Received Date:
24 August 2020 Revised Date:
28 September 2020 Available Online:
25 December 2020
Fund Project:
Project supported by the Youth Innovation and Technology Project of Shandong Province (No. 2019KJC021), the Natural Science Foundation of Shandong Province (No. ZR2018MB009) and the International Cooperation Project of Qinghai Province (No. 2018-HZ-806)
Abstract:N-Substituted-1, 2, 3-triazoles are an important class of nitrogen-containing hetrocyclic compounds, which exhibited wide applications in various fields such as medicinal chemistry, synthetic chemistry and materials. Therefore, their synthetic methods have attracted great attention of chemists. Herein, the recent research progress in the synthesis of N-substituted-1, 2, 3-triazoles is summarized. The synthetic routes and reaction mechanisms from raw materials such as azide compounds, diazo compounds, TsNHNH2, hydrazones and NH-1, 2, 3-triazoles are introduced and reviewed, respectively. Finally, the future development of this field is also prospected.
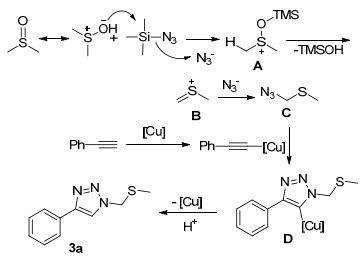
Scheme 12.
Copper catalyzed three-component reaction of alkynyl carboxylic acid, TMSN3 and ether to construct N1- and N2-oxyalkylated-1, 2, 3-triazoles
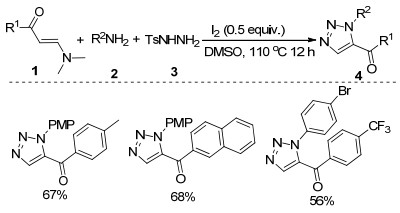
Scheme 14.
Copper catalyzed three-component reaction of acetylenic acid, azide and diselenide for the construction of N- alkyl-5-arylselanyl-1, 2, 3-triazoles
Scheme 20.
Copper catalyzed three-component reaction of aryl methyl ketone, N, N-dimethylformamide and organic azide to construct N1-alkyl/aryl-1, 2, 3-triazoles
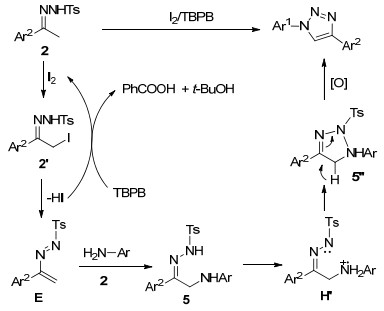
Scheme 50.
Cu-catalyzed oxidative [3+2] cycloaddition reaction of α-aminoketones and diazo compounds for the synthesis of N1-substituted-1, 2, 3-triazoles
(a) Chu, C.; Liu, R. Chem. Soc. Rev. 2011, 40, 2177. (b) Lau, Y. H.; Rutledge, P. J.; Watkinson, M.; Todd, M. H. Chem. Soc. Rev. 2011, 40, 2848. (c) Johnson, T. C.; Totty, W. G.; Wills, M. Org. Lett. 2012, 14, 5230.
(a) Wang, W.; Peng, X.; Wei, F.; Tung, C.-H.; Xu, Z. Angew. Chem., Int. Ed. 2016, 55, 649. (b) Cheung, K. P. S.; Tsui, G. C. Org. Lett.2017, 19, 2881.
[5]
(a) Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Chem. Rev.2013, 113, 4905. (b) Wang, K.; Chen, M.; Wang, Q.; Shi, X.; Lee, J. K. J. Org. Chem. 2013, 78, 7249.
[6]
(a) Jiang, Y.; Hansen. T. V. Bioorg. Med. Chem. Lett. 2011, 21, 1626. (b) Xu, X.; Guo, S.; Zhang, J.; Chen, Y.; Kang, Y.; Liu, N.; Liu J.; Luo, C.; Chen, S.; Chen, H. Chin. J. Org. Chem.2020, 40, 1345(in Chinese). (徐晓明, 郭思岐, 张静, 陈彦韬, 康亚青, 刘娜, 刘俊芳, 罗成, 陈示洁, 陈华, 有机化学, 2020, 40, 1345.)
[7]
(a) Quan, X. J.; Ren, Z. H.; Wang, Y. Y.; Guan, Z. H. Org. Lett. 2014, 16, 5728. (b) Chen, C. Y.; Lee, P. H.; Lin, Y. Y.; Yu, W. T.; Hu, W. P.; Hsu, C. C.; Lin, Y. T.; Chang, L. S.; Hsiao, C. T.; Wang, J. J.; Chung, M. L. Bioorg. Med. Chem. Lett. 2013, 23, 6854.
[8]
(a) Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed.2001, 40, 2004. (b) Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed.2002, 41, 2596. (c) Moses, J. E.; Moorhouse, A. D. Chem. Soc. Rev.2007, 36, 1249. (d) Tiwari, V. K.; Mishra, B. B.; Mishra, K. B.; Mishra, N.; Singh, A. S.; Chen, X. Chem. Rev.2016, 116, 3086. (e) Wang, C.; Zhou, F.; Zhou J. Chin. J. Org. Chem.2020, 40, 3065(in Chinese). (王才, 周峰, 周剑, 有机化学, 2020, 40, 3065.)
[9]
Kalisiak, J.; Sharpless, K. B.; Fokin, V. V. Org. Lett.2008, 10, 3171. doi: 10.1021/ol8006748
[10]
Zhang, Y.; Li, X.; Li, J.; Chen, J.; Meng, X.; Zhao, M.; Chen, B. Org. Lett.2012, 14, 26. doi: 10.1021/ol202718d
[11]
Kamijo, S.; Jin, T.; Huo, Z. B.; Yamamoto, Y. J. Am. Chem. Soc. 2003, 125, 7786. doi: 10.1021/ja034191s
[12]
Phanindrudu, M.; Tiwari, D. K.; Aravilli, V. K.; Bhardwaj, K. C.; Sabapathi, G.; Likhar, P. R. Eur. J. Org. Chem.2016, 27, 4629.
Luo, G. L.; Sun, C. Y.; Li, Y.; Li, X. X.; Zhao, Z. RSC Adv. 2018, 8, 27610. doi: 10.1039/C8RA04790A
[34]
Chen, Z.; Cao, G.; Song, J.; Ren, H. Chin. J. Chem.2017, 35, 1797. doi: 10.1002/cjoc.201700459
[35]
Hanselmann, R.; Job, G. E.; Johnson, G.; Lou, R.; Martynow, J. G.; Reeve, M. M. Org. Process Res. Dev.2010, 14, 152. doi: 10.1021/op900252a
[36]
van Berkel, S. S.; Brauch, S.; Gabriel, L.; Henze, M.; Stark, S.; Va-silev, D.; Wessjohann, L. A.; Abbas, M.; Westermann, B. Angew. Chem., Int. Ed.2012, 51, 5343. doi: 10.1002/anie.201108850
[37]
(a) Fçhlisch, B.; Flogaus, R. Synthesis1984, 734. (b) Bangalore, J.; Llavona, L.; Concellon, J. M.; Yus, M. J. Chem. Soc., Perkin Trans. 11991, 297.
Scheme 14
Copper catalyzed three-component reaction of acetylenic acid, azide and diselenide for the construction of N- alkyl-5-arylselanyl-1, 2, 3-triazoles
Scheme 20
Copper catalyzed three-component reaction of aryl methyl ketone, N, N-dimethylformamide and organic azide to construct N1-alkyl/aryl-1, 2, 3-triazoles
Scheme 50
Cu-catalyzed oxidative [3+2] cycloaddition reaction of α-aminoketones and diazo compounds for the synthesis of N1-substituted-1, 2, 3-triazoles


 下载:
下载:



















































 下载:
下载:























































 下载:
下载:
