
图式 1.
金鸡纳生物碱的结构与构型
Scheme 1.
Structures and configurations of cinchona alkaloids

自1737年Condamine发现从金鸡纳树中提取出的生物碱奎宁可作为治疗疟疾的特效药以后, 金鸡纳生物碱就开始受到人们的普遍关注[1]. 1820年, Pelletier和Caventou首先分离得到奎宁纯品[2], 随后Rabe等[3]于1907年确定了奎宁的分子结构, 其全合成工作首次于1944年由Woodward和Doering[4]报道. 1967年, Carter等[5]通过单晶衍射进一步确定了奎宁的结构.直到2001年, Stork等[6]利用手性的Taniguchi内酯作为初始原料历经15步反应后才得到光学纯的奎宁.研究者们陆续从金鸡纳属中提取出约30种天然生物碱, 并广泛用于医学、制药等领域.由于金鸡纳生物碱具有含量丰富、稳定性好以及优势的手性骨架, 金鸡纳生物碱及其衍生物被作为有机催化剂和手性配体广泛用于催化不对称合成, 已经成为不对称合成化学中的一个重要研究领域.
在有机小分子催化的不对称合成中, 自1853年[7]金鸡纳生物碱首次被报道可作为手性助剂参与不对称合成后, 金鸡纳生物碱作为有机催化剂或手性配体参与不对称合成已成为热门研究内容之一.这与其具有天然的手性环境结构及强大的后期可修饰能力密不可分(Scheme 1).可归纳为以下几点: (1)奎宁、喹啉环可产生具有类似酶的手性口袋提供手性环境; (2) R、OH和奎宁环等都具有可修饰性; (3) C8与C9的自由旋转创造动态环境可产生不同稳定性的构像; (4)奎宁环中的叔胺氮原子具有亲核特点, 可作为亲核试剂活化底物; (5)扁平的喹啉芳香环富电子可与缺电子分子形成电子给体供体复合物(EDA); (6) C9位上的羟基可通过氢键作用作为亲电试剂活化底物.在过去几十年, 通过对金鸡纳生物碱结构的修饰, 已经衍生出诸多具有代表性的金鸡纳碱衍生物, 如C9-胺基、脲/硫脲、酰胺、磺酰胺、方酰胺、醚等; C6'-羟基以及二聚或多聚高分子化合物(Scheme 2).关于这些衍生物的具体合成, Skarżewski等[8]在其2019年的综述中已做了非常详细地总结.本文将按照金鸡纳碱及其衍生物在不对称合成中的角色进行分类, 对其作为催化剂和配体参与的不对称反应进行归纳和总结, 并探讨了相关反应机理.

在不对称催化领域中, 有机小分子催化相较于金属催化和酶催化更具绿色经济以及可持续性, 一直以来都受到人们的广泛关注.有机催化的起源最早可以追溯到二十世纪初Knoevenagel等化学家报道的有机胺催化反应[9].随后, 有机小分子催化的不对称合成研究序幕慢慢被拉开, 有机胺、磷、硼试剂、脯氨酸、金鸡纳碱和杂环卡宾等以及各自的衍生物被发现可催化多类有机化学反应, 从而有效构建碳-碳键或碳-杂键.对于催化模式, 则主要包括烯胺催化、亚胺催化、SOMO (singly occupied molecular orbital)催化、氢键活化、卡宾催化以及相转移催化等[10], 而金鸡纳碱及其衍生物作为有机小分子催化中的重要分支, 尤其是自2000年以来已取得突飞猛进的发展.邓力教授作为该领域的开拓者和引领者之一, 在金鸡纳碱催化领域取得了重要突破, 也因此荣获了2020年“亚瑟•科普学者奖”.许多具有代表性的综述[11]在此之前已经详尽地总结了相关工作, 故本部分将对2016年以来发展的金鸡纳碱及其衍生物催化的不对称合成研究进行总结.
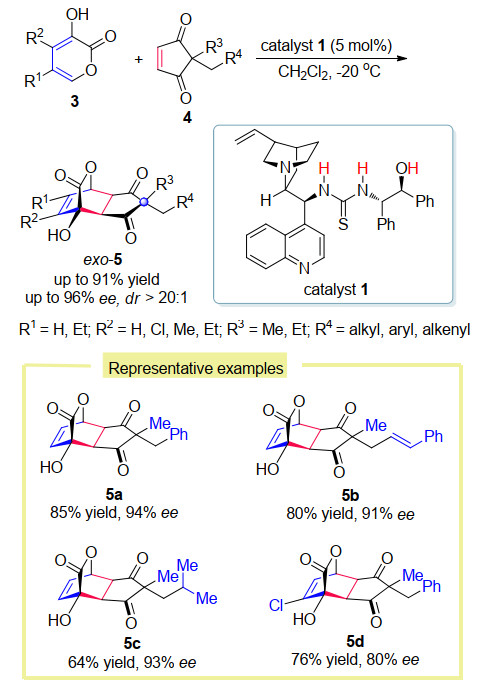
作为一种原子经济性的合成策略, 催化不对称Diels-Alder反应被广泛应用于构建结构多样的手性六元碳环或杂环化合物. 2017年, 王春江课题组[12]报道了在金鸡纳碱衍生的双功能胺-硫脲催化剂1的催化作用下, 通过去对称化策略实现了3-羟基-2-吡喃酮3与潜手性的1, 3-二酮4的不对称Diels-Alder反应, 从而构建了一系列具有五个手性中心的桥环内酯化合物5 (Scheme 3).在该反应中, 由于底物3-羟基-2-吡喃酮固有的缺电子性质, 以及环加成产物中四个连续手性中心和一个远端季碳中心的一次性构建, 使得该反应具有一定的挑战性.作者利用双功能胺-硫脲催化剂解决了这一难题, 且能以良好的收率、对映选择性以及非对映选择性得到外型产物.作者认为硫脲单元、羟基结合的多氢键供体与三级胺双活化模式是该反应能顺利进行的关键.利用这种合成策略可以方便地构建环状化合物, 这类结构在天然产物的全合成中有一定的应用潜力.
手性含氮杂环化合物广泛存在于许多天然产物和生物活性分子中, 也可以作为手性配体或催化剂被广泛应用于不对称合成反应中.因此, 手性含氮杂环化合物的合成一直受到科研工作者的密切关注. 2019年, 程道娟课题组[13]利用金鸡纳碱衍生的胺-硫脲双功能催化剂2, 以3H-吲哚6和硝基取代的烷烃7作为底物, 在温和的条件下实现了不对称Aza-Henry反应, 以中等到良好的收率和对映选择性成功地实现了非活化的五元环亚胺到手性含氮杂环8的转换(Scheme 4).在此反应中, 当R2是甲基时, 可能形成了环状三聚体, 反应不能够发生.但是当R2是正丁基时, 通过延长反应时间, 能够以51%的收率和80%的对映选择性得到目标产物.另一方面, 当选用1-硝基丙烷作为亲核试剂时, 该反应的效率和立体选择性明显降低, 可能是因为吲哚3-位的大基团阻碍了空间位阻较大的亲核试剂的进攻.作者认为, 催化剂2中奎宁环部分首先脱去底物7中的质子, 生成真正的亲核硝酸盐物种, 其自身被质子化.随后质子化的催化剂2通过氢键作用同时活化3H-吲哚6和真正的亲核物种硝酸盐, 这样, Aza-Henry加成选择性在底物3H-吲哚6的Si面进行, 从而生成R构型的产物.

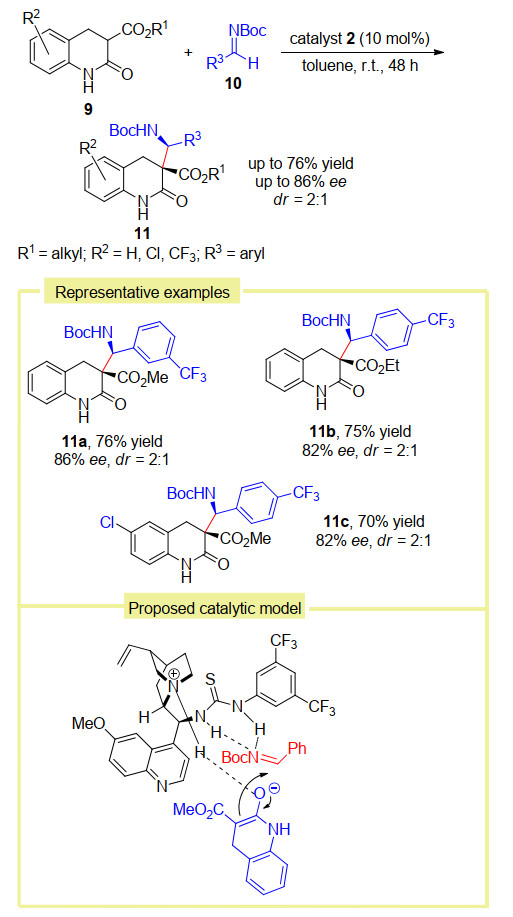
同年, Pan课题组[14]利用同样的催化剂2实现了二氢-3-烷氧羰基-2-喹诺酮9与Boc保护的亚胺10的不对称Mannich反应(Scheme 5).在该反应中, 反应条件温和, 操作简单, 具有较好的官能团兼容性, 最终能以中等到良好的收率和对映选择性, 以及中等的非对映选择性合成得到具有重要潜在生物活性的3, 3-二取代-二氢- 2-喹诺酮类化合物11.对于金鸡纳碱衍生的胺-硫脲催化剂的双活化模式, 作者认为, 催化剂2奎宁环片段中的三级胺作为布朗斯特碱活化底物9中烯醇互变所产生的羟基, 而硫脲单元作为氢键供体活化底物亚胺, 从而选择性地在亚胺的Si面发生亲核加成生成(S, S)构型的产物11.
在金鸡纳碱衍生的双功能催化剂中, 除C9位含有硫脲单元外, C9位含有磺酰胺和四方酰胺结构单元也是十分优良的氢键供体. 2018年, Funahashi课题组[15]利用金鸡纳碱衍生的磺酰胺催化剂3, 实现了2H-氮杂丙烯啶与硫醇13的不对称亲核加成反应, 以高收率和高对映选择性制备了一系列氮杂环丙烷类化合物14, 并且该产物在不损失对映选择性的条件下可以转化为噁唑啉或α-磺酰酯等具有季碳中心的手性化合物(Scheme 6).在该反应中, 初始原料叠氮丙烯酸酯12在溶剂二氯甲烷中封管加热即可原位生成2H-氮杂丙烯啶.随后在催化剂3的催化下, 硫醇作为亲核试剂对其进行亲核加成.作者认为该反应优异的收率和对映选择性正是得益于金鸡纳碱衍生的磺酰胺催化剂的双功能活化.另外, 催化剂3中的杂芳烃磺酰基在增强反应的对映选择性中起重要作用.

2016年, Itsuno课题组[16]合成了一类以金鸡纳碱衍生的磺酰胺为重复单元的手性聚合物, 并成功应用到环酐的不对称去对称化反应中(Scheme 7).以四氢呋喃作溶剂, 底物环酐15a与甲醇、苄醇、烯丙醇16a均能以中等到良好的收率和对映选择性得到去对称化的产物17a.随后, 该团队又合成了基于辛可尼啶的磺酰胺二聚体为重复单元的手性聚合物[17], 并成功地应用于β-酮酯15b和硝基苯乙烯16b的不对称Michael加成反应中.尽管此类手性聚合物不溶于常用的有机溶剂, 但在非均相反应条件下所表现出的催化活性和金鸡纳生物碱衍生的磺酰胺类催化剂在均相条件下所表现出的活性相当.并且, 聚合物催化剂4可以通过简单过滤从反应混合物中分离出来.将回收的聚合物粉末重新用于后续反应, 不会造成催化活性和产物的对映选择性损失.

多取代的四氢呋喃或吡喃广泛存在于天然产物和药物中.然而, 由于存在较大的空间位阻, 此类化合物的对映选择性合成仍具有很大的挑战性. 2016年, Ghorai课题组[18]报道了在金鸡纳碱衍生的四方酰胺催化剂5的作用下, 实现了对位取代的苯酚18氧化脱芳构化/不对称的Oxa-Michael加成串联反应(Scheme 8).在PhI(OAc)2为氧化剂的条件下, 底物18脱芳构化得到4-羟基环己二烯酮19.随后在催化剂5的作用下, 19发生对映选择性的分子内Oxa-Michael加成反应.利用这种两步一锅的策略能够高效地合成一系列四氢呋喃和四氢吡喃类化合物20.该反应具有出色的对映选择性, 底物普适性较好, 适用于α, β-不饱和的芳香酮、杂环酮以及脂肪酮.

2018年, Pan课题组[19]利用金鸡纳碱衍生的方酰胺催化剂6, 实现了γ/δ-羟基不饱和酮22与α-硝基酮21的不对称Michael加成/酰基转移串联反应(Scheme 9).在室温条件下, 以优异的收率和对映选择性制备了具有硝基、酮和酯等官能团的化合物23.作者认为, 双功能催化剂6中的奎宁环对底物21进行活化, 同时, 四方酰胺部分通过氢键作用和底物22进行结合.因此, 底物22中烯烃的Si面被催化剂所阻挡, 共轭加成只能选择性地在Re面进行, 产生中间体24.接着, 中间体24通过分子内的半缩酮化产生环状中间体25.最后, 经过一步逆Henry反应就能生成目标化合物23.

2-吡啶酮骨架广泛地存在于天然产物和药物中. 2019年, Hou和Tsai课题组[20]报道了双功能催化剂7催化的卤代2-羟基吡啶26和底物27的对映选择性氮杂-Michael加成反应, 以优异的收率和对映选择性得到氮取代的手性2-吡啶酮类化合物28 (Scheme 10).通过该方法合成的手性Michael加成产物, 经过几步简单的转化就能实现人鼻病毒蛋白抑制剂的合成. 2018年, 姜海洋课题组[21]利用金鸡纳碱衍生的伯胺催化剂以及辅助催化剂苯甲酸实现了1-硝基-2-苯基乙烯与2-甲基丙醛的不对称Michael加成反应, 并利用密度泛函方法对反应机理进行了理论计算研究.首先, 质子化的手性胺催化剂与底物醛生成关键的亚胺离子中间体, 随后经历亲核加成、质子转移和水解三个过程即可得到目标产物.理论研究表明, 反应的决速步以及立体选择性控制均在质子转移阶段控制.详细的机理研究使得人们对2-甲基丙醛参与的不对称Michael加成反应有了更深的理解.

2020年, 林宁和鲁桂课题组[22]利用金鸡纳碱衍生的方酰胺双功能催化剂6实现了对手性邻四取代二胺31的高效合成(Scheme 11).在传统的不对称Mannich反应中, 醛酮亚胺常作为亲电试剂与亲核试剂来构建碳碳键, 而在该反应中, 通过极性反转策略, 顺利地将亲电的环酮亚胺29转变为亲核试剂, 接着与另一分子亚胺30发生不对称交叉Mannich反应, 最终以优异的收率、对映选择性和非对映选择性得到目标产物.该手性催化剂在作为氢键供体催化剂之余, 也充当布朗斯特碱诱导底物发生极性反转.酮亚胺29失去质子后得到亲核性物种29-A, 29-A随后快速与共振式29-B达到平衡.通过对催化剂与溶剂的筛选, 可能的副反应均可以成功地避免, 且克量级反应也能顺利进行, 具有较大的应用价值.

金鸡纳生物碱及其衍生物除了作为双功能催化剂之外, 也可以作为布朗斯特碱或路易斯碱催化反应.另一方面, 近年来对于金鸡纳碱的修饰也不再局限于更换氢键供体单元, 而是另辟蹊径尝试设计出更加丰富多样的催化剂.例如, 2018年, Vilotijevic课题组[23]利用金鸡纳碱衍生的(DHQD)2PHAL催化剂8作为路易斯碱来活化N-甲硅烷基吡咯、吲哚和咔唑类潜亲核试剂33, 通过与烯丙基氟化物32的不对称亲核取代反应, 以高对映选择性实现了此类杂环的N-烯丙基化反应(Scheme 12).由于吡咯、吲哚化合物的N1, C2, C3位皆具有一定的亲核性, 与亲电试剂反应过程中存在SN2与SN2'竞争反应.因此, 对亲核、亲电试剂的选择显得尤为重要.在严格无氧的反应条件下, 该反应能以优异的收率和对映选择性得到目标产物34, 机理研究表明催化剂(DHQD)2- PHAL是作为路易斯碱催化该反应.

此外, 对于其他新型的金鸡纳碱类似物的催化剂, 也有零星报道. 2016年, Hiemstra课题组[24]通过对奎宁桥环扩环或缩环得到了一系列金鸡纳碱衍生的催化剂9. 2019年, Itsuno课题组[25]设计了一类金鸡纳碱衍生的交联手性多聚硅催化剂10, 这两类催化剂均可以运用到Michael加成反应中, 而催化剂10相对催化剂9效果更好. 2019年, Jindřich课题组[26]设计了一类金鸡纳碱衍生的α-和β-环糊精催化剂11, 并将其成功应用到不对称烯丙基氨化反应中.这些新型金鸡纳碱类似物和衍生物的催化剂不仅丰富了金鸡纳碱类催化剂的种类, 使得科研工作者在选择催化剂时有了更多选择, 同时也给化学家们带来新的启示, 有望在不久的将来设计出更多、更高效的有机催化剂(Scheme 13).

20世纪80年代, Sharpless课题组[27]在OsO4氧化烯烃的基础上, 利用金鸡纳碱衍生的(DHQ)2PHAL手性配体, 首次发展了烯烃的不对称双羟基化反应和胺羟基化反应, 这项开创性的工作给了科研工作者很大的启迪.化学家们考虑能否将金鸡纳生物碱及其衍生物作为一类优异的手性配体应用到其他类型反应中.金鸡纳生物碱因具有多个手性中心, 且奎宁和喹啉环的刚性结构使得该物质具有类似酶的手性口袋, 从而提供手性环境.另外, 奎宁桥环上的N原子以及C9位羟基氧原子均具有未配对的孤对电子, 可与具有空轨道的金属配位.值得一提的是, 金鸡纳生物碱具有多个可修饰位点, 通过合理的结构修饰即可达到对催化中心的空间和电子效应进行调控的目的.配体中的配位原子可以以阴离子的形式与金属成键, 或提供孤对电子与金属形成路易斯酸碱加合物, 而配位的方式主要有单齿配位、双齿配位和三齿配位, 这与配位的金属密切相关.本部分将按照配位方式展开介绍, 而早期发展的烯烃的不对称双羟基化反应将不作介绍.
2008年, 游劲松课题组[28]基于金属-有机自组装构建手性催化剂的策略, 设计了一类新型的辛可尼啶/钛/联萘酚络合物作为手性双功能催化剂, 并成功应用到亚磷酸二甲酯35和醛类化合物36的不对称氢磷酰化反应(Scheme 14).作者发现, 自组装催化剂的对映选择性控制和催化活性在很大程度上取决于联萘二酚37的电性.当其3, 3'-位的位阻增加时, 反而不利于此反应的进行.在这项工作中, 作者认为自组装构建的手性双功能催化剂中的钛金属中心作为路易斯酸, 通过与醛底物36配位使其活化.而金鸡纳生物碱中奎宁环的氮原子作为路易斯碱, 通过氢键作用活化亚磷酸酯, 从而使其选择性地从位阻较小的Si面发生反应.催化剂各组分之间的相互协同, 最后能以优异的收率和对映选择性得到具有重要生物活性与药物价值的α-羟基磷酸酯38.

2009年, 冯小明课题组[29]利用金鸡纳碱/四异丙氧基钛/联苯酚原位产生催化剂, 利用三甲基硅氰(TMS- CN, 40a)或者氰基甲酸乙酯(40b)作为氰基源, 以优异的收率和对映选择性实现了醛、酮、醛亚胺和酮亚胺的不对称氰基化反应(Scheme 15).值得注意的是, 氰基甲酸乙酯成功地应用于醛亚胺和酮亚胺的不对称Strecker反应中, 以优异的收率和对映选择性得到了一系列的α-氨基氰化物42a.在此反应中, 配体简单易得, 操作简便并且反应条件十分温和, 为制备具有重要应用价值的手性氰醇和α-胺基氰化物提供了一个有效的途径.通过控制实验和核磁实验分析, 作者发现金鸡纳生物碱和联苯酚中的所有羟基均和金属钛发生配位, 形成真正的催化剂物种.联苯酚配体3, 3'-位上的大位阻取代基对反应高对映选择性的实现至关重要.根据控制实验和核磁实验分析, 作者也确证了联苯酚配体在催化剂中绝对构型为S构型.另外, 作者证明了添加剂异丙醇(iPrOH)在催化剂的作用下, 能够和氰基源发生反应, 产生真正的氰基化试剂氢氰酸(HCN), 在此过程中, 金鸡纳生物碱中的三级胺对于氢氰酸的产生至关重要.此外, 其也作为路易斯碱来活化氢氰酸.

2013年, 冯小明和胡常伟等[30]将氰源替换为氢氰酸, 在类似的体系下同样研究了该反应(Scheme 16).而在该工作中, 作者借助密度泛函理论(DFT)计算和分层计算(ONIOM)模拟更进一步地解释了机理中存在的问题.作者提出氢氰酸的异构化过程是该反应的决速步, 而碳-碳键生成则是该反应过程中立体选择性的控制步骤.在不对称诱导过程中, 通过与中心金属钛的协同作用, 手性金鸡纳碱配体向轴向柔性非手性双酚配体进行手性转移, 这一机理细节的完善使得人们对该反应有了更深的了解.
2009年, 冯小明课题组[31]进一步拓展了该催化体系的实用性, 作者利用辛可尼/钛/(S)-联萘酚的催化体系实现了醛47与重氮乙酸乙酯48的不对称的Aldol反应, 得到一类具有生物活性醇的重要前体重氮-β-羟基酯50 (Scheme 17a). 2010年, 冯小明课题组[32]又利用类似的催化体系实现了氰基甲酸酯40b与α, β-不饱和羧酸酯51的不对称氰基化反应(Scheme 17b).在无溶剂和温和的反应条件下, 以优异的收率和对映选择性获得目标产物52, 且该产物具有较高的附加价值.

另外, 冯小明课题组[33]还发展了一种有效的奎宁- Al(OiPr)3络合物催化体系, 实现了丙二腈54与查尔酮53的不对称Michael加成反应(Scheme 18).并且产物可以进一步转为在生物化学和药学中十分有用的四氢吡喃类衍生物.作者认为奎宁在该反应中扮演了两个角色:一方面, 奎宁环中三级胺部分作为布朗斯特碱活化底物丙二腈; 另一方面, 羟基作为手性配体与三价铝作用.作者也给出了可能的催化活化模式.

2012年, Glorius课题组[34]报道了奎宁作为手性配体参与的钯催化环酮56和芳基溴代物或者碘代物57的不对称α-芳基化反应(Scheme 19).与传统常用的钯/联萘酚催化体系相比, 该反应具有更好的立体选择性, 其ee值最高可达到93%.此外, 他们提出了三种可能的作用模型, 而基于控制实验作者更认同B模型:奎宁作为单齿配体(氮原子与钯配位)的同时也通过弱相互作用与底物结合.

手性醇类化合物骨架广泛存在于许多天然产物和药物分子中, 而潜手性羰基化合物被认为是构建手性醇类化合物的最佳原料之一, 因而受到化学家广泛关注.在过去的二十年中, 陆续发展了手性二胺配体或者氮磷配体, 通过与钌、铑、铱、铝、钐等金属的结合实现羰基化合物的不对称氢转移化反应. 2003年, Arvidsson等[35]报道了以金属铱作为催化剂, 具有奎宁环结构单元的手性二胺4为外加配体, 异丙醇作为溶剂的条件下, 实现了苯乙酮59的高效还原, 其ee值最高可达95% (Scheme 20a).随后, 张生勇课题组[36]以[Ir(COD)Cl]2作为金属前体, 9-氨基金鸡纳生物碱作为配体, 异丙醇作为氢源, 也实现了芳基酮的不对称转移氢化反应.相比之前的工作, 产物的ee值有所上升, 但产率有所下降(Scheme 20b).

手性β-氨基磷酸及其衍生物由于具有独特的生物活性以及可作为配体参与不对称催化, 因此其立体选择性地合成受到了许多化学家们的密切关注. 2012年, Nakamura课题组[37]利用金鸡纳碱衍生的吡啶甲酰胺配体6, 在路易斯酸二乙基锌(Et2Zn)催化下, 实现了氮杂环丙烷61与亚磷酸酯62的不对称氢磷酰化反应.产物β-胺基亚磷酸酯类化合物63a经过简单的转换即可得到高附加值的β-氨基磷酸, 并且无对映选择性损失.此外, 他们结合ESI-MS分析所捕获到的关键中间体和产物的绝对构型, 给出了反应过程中可能的过渡态(Scheme 21).

2014年, Nakamura课题组[38]利用金鸡纳碱衍生的吡啶甲酰胺类配体7/Cu(OTf)2的催化体系, 顺利地实现了酮亚胺64与α-异氰酸酯65的不对称Mannich反应(Scheme 22).与之前工作不同的是, 金鸡纳碱衍生的吡啶甲酰胺配体中, 吡啶环4-位增加取代基可以明显改善反应的非对映选择性.通过筛选后发现, 4-位连有三氟甲基时效果最佳, 其dr值最高可达92:8.该反应以中等收率和高对映选择性, 高非对映选择性有效地合成了α, β-二氨基酸的重要前体手性咪唑啉类化合物66, 底物普适性良好, 适用于各种芳基烷基取代的酮亚胺.作者也给出了具体的催化模式.随后该团队[39]还实现了α-取代异氰酸酯与酮亚胺的直接不对称Mannich反应, 进一步拓展了该类催化体系的反应适用范围.

2013年, 宋玲课题组[40]设计了一类由金鸡纳碱衍生的新型手性磷酰胺配体8, 并将其成功应用到二乙基锌(Et2Zn)与醛36的不对称加成反应中(Scheme 23).通过对配体的筛选, 他们发现手性磷酰胺配体中喹啉环C6位甲氧基对于催化剂的催化效率和立体选择性有着重要的影响.在甲苯与二氯甲烷的混合溶剂中, 能以高达99%的收率和ee值得到具有重要价值的手性仲醇.对于可能的活化模式, 作者认为手性配体L8和二乙基锌(Et2Zn)反应生成手性L8-Zn复合物.该复合物是一种多功能的催化剂, 其含有N, N-螯合的锌金属中心作为路易斯酸来活化苯甲醛, 来自磷酰胺基团的P=O部分作为共轭的路易斯碱, 通过Zn—N—P=O键之间的共轭作用活化二乙基锌(Et2Zn).手性磷酰胺配体中喹啉环C6位甲氧基团作为第二个路易斯碱来协助P=O部分活化二乙基锌, 进一步增强其亲核性, 从而获得更高的反应性, 并使过渡态的构型更加刚性, 同时具有更好的对映选择性.

同年, 王永梅和黄耀东等[41]基于手性分子结合(非)手性分子构建手性配体这一策略, 从简单易得的原料出发, 只需一步, 就能够以相对较高的收率制备得到一系列新的金鸡纳碱衍生的联吡啶配体9, 并成功应用到铜催化的醛36的不对称Henry反应中(Scheme 24).该催化体系可以适用于多种芳香、杂芳香和脂肪醛, 以高收率和高对映选择性得到相应的硝基烷醇类化合物.此外, 他们基于1H NMR、DFT计算研究, 给出了可能的催化模式: Cu-L9复合物在平面上有四个强的配位点, 在顶端有两个弱的配位点, 手性配体联吡啶片段中两个氮原子占据两个相邻的强配位点.为了最大程度活化底物, 亲核试剂硝基甲烷7垂直于催化平面与铜作用, 而底物醛在平面上与作为路易斯酸的铜配位, 从而被活化.因为C6位上甲氧基的空间位阻效应, 底物醛中的大位阻基团远离甲氧基, 指向空位.同时, 铜配合物的顶端被硝基甲烷占据, 因此, 硝基甲烷从醛的Si面进攻, 进而得到R构型的产物.

2018年, 刘心元课题组[42]利用合理设计的金鸡纳碱衍生的手性磺酰胺化合物10作为配体, 以Togni’s试剂70为三氟甲基自由基源, 成功地实现了铜催化的烯基肟69的不对称自由基氧三氟甲基化反应, 以优异的收率和对映选择性直接获得具有α-四取代中心的含CF3的异噁唑啉产物(Scheme 25).该产物经过简单地转换即可合成高附加值的1, 3-氨基醇手性化合物.此外, 他们通过对照实验、FTIR光谱数据和高分辨质谱分析, 作者认为L10中了奎宁环上的叔胺和磺酰胺阴离子作为N, N-双齿配体, 与铜配位形成五元双螯合配合物.并在此基础上提出了可能的反应机理:铜和L10通过配体交换, 原位生成真正的活性催化剂双螯合的Cu(Ⅰ)络合物10-A, 与底物70发生单电子转移产生Cu(Ⅱ)络合物10-B和CF3自由基. CF3自由基迅速对底物69中的烯烃进行加成, 产生自由基中间体69-A, 69-A与Cu(Ⅱ)络合物10-B通过配体交换形成中间体69-B.随后发生分子内的氧化还原反应生成Cu(Ⅲ)物种69-C, 再经过还原消除得到最终产物71.但是作者也不能排除磺酰胺上的氮阴离子和铜单齿配位, L10中奎宁环上的叔胺作为碱协同活化肟羟基这一过程.

近年来, 自由基介导的末端烯烃的不对称双官能化反应取得了迅猛的发展, 但是, 对于自由基介导的非末端烯烃的不对称双官能团化反应(产生两个相邻的立体中心)仍然具有挑战性. 2020年, 刘心元课题组[43]利用该团队发展的铜-金鸡纳碱衍生的手性磺酰胺配体11络合物作为强的单电子还原催化剂, 实现了β, γ-不饱和酮肟72与磺酰氯73的不对称自由基氧磺酰化反应(Scheme 26).该方法反应条件温和, 适用于一系列含有端烯或非端烯的β, γ-不饱和酮肟, 以及(杂)芳基或烷基取代的磺酰氯, 以优异的收率、对映选择性和非对映选择性得到含有磺酰基取代的异噁唑啉类产物74.

通过机理实验和DFT理论计算, 作者提出了可能的反应机理(Scheme 27):原位产生的双螯合的Cu(Ⅰ)络合物11-A具有强的还原能力, 首先和底物磺酰氯73发生单电子转移产生相应的Cu(Ⅱ)络合物11-B和磺酰基自由基.接着, 11-B通过脱去底物72中的酮肟上的质子与其络合, 形成中间体72-A.随后, 磺酰基自由基对中间体72-A中的烯烃进行加成, 此过程是快速可逆的, 产生烷基自由基中间体72-B和72-B'.在Curtin-Hammett动力学控制下, 烷基自由基中间体72-B优先进行反应, 通过分子内的自由基取代反应, 立体选择性地形成C—O键, 产生络合72-C, 该络合物最终解离生成产物74和Cu(Ⅰ)络合物11-A.这一策略的成功实施将为自由基介导的、非末端烯烃的不对称双官能化反应的对映选择性及非对映选择性的精确立体控制打开一扇新的大门.

2019年, 何炜课题组[44]设计了一类新型金鸡纳碱衍生的噁唑啉配体12, 实现了钯催化含氮二烯芳香化合物75分子内的不对称Aza-Wacker氧化串联环化反应, 最终以良好的收率, 优异的对映选择性和非对映选择性得到目标产物76 (Scheme 28).值得一提的是, HNTf2对反应的立体选择性有着十分重要的影响, 可能的原因是: HNTf2可以稳定二价钯离子.该反应条件温和, 操作简单, 而绿色氧化剂氧气的使用使得该反应更加经济环保.

2009年, Skarzewski课题组[45]开发了金鸡纳碱衍生的手性硫醇配体13, 实现了苯甲醛77与硝基甲烷7的不对称Henry反应(Scheme 29).对于可能的作用模型, 作者认为手性硫醇配体占据赤道平面两个配位点.与此同时, 金属铜作为路易斯酸活化底物醛.硝基甲烷则在轴向位置与铜配位, 使得亲核试剂从空间位阻更小的Re面进攻醛羰基从而得到S构型的手性产物.

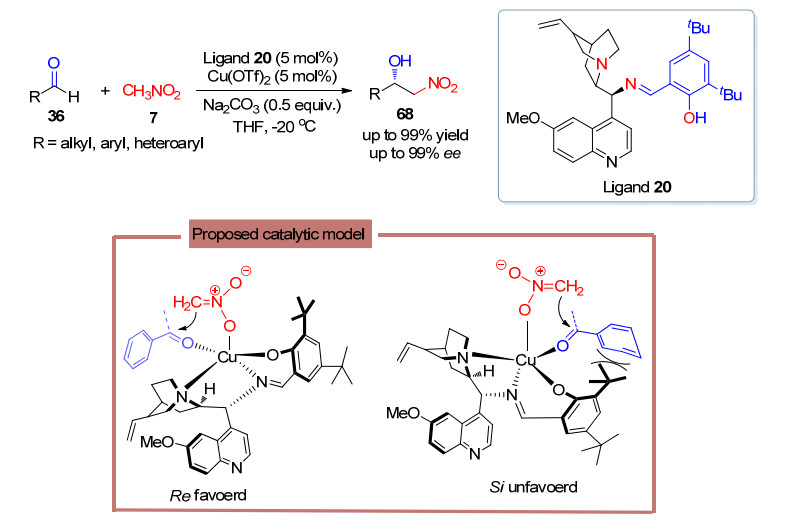
2011年, 何炜课题组[46]在此基础上开发了金鸡纳碱衍生的席夫碱手性氮氧配体14, 并成功应用到铜催化的不对称Henry反应中, 完成了对一系列邻硝基仲醇68的高效合成(Scheme 30).对于可能的作用模式, 作者认为两分子手性配体占据金属铜四个配位点, 同时金属铜作为路易斯酸活化底物, 而由于醛R1基团与喹啉环空间上的排斥作用, 从而使得亲核试剂将从空间位阻更小的Re-面进攻醛羰基.

2008年, 张生勇课题组[47]从商业可得的光学纯1, 2-二芳基乙二醇和金鸡纳碱为初始原料, 经过两步一锅简单的转化, 就能得到金鸡纳碱衍生的手性氮磷配体15, 这也是该类配体的首次合成(Scheme 31).作者也成功将该配体应用于钯催化的不对称烯丙基烷基化反应.该手性氮磷配体的合成, 是对钯催化烯丙基反应可选择配体的补充.

2011年, Dixon课题组[48]设计了一类金鸡纳碱衍生的新型手性膦胺配体16.在零下二十度, Ag2O作催化剂, AcOEt作溶剂的条件下, 成功实现了异氰酸酯82与醛36的不对称Aldol反应(Scheme 32).该反应适用于芳基醛和具有支链的脂肪醛, 能够以优异的收率、对映选择性和非对映选择构筑一系列具有重要生物活性的噁唑啉骨架.但是对于直链脂肪醛却不能兼容.同时, 降低此反应催化剂的用量对反应并无较大影响.对于可能的作用模式, 作者认为手性膦胺配体与一价银盐形成的复合物可作为布朗斯特碱/路易斯酸双功能催化剂, 从而实现对该反应的高效催化和手性控制. Dixon课题组[49-51]还把该催化体系分别成功应用于了α-取代异氰酸酯与酮的Aldol加成/环化反应以及酮亚胺的对映选择性Mannich加成/环化反应, 合成了一系列多官能化的咪唑啉产物.

2017年, Dixon和Paton等[52]基于烯胺催化与金属协同催化策略, 利用相似的手性膦胺配体17a实现了4-炔丙基胺环己酮84的环异构化反应(Scheme 33).在该反应中, 首先有机胺催化剂和羰基原位生成高反应活性的烯胺中间体, 随后与被银复合物活化的端炔发生分子内的亲核加成反应.基于之前的研究成果, 作者通过DFT计算进一步验证了可能的作用模式:手性膦胺配体中氮磷原子与银配位; 另一方面, 金属银作为π酸活化端炔.同时, 作者也发现酰胺与磺酰胺之间存在N—H…O的氢键作用.反应具有良好的官能团兼容性和底物适用范围, 原子经济性高.产物85作为合成药物吗啡的关键中间体, 因此, 该策略在有机合成中有较大的应用价值.

作者认为催化体系中两个手性配体之间存在着明显的匹配和不匹配效应, 如Scheme 34所示:当使用手性匹配的R构型的吡咯烷ent-12a和金鸡纳碱衍生的胺基膦配体17b时, 可以高效、高对映选择性的获得产物吗啡85a的对映体ent-85a (Scheme 34, 左上和右下比较).相反, 当使用手性不匹配的配体组合12a/17b或者ent-12a/17a时(Scheme 34, 右上和左下比较), 反应的效率和对映选择性均大幅降低.另外, 当作者使用非手性的吡咯烷12b和金鸡纳碱衍生的手性胺基膦配体17a时, 反应仅能以36%的对映选择性得到目标产物85a; 而当使用非手性的配体17c和手性配体12a时, 则能够以80%的对映体过量得到产物85a.此结果表明, 吡咯烷在此反应的对映选择性控制中发挥着主要作用.
具有轴手性的联芳基骨架, 尤其是芳基-杂芳基骨架, 存在于许多重要的天然产物和药物中, 也是许多配体和催化剂的关键结构. 2019年, 祝介平等[53]开发了一类高效的氧化银和手性膦胺配体18, 催化α-异氰基乙酸乙酯82与β-芳基-α, β-炔基酮86的不对称杂环反应(Scheme 35).该反应以高收率和高对映选择性实现了3-芳基吡咯类化合物87的轴手性构建.作者认为, 在此反应中, 配体上的酰胺氮、膦、炔酮物种上的羰基氧和异氰化物的碳原子和催化剂的银进行配位, 从而确定了两种反应物的空间位置.配体上质子化的奎宁环和酮氧之间的氢键作用, 可以进一步改善络合物的立体化学环境, 最终形成过渡结构86-A.由于空间位阻的原因, 烯醇化合物将从炔烃的背面进攻, 立体选择性地得到中间体86-B.随后, 发生分子内环化得到中间体86-C.中间体86-C发生质子化产生中间体86-D.最后经过1, 5-氢转移得到具有轴手性的3-芳基吡咯类化合物87.作者通过对底物电性的调控, 完美地避免了亲核试剂对炔酮的1, 2-加成反应.此外, 反应具有良好的官能团耐受性和底物适应性, 具有实际的应用价值.

2007年, Lin课题组[54]设计了金鸡纳碱衍生的席夫碱三齿配体19, 并将其成功地应用到有机金属试剂二乙基锌与芳香醛88的亲核加成反应中, 高效地构建了一系列1-芳基丙醇化合物89 (Scheme 36).对于可能的作用模式, 作者认为手性配体作为三齿配体与金属锌形成复合物, 与此同时, 奎宁环三级胺作为路易斯碱, 达到了路易斯酸-路易斯碱双功能活化的目的.该反应条件温和, 能以令人满意的收率和对映选择性得到目标化合物.

2012年, 张生勇和何炜课题组[55]将其团队开发的金鸡纳碱衍生的手性席夫碱配体14进行修饰后得到配体20, 并将其成功应用到醛36与硝基甲烷7的不对称Henry反应中(Scheme 37).相比之前的工作, 催化中心铜的空间和电子效应由于叔丁基的加入得到了调控, 反应的收率及对映选择性明显提升, 同时手性配体和催化剂的用量也均降至5 mol%.此外, 通过单晶衍射和NMR数据, 他们给出了催化模式.
Sonogashira偶联反应作为构建碳-碳键的一种经典方法, 受到了化学家们的广泛关注.然而, 不对称的Sonogashira偶联反应, 尤其是对C(sp3)—C(sp)碳-碳键的不对称构建在很大程度上仍未得到开发. 2019年, 刘心元课题组[56]报道了利用Cu(Ⅰ)/金鸡纳生物碱的P, N-配体催化体系, 基于自由基过程实现了卤代烃91与炔烃90的不对称Sonogashira C(sp3)—C(sp)偶联反应(Scheme 38).该反应操作简单, 反应条件温和, 对于一系列末端的炔烃和消旋的烷基卤化物具有很好的普适性(>120个实例).此外, 该方法也能很好地使用工业原料乙炔气体作为底物, 从而高度单一地合成有价值的手性末端炔烃类化合物(92a, 92b), 所获得手性端炔类化合物可以进一步发生后续反应, 从而以高收率和对映选择性产生环加成产物92c.该手性端炔类化合物可以进行第二次不对称的Sonogashira偶联反应, 以高非对映选择性提供产物92d.自由基前体片段不仅仅局限于外消旋的α-芳基和杂芳基取代的烷基卤化物, 烯丙基和α-炔基/氰基/酰胺类烷基溴化物也能很好地适用于该转化(92e, 92f, 92g, 92h).更重要的是, 此方法能够兼容具有生物活性和功能分子的核心骨架, 为药物和天然产物的合成和后期修饰提供了一种可行的方法.通过自由基捕获和自由基钟实验, 作者认为该反应体系涉及苄基自由基过程.

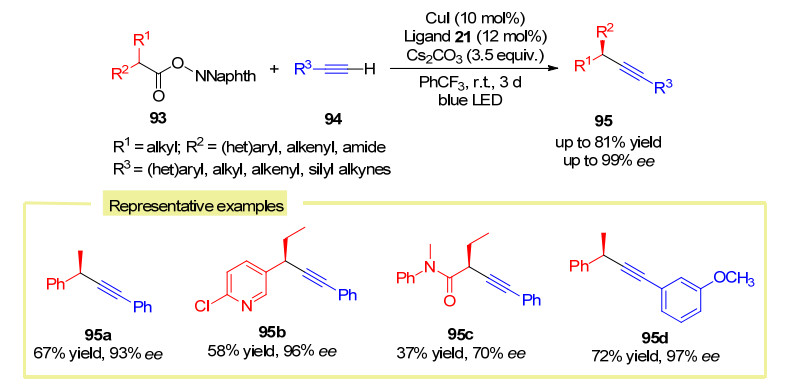
2020年, 刘心元课题组[57]利用消旋的N-羟基邻苯二甲酰亚胺类酯93作为烷基自由基前体, 在光引发铜催化的条件下实现了其与端炔94的不对称自由基脱羧C(sp3)—C(sp)偶联反应(Scheme 39).在该反应中, 端炔/铜(Ⅰ)/金鸡纳碱衍生的手性N, N, P-配体三组分在碱性条件下原位生成炔铜(Ⅰ)复合物, 该复合物可吸收蓝光到达激发态而充当具有强还原能力的光催化剂, 从而将底物N-羟基邻苯二甲酰亚胺类酯还原生成烷基自由基物种以及释放出一分子二氧化碳.该反应展示出优异的底物适用范围和官能团兼容性, 以中等到较好的产率和优秀的立体选择性得到相应的偶联产物.该工作也进一步拓展了该类催化体系的适用性, 弥补了以往的方法中较难适用于烷基卤代物以及不稳定前体等不足.
2019年, 刘心元课题组[58]基于之前的工作, 利用铜/金鸡纳生物碱的N, N, P-配体催化体系, 实现了远端未活化的C(sp3)—H键和末端炔烃的自由基不对称氧化C(sp3)—C(sp)交叉偶联反应, 具有高度的区域、化学和对映选择性(Scheme 40).使用N-氟代酰胺作为温和的酰胺氮自由基前体和氧化剂, 产生氮自由基以后通过分子内的1, 5-或者1, 6-氢迁移过程, 可以将远端未活化的C(sp3)—H键转化为相应的烷基自由基, 同时也有效避免了末端炔烃的自身的Glaser偶联反应.该反应适用于一系列取代的芳基和杂芳基炔类化合物.对于(杂)苄基和炔丙基的C(sp3)—H键也能够有效的兼容.基于机理探究实验和先前的文献报道, 作者提出了可能的反应机理:首先, 在碱的存在下, Cu(Ⅰ)与手性配体和末端炔烃发生反应, 产生相应的络合物97-B.随后, 络合物97-B与底物96发生单电子转移过程, 形成Cu(Ⅱ)络合物97-C和酰胺氮自由基物种97-A.接着, 酰胺氮自由基物种96-A通过分子内的1, 5或者1, 6氢迁移产生烷基自由基中间体96-B.然后, 烷基自由基中间体96-B被Cu(Ⅱ)络合物97-C捕获, 产生Cu(Ⅲ)络合物97-D.最后, 通过还原消除生成目标产物98和重新生成Cu(Ⅰ)络合物97-A.

2020年, 刘心元课题组[59]利用其团队开发的金鸡纳碱衍生的N, N, P-多齿手性配体21, 在铜催化的条件下, 从易于获得的烷基卤化物和末端炔烃出发, 实现了烯烃100分子间不对称自由基1, 2-烷基炔基化反应, 能够以优异的收率和对映选择性高效地制备了一系列手性炔化合物102 (Scheme 41).该反应底物范围广和优异官能团兼容性, 适用于各种(杂)芳基、炔基和酰胺取代的烯烃, (杂)芳基、烷基和甲硅烷基取代的炔烃, 以及各种取代的一级、二级和三级烷基自由基前体.重要的是该反应为烯烃的不对称1, 2-双官能化提供了一种可行的策略.基于控制实验、EPR光谱实验、NMR实验等其它机理研究, 作者提出了如下的反应过渡态.底物中相对较大的(杂)芳基与配体中的P-3, 5-二叔丁基苯基之间由于空间位阻导致Si-Ts不利, 从而产生绝对构型为R的产物.该策略与直接通过C(sp2/sp3)试剂实现烯烃的双官能化反应形成很好的互补, 具有较大的应用价值.

综上所述, 因其优势的手性骨架特点和丰富的可修饰潜力, 金鸡纳生物碱及其衍生物作为有机催化剂在不对称合成中已有了十分广泛的应用, 但其催化活化模式较为单一, 对反应底物适用范围有一定的局限性.而将其作为手性配体参与金属催化的不对称合成发展虽然起步较早, 但发展却相对缓慢, 反应类型受限, 仍然有非常大的发展空间.相信通过科研工作者的努力, 将可以开发出更加丰富多样的金鸡纳碱衍生的催化剂及手性配体, 进一步将其应用到各式各样的反应类型中, 进一步推动不对称合成的发展.
Yang, F.; Hanon, S.; Lam, P.; Schweitzer, P. Am. J. Med. 2009, 122, 317. doi: 10.1016/j.amjmed.2008.11.019
Haas, L. F. J. Neurol., Neurosurg. Psychiatry 1994, 57, 1333. doi: 10.1136/jnnp.57.11.1333
Rabe, P.; Ackerman, E.; Schneider, W. Chem. Ber. 1907, 40, 3655. doi: 10.1002/cber.190704003157
Woodward, R. B.; Doering, W. E. J. Am. Chem. Soc. 1945, 67, 860. doi: 10.1021/ja01221a051
Carter, O. L.; McPhail, A. T.; Sim, G. A. J. Chem. Soc. A 1967, 365. http://www.sciencedirect.com/science/article/pii/S0022328X00843740
Stork, G.; Niu, D.; Fujimoto, R. A.; Koft, E. R.; Balkovec, J. M.; Tata, J. R.; Dake, G. R. J. Am. Chem. Soc. 2001, 123, 3239. doi: 10.1021/ja004325r
Yeboah, E. O.; Yeboah, S.; Singh, G. Tetrahedron 2011, 67, 1725. doi: 10.1016/j.tet.2010.12.050
Boratyński, P. J.; Błajet, M. Z; Skarżewsk, J. Textbook of The Alkaloids:Chemistry and Biology, Elsevier, Poland, 2019, p. 29.
List, B. Angew. Chem., Int. Ed. 2010, 49, 1730. doi: 10.1002/anie.200906900
MacMillan, D. W. C. Nature 2008, 455, 304. doi: 10.1038/nature07367
(a) Kacprzak, K.; Gawronski, J. Synthesis 2001, 961.
(b) Tian, S. K.; Chen, Y.; Hang, J.; Tang, L.; McDaid, P.; Deng, L. Acc. Chem. Res. 2004, 37, 621.
(c) Singh, G.; Yeboah, E. Rep. Org. Chem. 2016, 6, 47.
Shi, L. M.; Dong, W. W.; Tao, H. Y.; Dong, X. Q.; Wang, C. J. Org. Lett. 2017, 19, 4532. doi: 10.1021/acs.orglett.7b02107
Shao, Y. D.; He, X. Y.; Han, D. D.; Yang, X. R.; Yao, H. B.; Cheng, D. J. Asian J. Org. Chem. 2019, 8, 2023. doi: 10.1002/ajoc.201900504
Mukhopadhyay, S.; Pan, S. C. Eur. J. Org. Chem. 2019, 2639.
Nakamura, S.; Hayama, D.; Miura, M.; Hatanaka, T.; Funahashi, Y. Org. Lett. 2018, 20, 856. doi: 10.1021/acs.orglett.7b04022
Takata, S.; Endo, Y.; Ullah, M. S.; Itsuno, S. RSC Adv. 2016, 6, 72300. doi: 10.1039/C6RA14535C
Endo, Y.; Takata, S.; Kumpuga, B. T.; Itsuno, S. ChemistrySelect 2017, 2, 10107. doi: 10.1002/slct.201702010
Reddy, R. R.; Gudup, S. S.; Ghora, P. Angew. Chem. Int. Ed. 2016, 55, 15115. doi: 10.1002/anie.201607039
Mondal, K.; Pan, S. C. J. Org. Chem. 2018, 83, 5301. doi: 10.1021/acs.joc.8b00436
Wu, Y. C.; Jhong, Y.; Lin, H. J.; Swain, S. P.; Tsaia, H. H. G.; Hou, D. R. Adv. Synth. Catal. 2019, 361, 4966. doi: 10.1002/adsc.201900997
姜海洋, 李强, 齐庆杰, 杨晨曦, 张丹, 有机化学, 2018, 38, 825. doi: 10.6023/cjoc201703037Jiang, H. Y.; Li, Q.; Qi, Q. J.; Yang, C. X.; Zhang, D. Chin. J. Org. Chem. 2018, 38, 825(in Chinese). doi: 10.6023/cjoc201703037
Zhu, W.-R.; Liu, K.; Huang, W.-H.; Huang, W.-J.; Chen, Q.; Weng, J.; Lin, N.; Lu, G. Org. Lett. 2020, 22, 5014. doi: 10.1021/acs.orglett.0c01578
Zi, Y.; Lange, M.; Schultz, C.; Vilotijevic, I. Angew. Chem. Int. Ed. 2019, 58, 10727. doi: 10.1002/anie.201903392
Breman, A. C.; Heijden, G. V. D.; Maarseveen, J. H. V.; Ingemann, S.; Hiemstra, H. Chem. Eur. J. 2016, 22, 14247. doi: 10.1002/chem.201601917
Kumpuga, B. T.; Itsuno, S. Asian J. Org. Chem. 2019, 8, 251. doi: 10.1002/ajoc.201800740
Tichá, I. C.; Hybelbauerová, S.; Jindřich, J. J. Org. Chem. 2019, 15, 830. http://www.ncbi.nlm.nih.gov/pubmed/31019575
Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483. doi: 10.1021/cr00032a009
Yang, F.; Zhao, D. B.; Lan, J. B.; Xi, P. H.; Yang, L.; Xiang, S. H.; You, J. S. Angew. Chem. Int. Ed. 2008, 47, 5646. doi: 10.1002/anie.200801766
Wang, J.; Wang, W. T.; Li, W.; Hu, X. L.; Shen, K.; Tan, C, ; Liu, X. H.; Feng, X. M. Chem. Eur. J. 2009, 15, 11642. doi: 10.1002/chem.200900936
Su, Z. S.; Li, W. Y.; Wang, J.; Hu, C. W.; Feng, X. M. Chem. Eur. J. 2013, 19, 1637. doi: 10.1002/chem.201202237
Wang, W. T.; Shen, K.; Hu, X. L.; Wang, J.; Liu, X. H.; Feng, X. M. Synlett 2009, 1655.
Wang, J.; Li, W.; Liu, Y. L.; Chu, Y. Y.; Lin, L. L.; Liu, X. H.; Feng, X. M. Org. Lett. 2010, 12, 1280. doi: 10.1021/ol100169r
Shi, J.; Wang, M.; He, L.; Zheng, K.; Liu, X. H.; Lin, L. L.; Feng, X. M. Chem. Commun. 2009, 31, 4711.
Richter, C.; Ranganath, K. V. S.; Glorius, F. Adv. Synth. Catal. 2012, 354, 377. doi: 10.1002/adsc.201100669
Hartikka, A.; Modin, S. A.; Arvidsson, P. G. Org. Biomol. Chem. 2003, 1, 2522. doi: 10.1039/b304060g
He, W.; Liu, P.; Zhang, B. L.; Sun, X. L.; Zhang, S. Y. Appl. Organomet. Chem. 2006, 20, 328. doi: 10.1002/aoc.1055
Hayashi, M.; Shiomi, N.; Funahashi, Y.; Nakamura, S. J. Am. Chem. Soc. 2012, 134, 19366. doi: 10.1021/ja309963w
Hayashi, M.; Iwanaga, M.; Shiomi, N.; Nakane, D.; Masuda, H.; Nakamura, S. Angew. Chem. Int. Ed. 2014, 53, 8411. doi: 10.1002/anie.201404629
Yamaji, R.; Iwanaga, M.; Nakamura, S. Chem. Commun. 2016, 52, 7462. doi: 10.1039/C6CC02911F
Shen, B.; Huang, H. Y.; Bian, G. L.; Zong, H.; Song, L. Chirality 2013, 25, 561. doi: 10.1002/chir.22171
Liu, M.; Ma, S. J.; Tian, Z. Q.; Wu, H.; Wu, L. L.; Xu, X. F.; Huang, Y. D.; Wang, Y. M. Tetrahedron:Asymmetry 2013, 24, 736. doi: 10.1016/j.tetasy.2013.05.009
Li, X. T.; Gu, Q. S.; Dong, X. Y.; Meng, X.; Liu, X. Y. Angew. Chem. Int. Ed. 2018, 57, 7668. doi: 10.1002/anie.201804315
Li, X. T.; Lv, L.; Wang, T.; Zhang, X. H.; Cheng, G. J.; Liu, X. Y. Chem 2020, 6, 1692. doi: 10.1016/j.chempr.2020.03.024
Tian, Q. Q.; Liu, Y. L.; Wang, X. Y.; Wang, X.; He, W. Eur. J. Org. Chem. 2019, 24, 3850.
Zielińska, B. M.; Skarżewski, J. Tetrahedron:Asymmetry 2009, 20, 1992. doi: 10.1016/j.tetasy.2009.07.020
Wei, Y.; Yao, L.; Zhang, B. L.; He, W.; Zhang, S. Y. Tetrahedron 2011, 67, 8552. doi: 10.1016/j.tet.2011.08.076
Wang, Q. F.; He, W.; Liu, X. Y.; Chen, H.; Qin, X. Y.; Zhang, S. Y. Tetrahedron:Asymmetry 2008, 19, 2447. doi: 10.1016/j.tetasy.2008.10.030
Sladojevich, F.; Trabocchi, A.; Guarna, A.; Dixon, D. J. J. Am. Chem. Soc. 2011, 133, 1710. doi: 10.1021/ja110534g
Campa, R.; Dixon, D. J. Angew. Chem. Int. Ed. 2015, 54, 4895. doi: 10.1002/anie.201411852
Ortin, I.; Dixon, D. J. Angew. Chem. Int. Ed. 2014, 53, 3462. doi: 10.1002/anie.201309719
Campa, R.; Yamagata, A. D. G.; Ortin, I, Franchino, A.; Thompson, A. L.; Odell, B.; Dixon, D. J. Chem. Commun. 2016, 52, 10632. doi: 10.1039/C6CC04132A
Manzano, R.; Rubén; Datta, S.; Paton, R.; Dixon, D. J. Angew. Chem. Int. Ed. 2017, 56, 5834. doi: 10.1002/anie.201612048
Zheng, S. C.; Wang, Q.; Zhu, J. P. Angew. Chem. Int. Ed. 2019, 58, 1494. doi: 10.1002/anie.201812654
Casarotto, V.; Li, Z. T.; Boucau, J.; Lin, Y. M. Tetrahedron. Lett. 2007, 48, 5561. doi: 10.1016/j.tetlet.2007.05.130
Yao, L.; Wei, Y.; Wang, P. G.; He, W.; Zhang, S. Y. Tetrahedron 2012, 68, 9119. doi: 10.1016/j.tet.2012.08.029
Dong, X. Y.; Zhang, Y. F.; Ma, C. L, ; Gu, Q. S.; Wang, F. L.; Li, Z. L.; Jiang, S. P.; Liu, X. Y. Nat. Chem. 2019, 11, 1158. doi: 10.1038/s41557-019-0346-2
Xia, H. D.; Li, Z. L.; Gu, Q. S.; Dong, X. Y.; Fang, J. H.; Du, X. Y.; Wang, L. L.; Liu, X. Y. Angew. Chem. Int. Ed. 2020, 59, 16926. doi: 10.1002/anie.202006317
Zhang, Z. H.; Dong, X. Y.; Du, X. Y.; Gu, Q. S.; Li, Z. L.; Liu, X. Y. Nat. Commun. 2019, 10, 5689. doi: 10.1038/s41467-019-13705-1
Dong, X.-Y.; Cheng, J.-T.; Zhang, Y.-F.; Li, Z.-L.; Zhan, T.-Y.; Chen, J.-J.; Wang, F.-L.; Yang, N.-Y.; Ye, L.; Gu, Q.-S.; Liu, X.-Y. J. Am. Chem. Soc. 2020, 142, 9501. doi: 10.1021/jacs.0c03130

图式 2 金鸡纳碱衍生物催化剂的代表性例子
Scheme 2 Representative examples of cinchona alkaloid-de- rived catalysts

图式 3 金鸡纳碱衍生的胺-硫脲催化的不对称Diels-Alder反应
Scheme 3 Asymmetric Diels-Alder reaction catalyzed by cinchona alkaloid-derived amine-thiourea

图式 4 金鸡纳碱衍生的胺-硫脲催化的不对称Aza-Henry反应
Scheme 4 Asymmetric aza-Henry reaction catalyzed by amine- thiourea from cinchona alkaloid

图式 5 金鸡纳碱衍生的胺-硫脲催化的不对称Mannich反应
Scheme 5 Asymmetric Mannich reaction catalyzed by cinchona alkaloid-derived amine-thiourea

图式 6 金鸡纳碱衍生的磺酰胺催化的不对称亲核加成
Scheme 6 Asymmetric nucleophilic addition catalyzed by cinchona alkaloid-derived sulfonamide

图式 7 金鸡纳碱衍生的多聚磺酰胺催化的不对称亲核加成反应
Scheme 7 Asymmetric nucleophilic addition catalyzed by cinchona alkaloid-derived sulfonamide polymers

图式 8 金鸡纳碱衍生的方酰胺催化的不对称氧化脱芳/Oxa- Michael加成反应
Scheme 8 Asymmetric oxidative dearomatization/Oxa-Michael addition catalyzed by squaramide from cinchona alkaloid

图式 9 金鸡纳碱衍生的四方酰胺催化的不对称多米诺Michael/酰基转移反应
Scheme 9 Domino Michael/acyl transfer reaction catalyzed by cinchona alkaloid-derived squaramide

图式 10 金鸡纳碱衍生的四方酰胺催化的不对称氮杂-Mi-chael加成反应
Scheme 10 Asymmetric aza-Michael addition catalyzed by squaramide from cinchona alkaloid

图式 11 金鸡纳碱衍生的四方酰胺催化的不对称极性反转交叉Mannich反应
Scheme 11 Asymmetric umpolung cross-Mannich reaction catalyzed by cinchona alkaloid-derived squaramide

图式 12 金鸡纳碱衍生的(DHQD)2PHAL催化的不对称亲核加成
Scheme 12 Asymmetric nucleophilic addition catalyzed by (DHQD)2PHAL

图式 13 奎宁环修饰的奎尼丁类似物新型手性催化剂
Scheme 13 New catalysts of quinidine analogues with ring modifications in the quinuclidine scaffold

图式 14 钛(Ⅳ)络合物催化醛的氢磷酰化反应
Scheme 14 Asymmetric hydrophosphonylation catalyzed by titanium complex

图式 17 钛催化的不对称Aldol和Michael加成反应
Scheme 17 Asymmetric aldol, Michael reaction catalyzed by titanium

图式 18 铝-喹宁络合物催化的Michael加成反应
Scheme 18 Michael addition reaction catalyzed by quinine-Al- (OiPr)3 complex

图式 19 钯/金鸡纳碱络合物催化环酮的不对称α-芳基化反应
Scheme 19 Asymmetric α-arylation of cycloketones catalyzed by palladium/cinchona alkaloid

图式 20 铱催化羰基化合物的不对称转移氢化反应
Scheme 20 Asymmetric transfer hydrogenation reaction of carbonyl compounds catalyzed by iridium

图式 21 锌/吡啶甲酰胺络合物催化氮杂环丙烷的氢磷酰化反应
Scheme 21 Asymmetric hydrophosphorylation of aziridines catalyzed by Zn(Ⅱ)-cinchona alkaloid amide

图式 22 铜(Ⅱ)/吡啶甲酰胺络合物催化不对称Mannich反应
Scheme 22 Asymmetric Mannich reaction catalyzed by Cu(Ⅱ)- cinchona alkaloid amide

图式 23 磷酰胺-锌(Ⅱ)复合物催化二乙基锌与醛的不对称加成反应
Scheme 23 Asymmetric additions of diethylzinc to aldehydes catalyzed phosphoramide-Zn(Ⅱ) complexes

图式 24 铜/含金鸡纳碱联吡啶络合物催化的不对称Henry反应
Scheme 24 Asymmetric Henry reaction catalyzed by copper/ cinchona alkaloid-derived bipyridine

图式 25 铜/金鸡纳碱磺酰胺催化烯基肟的不对称氧三氟甲基化反应
Scheme 25 Asymmetric oxytrifluoromethylation of alkenyl oximes catalyzed by copper/cinchona alkaloid-based sulfonamide

图式 26 非对映选择性和对映选择性的β, γ-不饱和酮肟的自由基氧磺酰化反应
Scheme 26 Diastereo- and enantio-selective radical oxysulfonylation reaction of β, γ-unsaturated ketoximes

图式 27 可能的机理及Curtin-Hammett原理
Scheme 27 Plausible reaction mechanism and Curtin-Hammett principle

图式 28 钯/金鸡纳碱噁唑啉络合物催化分子内的Aza-Wacker氧化串联环化反应
Scheme 28 Enantioselective intramolecular aza-Wacker oxidation cyclization reaction catalyzed by palladium/cinchona alkaloid-de- rived oxazoline complex

图式 29 铜催化的不对称Henry反应
Scheme 29 Asymmetric Henry reaction catalyzed by copper(Ⅱ) complex

图式 30 铜/金鸡纳碱席夫碱络合物催化的不对称Henry反应
Scheme 30 Asymmetric Henry reaction catalyzed by copper(Ⅱ)/cinchona alkaloid-derived Schiff base

图式 31 钯/金鸡纳碱衍生P, N配体催化不对称烯丙基烷基化反应
Scheme 31 Asymmetric allylic alkylation catalyzed by palladium/cinchona alkaloid-derived P, N ligand

图式 32 银催化的异氰乙酸乙酯不对称Aldol反应/环化
Scheme 32 Asymmetric aldol reaction/cyclization of isocyano- acetates catalyzed by silver(Ⅰ)/amino-phosphine

图式 33 银与胺共催化烷基环己酮的去对称化环异构化反应
Scheme 33 Desymmetrizing cycloisomerization of alkyl cyclohexanones catalyzed by silver and amine

图式 35 银催化合成轴手性3-芳基吡咯类化合物
Scheme 35 Synthesis of axially chiral 3-arylpyrroles catalyzed by silver

图式 36 席夫碱-锌(Ⅱ)复合物催化二乙基锌与醛的不对称加成反应
Scheme 36 Asymmetric addition of diethylzinc to aldehydes catalyzed by Schiff base-Zn(Ⅱ) complex

图式 37 铜/金鸡纳碱席夫碱催化的不对称的Henry反应
Scheme 37 Asymmetric Henry reaction catalyzed by copper(Ⅱ)/cinchona alkaloid-derived Schiff base

图式 38 铜催化的不对称Sonogashira偶联反应
Scheme 38 Asymmetric Sonogashira coupling catalyzed by copper/cinchona alkaloid-based P, N-ligand

图式 39 光驱动铜催化不对称自由基脱羧偶联反应
Scheme 39 Photoinduced copper-catalyzed asymmetric radical decarboxylation coupling reaction

图式 40 铜催化不对称类Sonogashira-类型氧化偶联反应
Scheme 40 Asymmetric Sonogashira-type oxidative cross-coupling catalyzed by copper

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



 下载:
下载:




 下载:
下载: