+Na]+ 314.94941, found 314.94973.
3-Chloro-6-iodo-2-methoxybenzoic acid (2f): Yield 69%, white oil. 1H NMR (500 MHz, DMSO-d6) δ: 12.67 (s, 1H), 7.60 (d, J=8.5 Hz, 1H), 7.27 (d, J=8.5 Hz, 1H), 3.80 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 167.5, 152.2, 138.4, 135.5, 132.1, 127.4, 90.5, 62.1; HRMS calcd for C8H6ClIO3Na [M+Na]+ 334.89478, found 334.89529.
3.3 General procedure of the synthesis of compounds 5a~5j
To a stirred solution of 4a (1 mmol) in anhydrous CH2Cl2 (10 mL) was added BBr3 (2 mmol) at -78 ℃. The reaction mixture was stirred at room temperature for 1~2 h, followed by pouring into ice-cold H2O and extracted twice with CH2Cl2; the organic phases were washed with water, dried over Na2CO3, filtered and evaporated. The residue was purified by recrystallization from EtOAc/heptane to give 5a in 94% yield.
5-Fluoro-3-hydroxy-1, 1'-biphenyl-2-carboxylic acid (5a): Yield 92%, white solid. m.p. 186~188 ℃ (recry-stallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 7.96~7.83 (m, 1H), 7.58~7.41 (m, 4H), 7.38 (d, J=6.0 Hz, 1H), 6.99~6.80 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 171.1, 164.1, 163.3 (d, JC-F=185 Hz), 162.2 (d, JC-F=14 Hz), 162.1, 162.0, 159.9 (d, JC-F=15 Hz), 134.7, 134.2, 132.5 (d, JC-F=7 Hz), 131.4 (d, JC-F=6 Hz), 128.7, 128.6, 128.6, 127.7, 127.5, 120.4, 120.4, 120.3, 120.2, 112.7, 110.5, 105.2 (d, JC-F=25 Hz), 104.6 (d, JC-F=26 Hz); HRMS calcd for C13H9FO3Na: [M+Na]+ 255.04334, found 255.04361.
4', 5-Difluoro-3-hydroxy-1, 1'-biphenyl-2-carboxylic acid (5b): Yield 90%, white solid. m.p. 219~220 ℃(recrystallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 7.90 (d, J=9.0 Hz, 1H), 7.63~7.50 (m, 2H), 7.30 (dd, J=8.4, 8.4 Hz, 2H), 6.98~6.85 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 171.0, 163.3 (d, JC-F=153 Hz), 162.3 (d, JC-F=14 Hz), 161.3 (d, JC-F=144 Hz), 132.5 (d, JC-F=6 Hz), 130.7 (d, JC-F=2 Hz), 130.6 (d, JC-F=2 Hz), 119.3 (d, JC-F=15 Hz), 115.5 (d, JC-F=22 Hz), 110.6, 104.5 (d, JC-F=26 Hz); HRMS calcd for C13H8F2O3Na [M+Na]+ 273.03392, found 273.03427.
5-Fluoro-3-hydroxy-4'-methyl-(1, 1'-biphenyl)-2-car-boxylic acid (5c): Yield 92%, white solid. m.p. 221~222 ℃ (recrystallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 7.87 (d, J=10.5 Hz, 1H), 7.48~7.37 (m, 2H), 7.29 (d, J=7.5 Hz, 2H), 7.00~6.79 (m, 1H), 2.36 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 171.2, 163.2 (d, JC-F=252 Hz) 162.8, 162.1, 159.9, 159.7, 137.2, 136.9, 132.4 (d, JC-F=6 Hz), 131.8, 131.3 (d, JC-F=6 Hz), 129.4, 128.5, 128.5, 120.4 (d, JC-F=15 Hz), 112.7, 110.6, 105.3 (d, JC-F=25 Hz), 104.6 (d, JC-F=26 Hz), 20.8; HRMS calcd for C14H11FO3Na [M+Na]+ 269.05899, found 269.05871.
5-Fluoro-3-hydroxy-4'-trifluoromethyl-1, 1'-biphenyl-2-carboxylic acid (5d): Yield 85%, white solid. m.p. 259~260 ℃ (recrystallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 7.96 (d, J=9.0 Hz, 1H), 7.81 (d, J=8.0 Hz, 2H), 7.74 (d, J=8.0 Hz, 2H), 6.88 (d, J=12.5 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 170.8, 170.3, 164.0, 163.8, 162.4 (d, JC-F=253 Hz), 138.9, 132.4 (d, JC-F=6 Hz), 129.2, 129.2, 127.7 (q, JC-F=32 Hz), 125.5, 125.4, 123.2 (q, JC-F=272 Hz), 117.3 (d, JC-F=14 Hz), 113.0, 104.2 (d, JC-F=25 Hz); HRMS calcd for C14H8F4O3Na [M+Na]+ 323.03073, found 323.03101.
4'-(tert-Butyl)-5-fluoro-3-hydroxy-1, 1'-biphenyl-2-carboxylic acid (5e): Yield 87%, white solid. m.p. 208~209 ℃ (recrystallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 7.89 (d, J=11.5 Hz, 1H), 7.54~7.36 (m, 4H), 7.00~6.74 (m, 1H), 1.31 (s, 9H); 13C NMR (125 MHz, DMSO-d6) δ: 171.1, 163.3 (d, JC-F=193 Hz), 162.0, 161.9, 159.8, 150.1, 149.8, 132.3 (d, JC-F=6 Hz), 131.8, 131.2, 128.2, 125.4, 125.4, 120.2 (d, JC-F=15 Hz), 120.0, 112.6, 110.4, 105.2 (d, JC-F=25 Hz), 104.5 (d, JC-F=26 Hz) 34.3, 31.1, 31.0; HRMS calcd for C17H17FO3Na [M+Na]+ 311.10594, found 311.10617.
5-Chloro-3-hydroxy-1, 1'-biphenyl-2-carboxylic acid (5f): Yield 90%, white solid. m.p. 210~212 ℃ (recrystallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 7.73 (s, 1H), 7.43 (d, J=6.0 Hz, 2H), 7.41~7.36 (m, 3H), 7.18 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 170.9, 160.4, 137.7, 132.4, 131.1, 129.3, 128.2, 127.6, 118.0, 112.8; HRMS calcd for C13H9ClO3Na [M+Na]+ 271.01379, found 271.01398.
3-Hydroxy-5-trifluoromethyl-1, 1'-biphenyl-2-carbo-xylic acid (5g): Yield 87%, white solid. m.p. 158~159 ℃ (recrystallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 10.74 (s, 1H), 7.51~7.39 (m, 5H), 7.20 (s, 1H), 7.11 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 167.9, 154.7, 140.8, 138.8, 130.2 (q, JC-F=32 Hz), 128.5, 128.4, 128.2, 128.1, 125.6 (q, JC-F=244 Hz), 116.3, 116.3, 110.9, 110.8; HRMS calcd for C14H9F3O3Na [M+Na]+ 305.04015, found 305.04045.
3-Hydroxy-5-methyl-1, 1'-biphenyl-2-carboxylic acid (5h): Yield 93%, white solid. m.p. 231~234 ℃ (recrys-tallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 7.61~7.53 (m, 1H), 7.42 (d, J=7.0 Hz, 2H), 7.38~7.27 (m, 3H), 6.96~6.78 (m, 1H), 2.22 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 171.8, 163.7, 160.3, 158.2, 143.2, 142.3, 140.6, 140.2, 132.6, 130.8, 129.9, 129.1, 129.1, 128.2, 128.2, 126.8, 126.7, 119.4, 118.3, 113.4, 111.5, 20.6; HRMS calcd for C14H13O3 [M+H]+ 229.08647, found 229.08677.
3-Hydroxy-1, 1'-biphenyl-2-carboxylic acid (5i): Yield 93%, white solid. m.p. 210~211 ℃ (recrystallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 8.06 (s, 1H), 7.90~7.73 (m, 1H), 7.70~7.57 (m, 2H), 7.53~7.41 (m, 2H), 7.35 (s, 1H), 7.14~6.94 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 171.8, 163.6, 160.6, 158.6, 139.6, 139.0, 133.9, 133.1, 131.3, 131.1, 129.0, 128.9, 128.0, 127.1, 127.0, 126.8, 126.2, 126.1, 118.8, 117.8, 115.6, 113.3; HRMS calcd for C13H10O3Na [M+Na]+ 237.05276, found 237.05305.
4-Chloro-3-hydroxy-1, 1'-biphenyl-2-carboxylic acid (5j): Yield 83%, white solid. m.p. 278~280 ℃(recrystallization from EtOAc/n-heptane); 1H NMR (500 MHz, DMSO-d6) δ: 7.67~7.47 (m, 1H), 7.45~7.32 (m, 3H), 7.32~7.17 (m, 2H), 6.90~6.63 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 168.8, 161.3, 155.2, 150.8, 143.8, 141.5, 139.7, 139.5, 132.9, 130.5, 128.3, 128.2, 128.1, 127.5, 127.4, 126.6, 123.3, 122.5, 122.0, 121.3, 120.4, 115.3; HRMS calcd for C13H9ClO3Na [M+Na]+ 271.01379, found 271.01402.
Supporting Information NMR spectra of all compounds is available free of charge via the Internet at http://sioc-journal.cn.
3.3 General procedure for the synthesis of compounds 4a~4j
Aryl iodide 2 (1.0 mmol) and 3 (1.0 mmol) were dissolved in DME (3 mL) and H2O (0.6 mL) in a microwave tube vial under a nitrogen atmosphere. Pd(PPh3)4 (0.015 mmol) and potassium carbonate (4.0 mmol) were added, and the reaction mixture was irradiated in a microwave apparatus at 110 ℃ for 6~13 min. After the reaction mixture was cooled to ambient temperature, the product was concentrated, and the crude mixture was purified by silica gel column chromatography using petroleum ether/acetone (V/V=20/5 to 20/8) as eluent to give the compounds 4 in good yields.
5-Fluoro-3-methoxy-[1, 1'-biphenyl]-2-carboxylic acid (4a): Yield 95%, white solid. m.p. 191~193 ℃ (recrys-tallization from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.71 (s, 1H), 7.79 (d, J=9.2 Hz, 1H), 7.49 (d, J=8.0 Hz, 2H), 7.44 (dd, J=7.6, 7.6 Hz, 2H), 7.40~7.33 (m, 1H), 7.14 (d, J=13.2 Hz, 1H), 3.87 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 166.1, 161.6 (d, JC-F=252 Hz) 159.6 (d, J=11 Hz) 134.2, 133.1 (d, JC-F=6 Hz) 128.7, 128.5, 128.5, 127.6, 119.8 (d, JC-F=14 Hz), 117.8 (d, JC-F=3 Hz), 101.3 (d, J=27 Hz) 56.5; HRMS calcd for C14H11FO3Na [M+Na]+ 269.05899, found 269.05913.
4', 5-Difluoro-3-methoxy-[1, 1'-biphenyl]-2-carboxylic acid (4b): Yield 92%, white solid. m.p. 148~149 ℃ (recrystallization from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.73 (s, 1H), 7.78 (d, J=9.2 Hz, 1H), 7.54 (dd, J=7.2, 5.2 Hz, 2H), 7.27 (dd, J=8.8, 8.8 Hz, 2H), 7.14 (d, J=13.2 Hz, 1H), 3.86 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 166.1, 161.7 (d, JC-F=245 Hz), 161.5 (d, JC-F=252 Hz), 159.6 (d, JC-F=11 Hz), 133.1 (d, JC-F=6 Hz), 130.6 (d, JC-F=3 Hz), 130.5 (d, JC-F=3 Hz), 118.8 (d, JC-F=14 Hz), 117.8 (d, JC-F=3 Hz), 115.6 (d, JC-F=22 Hz), 101.3 (d, JC-F=27 Hz). 56.5; HRMS calcd for C14H10F2O3Na [M+Na]+ 287.04957, found 287.04996.
5-Fluoro-3-methoxy-4'-methyl-1, 1'-biphenyl-2-carbo-xylic acid (4c): Yield 93%, white solid. m.p. 193~194 ℃ (recrystallizetion from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.69 (s, 1H), 7.77 (d, J=9.2 Hz, 1H), 7.38 (d, J=8.0 Hz, 2H), 7.25 (d, J=8.0 Hz, 2H), 7.11 (d, J=13.2 Hz, 1H), 3.86 (s, 3H), 2.34 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 166.1, 161.6 (d, JC-F=252 Hz), 159.4 (d, JC-F=11 Hz), 137.0, 133.0 (d, JC-F=6 Hz), 131.2, 129.3, 128.3, 128.3, 119.8 (d, JC-F=14 Hz), 117.7 (d, JC-F=3 Hz), 101.3 (d, JC-F=28 Hz), 56.4, 20.7; HRMS calcd for C15H13FO3Na [M+Na]+ 283.07464, found 283.07447.
5-Fluoro-3-methoxy-4'-trifluoromethyl-1, 1'-biphenyl-2-carboxylic acid (4d): Yield 90%, white solid. m.p. 151~152 ℃ (recrystallization from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.77 (s, 1H), 7.87 (d, J=9.2 Hz, 1H), 7.80 (d, J=8.4 Hz, 2H), 7.74 (d, J=7.6 Hz, 2H), 7.19 (d, J=13.2 Hz, 1H), 3.89 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 166.5, 162.1 (d, JC-F=253 Hz), 160.7 (d, JC-F=11 Hz), 138.7, 133.6 (d, JC-F=5.7 Hz), 129.8, 129.7, 128.5 (q, JC-F=32 Hz), 125.9, 125.8, 124.7 (q, JC-F=272 Hz), 118.7 (d, JC-F=14 Hz), 118.5 (d, JC-F=3 Hz), 107.1 (d, JC-F=22 Hz), 101.9 (d, JC-F=27 Hz), 57.0; HRMS calcd for C15H10F4O3Na [M+Na]+ 337.04638, found 337.04677.
4'-(tert-Butyl)-5-fluoro-3-methoxy-1, 1'-biphenyl-2-carboxylic acid (4e): Yield 95%, white solid. m.p. 137~138 ℃ (recrystallization from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.68 (s, 1H), 7.78 (d, J=9.3 Hz, 1H), 7.46 (d, J=8.4 Hz, 2H), 7.42 (d, J=8.4 Hz, 2H), 7.12 (d, J=13.2 Hz, 1H), 3.86 (s, 3H), 1.31 (s, 9H); 13C NMR (125 MHz, DMSO-d6) δ: 166.1, 161.6 (d, JC-F=252 Hz), 159.4 (d, JC-F=11 Hz), 150.1, 133.0 (d, JC-F=6 Hz), 131.2, 128.2, 128.1, 125.4, 119.7 (d, JC-F=14 Hz), 117.7 (d, JC-F=3 Hz), 101.3 (d, JC-F=28 Hz), 56.4, 34.3, 31.0; HRMS calcd for C18H19FO3Na [M+Na]+ 325.12159, found 325.12188.
5-Chloro-3-methoxy-1, 1'-biphenyl-2-carboxylic acid (4f): Yield 93%, white solid. m.p. 203~204 ℃ (recrystallization from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.83 (s, 1H), 7.62 (s, 1H), 7.43 (dd, J=8.0, 6.8 Hz, 2H), 7.41~7.36 (m, 3H), 7.31 (s, 1H), 3.88 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 166.2, 158.0, 137.7, 135.5, 133.1, 131.6, 129.2, 128.2, 127.6, 120.3, 114.1, 56.4; HRMS calcd for C14H11ClO3Na [M+Na]+ 285.02944, found 285.02931.
3-Methoxy-5-trifluoromethyl-1, 1'-biphenyl-2-carbo-xylic acid (4g): Yield 89%, white solid. m.p. 135~136 ℃ (recrystallization from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 13.26 (s, 1H), 7.48~7.36 (m, 6H), 7.26 (s, 1H), 3.92 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 167.4, 156.1, 140.1, 138.2, 130.5 (q, JC-F=32 Hz), 128.5, 128.2, 128.2, 123.7 (q, JC-F=273 Hz), 118.2, 118.1, 107.2, 107.2, 56.5; HRMS calcd for C15H12F3O3 [M+H]+ 297.07385, found 297.07431.
3-Methoxy-1, 1'-biphenyl-2-carboxylic acid (4h): Yield 96%, white solid. m.p. 162~163 ℃ (recrystallization from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.70 (s, 1H), 7.87 (d, J=2.0 Hz, 1H), 7.78 (dd, J=8.8, 2.4 Hz, 1H), 7.61 (d, J=7.2 Hz, 2H), 7.42 (dd, J=7.6, 7.6 Hz, 2H), 7.31 (dd, J=7.2, 7.2 Hz, 1H), 7.19 (d, J=8.8 Hz, 1H), 3.85 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 167.3, 157.6, 138.9, 132.0, 131.0, 129.0, 128.6, 127.1, 126.2, 121.8, 113.1, 55.9; HRMS calcd for C14H12O3Na [M+Na]+ 251.06841, found 251.06874.
3-Methoxy-5-methyl-1, 1'-biphenyl-2-carboxylic acid (4i): Yield 91%, white solid. m.p. 194~195 ℃ (recrystallization from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 12.43 (s, 1H), 7.48 (s, 1H), 7.41 (dd, J=7.2, 7.2 Hz, 2H), 7.34 (dd, J=7.2, 7.2 Hz, 1H), 7.29 (d, J=8.0 Hz, 2H), 7.05 (s, 1H), 3.84 (s, 3H), 2.27 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 166.8, 157.5, 140.8, 140.1, 133.2, 132.1, 129.0, 128.3, 126.87, 118.3, 114.4, 55.9, 40.0, 20.6; HRMS calcd for C15H14O3Na [M+Na]+ 265.08406, found 265.08441.
4-Chloro-3-methoxy-1, 1'-biphenyl-2-carboxylic acid (4j): Yield 90%, white solid. m.p. 171~172 ℃ (recrys-tallization from ethyl acetate/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ: 13.31 (s, 1H), 7.58 (d, J=8.4 Hz, 1H), 7.45~7.35 (m, 5H), 7.18 (d, J=8.4 Hz, 1H), 3.85 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 167.6, 151.8, 138.5, 138.5, 131.4, 130.7, 128.5, 128.1, 128.0, 126.5, 126.0, 61.8; HRMS calcd for C14H12ClO3 [M+H]+ 263.04750, found 263.04773.
-
-
[1]
(a) Namki, C. ; Hee, J. Y. ; Hong, P. K. ; Sang, H. S. J. Nat. Prod. 2013, 76, 2291. (b) Gang, S. ; Wang, M. W. ; Welch, T. R. ; Blagg, B. S. J. J. Org. Chem. 2006, 71, 7618. (c) Delgado, E. J. J. Mol. Model. 2010, 16, 1421. (d) Abe, H. ; Nishioka, K. ; Takeda, S. ; Arai, M. ; Takeuchi, Y. ; Harayama, T. Tetrahedron Lett. 2005, 46, 3197. (e) Lin, C. N. ; Huang, P. L. ; Lu, C. M. ; Yen, M. H. , Wu, R. R. Phytochemistry 1997, 44, 1359. (f) Yang, G. F. ; Yang, H. Z. J. Cent. China Norm. Univ. , Nat. Sci. 2001, 35, 40 (in Chinese). (杨光富, 杨华铮, 华中师范大学学报(自然科学版), 2001, 35, 40. )(g) Yang, G. F. ; Liu, H. Y. ; Lu, R. J. ; Yang, H. Z. Chem. J. Chin. Univ. 1998, 19, 222 (in Chinese). (杨光富, 刘华银, 陆荣键, 杨华铮, 高等学校化学学报, 1998, 19, 222. )
-
[2]
(a) He, Y. Z. ; Li, Y. X. ; Zhu, X. L. ; Yang, G. F. J. Chem. Inf. Model. 2007, 47, 2335. (b) Li, Y. X. ; Luo, Y. P. ; Xi, Z. ; Yang, G. F. J. Agric. Food Chem. 2006, 54, 9135. (c) Chen, C. N. ; Chen, Q. ; Liu, Y. C. ; Yang, G. F. Bioorg. Med. Chem. 2010, 18, 4897. (d) Xiong, Y. ; Liu, J. J. ; Yang, G. F. J. Comput. Chem. 2010, 31, 1592. (e) Chen, C. N. ; Lv, L. L. ; Ji, F. Q. ; Yang, G. F. Bioorg. Med. Chem. 2009, 17, 3011.
-
[3]
(a) Ji, F. Q. ; Niu, C. W. ; Yang, G. F. ; Xi, Z. Chem. Med. Chem. 2008, 3, 1203. (b) Liu, Y. C. ; Qu, R. Y. ; Chen, Q. ; Yang, J. F. ; Niu, C. W. ; Xi, Z. ; Yang, G. F. J. Agric. Food Chem. 2016, 64, 4845. (c) Yang, G. F. ; Liu, Y. C. ; Chen, Q. ; Xi, Z. ; Niu, C. CN 104650084, 2015 [Chem. Abstr. 2015, 903779]. (d) Yang, G. F. ; Liu, Y. C. ; Chen, Q. CN 104140397, 2013[Chem. Abstr. 2014, 1908130].
-
[4]
(a) Chan, T. H. ; Brownbridge, P. J. Am. Chem. Soc. 1980, 102, 3534. (b) Brownbridge, P. ; Chan, T. H. ; Brook, M. A. ; Kang, G. J. Can. J. Chem. 1983, 61, 688. (c) Feist, H. ; Langer, P. Synthesis 2007, 3, 327. (d) Stefan, B. ; Nazken, K. K. ; Zharylkasyn, A. A. ; Alexander, V. ; Peter, L. Tetrahedron 2012, 68, 3654. (e) Matthias, L. ; Muhammad, S. ; Alexander, V. ; Christine, F. ; Peter, L. Eur. J. Org. Chem. 2010, 5118. (f) Mohanad, S. ; Olumide, F. ; Abdolmajid, R. ; Mathias, L. ; Stefanie, R. ; Muhammad, S. ; Alexander, V. ; Christine, F. ; Peter, L. Eur. J. Org. Chem. 2010, 3732. (g) Ibrar, H. ; Mirza, A. Y. ; Matthias, L. ; Thomas, P. ; Christine, F. ; Helmar, G. ; Peter, L. Eur. J. Org. Chem. 2008, 503. (h) Satya, P. S. ; Rakesh, T. ; Akhilesh, K. V. Tetrahedron 2012, 68, 9035. (i) So, W. Y. ; Byung, S. K. ; Arun, R. J. J. Am. Chem. Soc. 2012, 134, 11308. (j) Hao, X. ; Shang, f. L. ; Hong, X. L. ; Hua, F. ; Yu, Y. J. Chem. Commun. 2010, 7617. (k) Patricia, G. ; Manuel, A. ; Fernndez, R. ; Enrique, A. Angew. Chem. , Int. Ed. 2009, 48, 5534.
-
[5]
(a) Liu, Y. C. ; Huang, Z. Y. ; Chen, Q. ; Yang, G. F. Tetrahedron 2013, 69, 9025. (b) Qu, R. Y. ; Liu, Y. C. ; Wu, Q. Y. ; Chen, Q. ; Yang, G. F. Tetrahedron 2015, 71, 8123.
-
[6]
(a) Leadbeater, N. E. Chem. Commun. 2005, 2881. (b) Lussier, T; Herve, G; Enderlin, G; Len, C. RSC Adv. 2014, 4, 462183. (c) Kappe, C. O. Angew. Chem. , Int. Ed. 2004, 43, 6250. (d) Zhou, Z. Z. ; Zhao, P. L. ; Huang, W. ; Yang, G. F. Adv. Synth. Catal. 2006, 348, 63. (e) Zhou, Z. Z. ; Ji, F. Q. ; Cao, M. ; Yang, G. F. Adv. Synth. Catal. 2006, 348, 1826. (f) Liu, Y. C. ; Ye, C. J. ; Chen, Q. ; Yang, G. F. Tetrahedron Lett. 2013, 54, 949. (g) Liu, Y. C. ; Qu, R. Y. ; Chen, Q. ; Yang, G. F. Tetrahedron 2014, 70, 2746. (h) Hua, C. ; Wu, Q. Y. ; Han, F. ; Yang, G. F. Chin. Chem. Lett. 2014, 25, 705.
-
[7]
Mei, T. S.; Giri, R.; Maugel, N.; Yu, J. Q. Angew. Chem., Int. Ed. 2008, 47, 5215. doi: 10.1002/anie.v47:28
-
[1]
-
Table 1. Optimization of the reaction conditionsa
Entry Temp./℃ Pd(PPh3)4/mol% Base (equiv.) Time/min Yieldb/% 1 110c 3 NaHCO3(2) 15 30 2 110 3 NaOH(2) 15 20 3 110 3 K2CO3(2) 15 45 4 110 3 K2CO3(4) 15 69 5 110 2 K2CO3(4) 10 83 6 110 1.5 K2CO3(4) 10 94 7 110 1.5 K2CO3(4) 8 95 8 110 1.0 K2CO3(4) 10 75 9 110 0.5 K2CO3(4) 15 53 10 120 1.5 K2CO3(4) 15 89 11 100 1.5 K2CO3(4) 15 35 12c 110 1.5 K2CO3(4) 15 57 aUnless otherwise noted, all the reactions were preceded in the seal microwave tube and carried out at 1 mmol of scale in the solution of dimethoxyethane (DME) (3 mL) and H2O (0.6 mL) protected by N2. b Isolated yields.c Conventional heating reaction was preceded in the seal tube, DME (10 mL) and H2O (2 mL) was used.  下载: 导出CSV
下载: 导出CSV
Table 2. The reaction with aryl iodide (2) and various arylboronic acids (3)a
Entry R1(2) R2 (3) Product Time/min Yieldb/% 1 5-F (2a) H (3a) 4a 8 95 2 5-F (2a) 4'-F (3b) 4b 7.5 92 3 5-F (2a) 4'-CH3(3c) 4c 8 93 4 5-F (2a) 4'-CF3(3d) 4d 12 90 5 5-F (2a) 4'-t-Bu (3e) 4e 8 95 6 5-Cl (2b) H (3a) 4f 7 93 7 5-CF3 (2c) H (3a) 4g 11 89 8 H (2d) H (3a) 4h 6 96 9 5-CH3 (2e) H (3a) 4l 7 91 10 4-Cl (2f) H (3a) 4j 13 90 aReaction conditions: aryl iodide (2) (1.0 mmol), arylboronic acids (3) (1.0 mmol), Pd(PPh3)4 (1.5 mol%), and K2CO3 (4.0 mmol, 4 equiv.) in DME (3 mL) and H2O (0.6 mL) at 110 ℃under N2protected. bIsolated yield.
下载: 导出CSV
Table 3. The synthesis of substituted 6-arylsalicylates through demethylation reactiona
Entry R1 R2 Product Yieldb/% 1 5-F H 5a 92 2 5-F 4'-F 5b 90 3 5-F 4'-CH3 5c 92 4 5-F 4'-CF3 5d 85 5 5-F 4'-t-Bu 5e 87 6 5-Cl H 5f 90 7 5-CF3 H 5g 87 8 H H 5h 93 9 5-CH3 H 5l 93 10 4-Cl H 5j 83 aUnless otherwise noted, all the reactions were carried out 1 mmol scale in the solution of dry dichloromethane (DCM) with 2 equiv. of BBr3 in -78 ℃. bIsolated yield.
下载: 导出CSV
-


 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 7
- 文章访问数: 1642
- HTML全文浏览量: 194
 下载:
下载:
