
图式 1.
含有甲基基团的代表性药物分子
Scheme 1.
Representative drug molecules containing methyl groups

甲基官能团虽然体积小且极性弱, 却能对诸多生物过程产生重要影响.在有机小分子中引入甲基有助于提高其结合亲和力、生物溶解度和代谢稳定性, 从而极大地改变生物活性分子的药理特性.甲基可以通过构象上的细微改变影响药物分子的溶解性, 对蛋白质的选择性及其药物代谢的半衰期等都有重要影响[1]; 该效应被称为“神奇的甲基效应”[2].在已上市的药物分子或者前体药物中, 含有芳香烃甲基结构的小分子普遍存在(Scheme 1)[3].例如自噬抑制剂硫坎酮(Lucanthone), 抗炎药罗美昔布(Lumiracoxib)、美洛昔康(Meloxicam)、塞来昔布(Celecoxib), 抗白血病药物格列卫(Gleevec), 选择性磷酸二酯酶抑制剂(GSK256066), 胃病药物埃索美拉唑(Nexium), 精神疾病治疗药物奥氮平(Olanzapine)等都含有一个或者多个芳甲基.另一方面, 甲基在有机合成中被作为灵活的有效官能团, 可以通过氧化反应等途径转化为一系列官能团, 例如醛基、羧基和卤素等官能团[4].基于甲基在现代药物化学及有机合成化学中的重要作用, 发展甲基类化合物的温和、高效、绿色的合成方法一直是有机化学家及药物化学家们研究的热点.
在过去的几十年中, 过渡金属催化的交叉偶联反应已成为现代有机合成中构建碳-碳键的有力工具, 在芳香烃化合物甲基化反应应用方面得到很大的发展[5].近年来, 可见光促进的氧化还原反应得到了不断发展[6], 该类反应已经成为有机合成化学的一个重要工具.这一新颖的催化模式利用可见光能量作为反应活化能, 得到高活性反应中间体, 从而实现多类型重要转化.更为重要的是, 可见光促进的反应可以为有机反应提供新的反应途径, 利用温和的反应条件, 实现高效高选择性转化.随着光催化与过渡金属催化(镍催化、铜催化等)的有机融合, 一些传统上的挑战性偶联反应, 包括sp2-sp3偶联可以利用廉价催化剂来实现, 这给芳基甲基化反应的发展提供了新的发展机会.近来, 已有多例利用可见光氧化还原催化体系实现了芳香烃的甲基化反应的报道, 为向芳烃中引入甲基提供了新颖、高效的途径.本文系统地阐述了可见光促进的芳香烃甲基化反应的研究进展, 试图按照偶联反应、自由基加成反应两种不同途径来分类阐述, 并讨论了部分反应的反应机理.
芳基卤化物是一类广泛易得的在合成中通用的芳基偶联子, 在过渡金属催化交叉偶联反应中有着广泛的应用.因此, 基于芳基卤化物参与的甲基直接偶联反应具有重要应用价值, 受到了化学家的广泛关注.随着光催化/过渡金属催化协同模式的发展, 光促的甲基偶联反应也开始取得突破.尤为突出的是, 利用镍催化剂易与自由基相互作用, 可实现形式上的单电子氧化加成模式[7], 并且高价镍中间体还原消除快, 不易发生β-氢消除副反应, 光催化和镍催化的协同模式为Csp2—Csp3偶联提供了有效的解决方案[8].
2016年, MacMillan课题组[9]报道了可见光与金属镍协同催化芳基溴化合物的亲电交叉偶联反应, 并实现了一例甲基化反应(Scheme 2).该反应使用[Ir[dF(CF3)-ppy]2(dtbbpy)]PF6及NiCl2•dtbbpy作为催化剂, 以对甲基苯磺酸甲酯(MeOTs)作为甲基化试剂实现了对溴苯甲酸酯的甲基化.在该反应过程中, 对甲基苯磺酸甲酯(MeOTs)与溴化锂(LiBr)现场生成一溴甲烷(CH3Br), 在可见光氧化还原条件下生成的硅自由基参与下, 生成高活性的甲基自由基, 继而与芳基镍物种发生偶联反应得到相应的芳基甲基化产物.
作者所使用的磺酸酯试剂相比传统的偶联反应所使用的有机硼酸试剂、格氏试剂及有机锌试剂等易于制备, 且在水氧条件下稳定[10].在该光氧化还原循环中(Scheme 3), 芳基NiⅡ配合物6有可以解离的配体溴负离子(Br-), 其还原电势为E1/2red=+0.80 V (vs. SCE in DME).激发态的*IrⅢ催化剂(E1/2red=[*IrⅢ/IrⅡ]=+1.21 V, vs. SCE in CH3CN)可以将溴负离子氧化, 生成溴自由基4.溴自由基4与三(三甲基硅基)硅烷(TTMMS)反应生成更为稳定的硅基自由基9.接下来, 由于Si—Br键的键解离能(401 kJ/mol, Me3Si—Br)相对于C—Br键的键解离能(288 kJ/mol, 溴乙烷)更高, 硅自由基可以攫取一溴甲烷的溴原子, 得到亲核性的甲基自由基.同时, Ni0配合物5与芳基溴代物1氧化加成生成芳基NiⅡ配合物6.甲基自由基与该中间体发生自由基加成反应得到(甲基)(芳基)NiⅢ中间体7, 进一步还原消除得到NiⅠ配合物8和甲基化产物2.随后NiⅠ配合物与还原性的IrⅡ催化剂发生单电子转移反应得到IrⅢ催化剂和Ni0配合物5, 完成两个催化循环.该反应利用镍催化剂能够有效与甲基自由基发生单电子加成, 与铱催化剂形成双催化循环, 实现了可见光促进的双亲电试剂交叉偶联的甲基化反应, 为芳香烃甲基化方法的进一步研究提供了新思路.

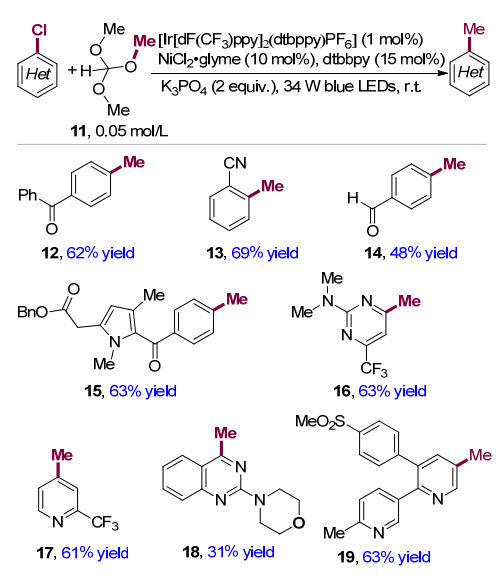
2020年, Doyle课题组[11]利用原甲酸三甲酯11作为甲基化试剂, 实现了可见光与金属镍协同催化的芳基氯化物的甲基化反应(Scheme 4), 该反应条件温和, 具有广泛的底物适用性和良好的官能团耐受性.该方法可以将含氯亲电试剂的位点选择性甲基化, 且所使用的原甲酸三甲酯是丰富、无毒且与官能团兼容性好的甲基化试剂.相对于采用亲核或者亲电的甲基化试剂, 该反应对含有较为敏感官能团的底物也有很好的兼容性, 比如羰基12, 氰基13, 醛基14等.另外作者对杂环芳基氯代物的反应适用性进行了考察, 吡啶类17、嘧啶类16、喹唑啉类18, 另外具有生物活性的杂芳基氯代物依托考昔(etoricoxib) 19的甲基化反应都有良好的收率, 进一步拓展了该方法的适用范围.
作者提出的反应机理如Scheme 5所示.首先芳基氯化物20与Ni0配合物21氧化加成生成芳基NiⅡ配合物22.该络合物被激发态的*IrⅢ催化剂氧化为芳基NiⅢ络合物23, 并快速释放氯自由基, 氯自由基进一步与原甲酸三甲酯11发生氢转移过程(HAT)得到甲基自由基.甲基自由基随后被NiⅡ配合物25捕获, 得到NiⅢ配合物28, 之后发生还原消除得到甲基化产物30及NiⅠ配合物29.得到的NiⅠ配合物29可以将二价铱配合物氧化成三价铱配合物, 同时NiⅠ配合物29被还原到Ni0配合物21, 完成催化循环.该方法为获取甲基自由基以及其他脂肪族自由基提供了新的方法, 有望在化学合成及药物合成中进一步广泛应用.

最近, Ohmiya课题组及其合作者[12]利用甲基硼络合物发展了光激发镍催化芳基卤化物的直接甲基化反应.该反应基于有机硼试剂的光物理性质[13], 可见光激发有机硼酸络合物, 被激发的有机硼酸复合物可以发生电子转移或者发生碳−硼键均裂得到甲基自由基等多种烷基自由基.作者利用甲基锂制备的有机硼酸络合物, 在光激发下可以得到甲基自由基, 并在镍催化作用下实现了高效的甲基−芳基交叉偶联反应.该反应无需额外使用光氧化还原催化剂, 从而避免了部分光氧化还原催化剂本身的氧化还原过程可能会带来副反应使化学转化变得复杂的问题.反应中使用的有机硼酸络合物在反应后得到的硼基底化合物(boracene)可以回收后再与甲基亲核试剂(MeLi)反应得到新的有机硼酸甲基络合物, 从而重复使用.

作者对有机硼酸络合物的制备方法以及反应机理进行了介绍(Scheme 7).使用有机硼化合物(boracene)与甲基锂反应, 制备得到有机硼酸的甲基络合物31, 该甲基络合物在空气及潮湿的环境中稳定.络合物31在可见光下激发形成激发态中间体37, 该物种具有强还原性, 可以将NiⅠ配合物还原为Ni0配合物, 并生成自由基阳离子38.中间体38极度不稳定, 快速释放甲基自由基, 并回收有机硼化合物36 (boracene).对于镍催化的循环, 可以通过两种反应路径得到目标产物43.一种路径是芳基溴化物先与Ni0配合物氧化加成得到Ar— NiⅡ—Br配合物41, 然后与甲基自由基反应; 另一种路径是甲基自由基先与Ni0配合物结合得到NiⅠ配合物40, 再与芳基卤化物氧化加成.两种路径都会得到共同的反应中间体42.最后该反应中间体42发生还原消除得到交叉偶联的芳基甲基化产物43以及NiⅠ物种.作者开发了一种无需光氧化还原催化剂, 直接通过光激发π共轭的有机硼酸络合物产生自由基来实现交叉偶联反应.在该反应中芳基硼36 (boracene)还可以回收利用, 极大地体现了该反应的应用价值.

亲核性碳中心自由基能够与缺电性芳杂环发生加成反应, 在氧化条件下进一步实现取代反应, 自Minisci等[14]发现这一反应途径以来, 已经发展了众多的自由基介导的芳环取代反应.随着光促氧化还原模式的发展, 自由基的产生途径变得更加多样更具实用性, 光促Minisci反应取得了较大发展, 利用甲基自由基的亲核性和高反应性, 这一光促自由基加成途径也为芳杂环中甲基官能团的引入提供了有效手段.
2014年, 默克公司的Matthew Tudge等[15]报道了可见光促进的杂芳烃甲基化反应.该方法使用过氧乙酸叔丁酯作为甲基化试剂, 以[Ir[dF(CF3)ppy]2(dtbbpy)]-PF6作为光催化剂, 通过光催化循环生成的甲基自由基与质子化的杂芳烃发生自由基加成反应, 进而得到甲基化的杂芳烃产物.该反应有着很好的底物适用性及官能团兼容性, 多种常见药物分子均能很好参与反应, 高效构建相应的甲基化衍生物.在一些特定底物(45, 48)中, 作者也发现了双甲基产物.

作者提出的反应机理如Scheme 9所示.金属IrⅢ的配合物在可见光照射下被激发为*IrⅢ, 然后与质子化的过氧乙酸叔丁酯49发生单电子转移, 被氧化为IrⅣ配合物, 同时生成叔丁氧基自由基中间体50.中间体50不稳定, 易进一步分解得到甲基自由基及一分子丙酮[16].质子化的杂芳烃51与甲基自由基发生加成反应得到自由基正离子中间体52, 该中间体52在光催化循环中发生单电子转移, 进而得到杂芳烃甲基化的目标产物53, 同时四价的IrⅣ配合物被还原为IrⅢ的配合物, 完成光催化循环.同时作者也注意到酸性条件对这一反应实现的重要性, 过氧乙酸叔丁酯的电位(Eo=-1.95V vs. SCE)[17]与[Ir[dF(CF3)ppy]2(dtbbpy)]PF6的电位(Eo= -0.89 V vs. SCE)不匹配, 这表明在热力学上激发态的*IrⅢ配合物无法将过氧乙酸叔丁酯还原, 但是酸性条件下, 质子化的过氧化乙酸叔丁酯可能会降低单电子转移过程的能垒, 促使该反应在动力学上的可行性[18].

2015年, MacMillan课题组[19]报道了使用廉价易得的甲醇作为甲基化试剂, 以[Ir(ppy)2(dtbbpy)]PF6作为光催化剂, 在可见光条件下实现了杂芳烃C—H键官能团化反应, 成功地在杂芳烃上引入甲基基团(Scheme 10).该方法反应条件温和, 具有收率高、底物范围较广等优点.吡啶类化合物含有各类官能团, 比如酰胺55, 苯基56, 三氟甲基57等都可以较高的收率得到目标产物.对于异喹啉类化合物59, 60, 酞嗪类化合物58, 菲啶类化合物61都能够很好的反应.同时, 作者将甲醇换成乙醇、异丙醇等, 可以成功实现了杂芳烃的乙基化、异丙基化反应等.作者还对依立卢(Fasudil), 一个蛋白激酶抑制剂和血管舒张剂药物进行了甲基化修饰反应, 并以82%的收率成功得到目标产物67, 显示出该方法在药物中应用的巨大潜力.
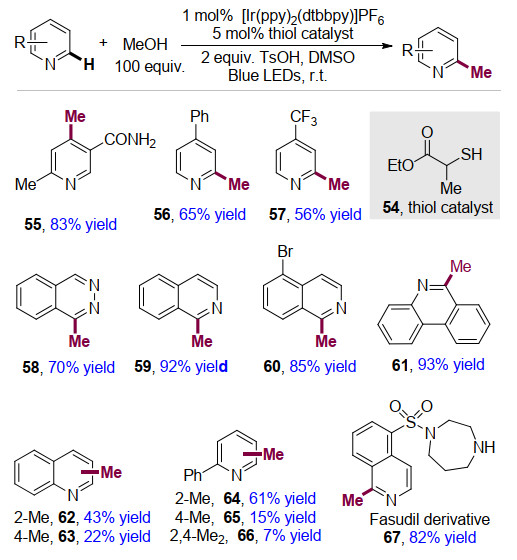
作者提出了可能的反应机理(Scheme 11)[20].有机催化剂(thiol catalyst) 72在参与光催化循环时发生单电子转移, 生成自由基中间体73, 该中间体73与甲醇发生氢原子转移, 生成羟甲基自由基75.该烷基自由基75进而与质子化杂芳环68发生自由基加成反应, 得到自由基正离子中间体69.正离子中间体69不稳定, 易脱质子得到自由基中间体70.然后该自由基中间体经历一个旋转中心迁移过程(SCS: Spin-center shift)脱去一分子水, 生成自由基中间体71.最后中间体71在光催化循环过程中发生单电子转移, 被还原为碳负离子, 并与体系中的质子结合, 得到杂芳基甲基化的目标产物76.

2016年, 陈弓课题组[21]报道了使用甲基硼酸[22]作为甲基化试剂, 使用[Ru(bpy)3]Cl2作催化剂, BI-OAc作为氧化剂, 在可见光促进条件下实现氮杂芳环C—H键的甲基化.
作者提出的反应机理如Scheme 12所示, 在光照条件下RuⅡ催化剂受激发, 得到激发态的*RuⅡ催化剂.激发态的*RuⅡ催化剂与体系中的氧化剂BI-OAc发生单电子转移, 进而发生碘氧键断裂生成邻碘苯甲酸自由基中间体80.随后, 该自由基与甲基硼酸反应经过反应中间体81得到甲基自由基.甲基自由基与质子化的氮杂芳烃化合物83发生加成反应, 再与RuⅢ催化剂发生单电子转移, 最终得到C—H键甲基化的产物85, 完成光氧化还原循环.

2017年, 徐华建课题组[23]报道了使用甲基硼酸钾盐作为甲基化试剂, 使用Ru作光催化剂, 在可见光条件下的吡啶氮氧化物的C2位C—H键甲基化的反应, 反应条件温和.作者提出的反应机理如Scheme 13所示, RuⅡ催化剂在可见光照射下受激发, 得到激发态的*RuⅡ催化剂.随后受激发的*RuⅡ催化剂与氧化剂BI—OAc发生单电子转移, 得到邻碘苯甲酸自由基80及RuⅢ催化剂.得到的RuⅢ催化剂与甲基三氟硼酸钾盐87发生单电子转移, 得到甲基自由基.甲基自由基与吡啶氮氧化物89发生加成反应得到自由基阳离子90.最后该自由基阳离子被前面所得到的邻碘苯甲酸自由基80氧化, 得到甲基化的目标产物91.

2017年, 李朝军课题组[24]报道了使用甲醇作为甲基化试剂和溶剂, 在室温光照条件下实现杂芳环甲基化反应.该反应使用二氯甲烷作为共溶剂, 三氟乙酸作为添加剂, 无需添加其他的光催化剂或者金属试剂.该方法体系对水和空气均不敏感, 可以对具有不同官能团的杂芳烃底物甲基化.在该方法中, 甲醇在反应中产生羟甲基自由基, 羟甲基自由基对质子化的杂芳烃底物亲核进攻, 通过脱水, 去质子化和互变异构芳构化得到甲基化的产物(Scheme 14).

2018年, Dhar课题组[25]报道了利用乙酸为甲基化试剂, 使用4-CzIPN作为光催化剂, 实现了4-甲基喹啉的甲基化反应.该反应采用N-羟基邻苯二甲酰亚胺活化乙酸, 一锅法生成高活性前体103作为真正的甲基化试剂.作者提出的反应机理如Scheme 15所示. N-羟基邻苯二甲酰亚胺预处理的乙酸(NAP)与光照下受激发的光催化剂发生单电子转移过程, 脱除一分子CO2和邻苯二甲酰亚胺后生成甲基自由基.甲基自由基进攻质子化的杂芳烃83, 得到自由基正离子84, 之后去质子化并与光催化剂发生单电子转移得到甲基化的杂芳烃产物85.

2018年, Glorius课题组[26]报道使用二甲基亚砜(DMSO)作甲基化试剂, 铱配合物作为光催化剂, 在可见光条件下实现了杂芳烃的甲基化反应.反应条件温和、底物适用性广泛, 一系列杂芳烃均能有效转化.反应中使用的DMSO是一种廉价的非质子溶剂, 也是有机合成中常用的试剂.该反应也可以通过氘代DMSO作为反应试剂在杂芳烃中引入氘代甲基(Scheme 16).

作者提出的反应机理如Scheme 17所示.二甲基亚砜(DMSO) 110与苯基膦酰二氯(PhPOCl2) 112反应形成硫嗡离子中间体114, 其被单电子还原后得到中间体115, 之后发生碳硫键均裂得到甲基自由基. IrⅢLn催化剂在光照下得到受激发的*IrⅢLn催化剂, 与质子化的杂芳烃发生单电子转移, 得到电子穿梭体117及IrⅣLn催化剂.反应中得到的甲基自由基与质子化的杂芳烃自由基加成得到118, 118和光催化剂IrⅣLn发生单电子转移得到甲基化的目标产物119及IrⅢLn催化剂以完成该催化循环.

2018年, 左智伟课题组[27]报道了使用甲烷作为甲基化试剂, 在三氯化铈和三氯乙醇的协同催化作用下, 通过光促氧化还原将光能转化为化学能, 促使室温条件下甲烷的活化, 得到甲基化的目标产物59.该反应采用烷氧自由基参与的氢转移途径选择性活化甲烷为高反应性的甲基自由基, 进而发生杂芳烃的自由基甲基化反应.该反应可能的机理如Scheme 18所示, 在该反应中三氯乙醇124与四价铈形成复合物, 通过光激发配体到金属电子转移过程得到烷氧自由基123进一步发生与甲烷反应, 攫取甲烷上的氢原子, 得到甲基自由基, 继而与质子化的异喹啉发生加成, 进一步单电子氧化得到甲基化的产物.

2019年, Barriault课题组[28]报道使用甲醇做甲基化试剂, 铱配合物为催化剂, 在可见光条件下实现了杂芳烃的甲基化反应.与之前MacMillan课题组[4]所报道的方法不同的是, 该反应并未使用硫醇做共催化剂, 而通过盐酸在光氧化条件下生成的氯自由基作为氢转移物种.作者提出了可能的反应机理(Scheme 19), 氯自由基与甲醇发生氢原子转移, 生成羟甲基自由基75并与被质子化的杂芳烃51发生自由基加成反应, 得到自由基正离子中间体69.自由基正离子中间体与光催化剂发生单电子转移得到中间体127, 然后该中间体脱去一分子水, 并发生质子迁移最终得到甲基化的杂芳烃产物76.

2019年, 金健课题组[29]报道使用乙酸作甲基化试剂, 硫酸亚铁(FeSO4)作催化剂, 在可见光条件下实现杂芳烃甲基化的反应.该反应是通过铁介导的氧化过程, 通过脱羧产生烷基自由基, 进而引入烷基基团.作者提出的反应机理如Scheme 20所示, 反应中生成三价铁盐与乙酸的配合物130, 并在可见光的照射下发生分子内电子转移, 生成羧基自由基131.随后该自由基中间体发生分解脱除二氧化碳得到甲基自由基, 所得到的甲基自由基与质子化的杂芳烃51发生加成反应得到自由基物种133.自由基物种133进一步氧化芳构化得到目标产品.

可见光促进的芳基甲基化反应充分利用了甲基自由基这一高活性中间体的亲核性, 能够实现芳基卤化物的偶联反应、杂环芳烃甲基化反应.在过去的十年中, 已有众多光促甲基化反应涌现.可见光促进的芳烃甲基化反应条件温和、官能团兼容性好、底物适用性广, 为芳烃上引入甲基基团提供了新颖、高效、温和的途径, 也拓展了甲基化试剂, 是对过渡金属催化芳基甲基化反应的很好的补充.值得注意的是, 目前亦有将光促氧化还原可以应用到烷烃的C(sp3)—H键甲基化反应的报道[30], 进一步凸显了光促甲基化反应的巨大潜力及丰富的应用前景.随着药物化学家及有机合成化学家对采用光促进的策略实现甲基化反应的高效、绿色合成方法的不断探索, 必将涌现出一系列更加高效和实用的光促甲基化方法, 挖掘出更多的绿色、适用性更广的甲基化试剂, 并应用到更多的药物分子及天然产物的后期修饰中, 为药物分子的发现与发展提供更多快速可靠的方法.
(a) Friesen, R. W.; Brideau, C.; Chan, C. C.; Charleson, S.; Deschanes, D.; Dub, D.; Ethier, D.; Fortin, R.; Gauthier, J. Y.; Girard, Y.; Gordon, R.; Greig, G. M.; Riendeau, D.; Savoie, C.; Wang, Z.; Wong, E.; Visco, D.; Xu, L.-J.; Young, R. N. Bioorg. Med. Chem. Lett. 1998, 8, 2777.
(b) Li, L.; Beaulieu, C.; Carriere, M. C.; Denis, D.; Greig, G.; Guay, D.; ONeill, G.; Zamboni, R.; Wang. Z. Bioorg. Med. Chem. Lett. 2010, 20, 7462.
(c) Ginnings, P. M.; Baum. R. J. Am. Chem. Soc. 1937, 59, 1111.
Schönherr, H.; Cernak, T. Angew. Chem., Int. Ed. 2013, 52, 12256. doi: 10.1002/anie.201303207
(a) McGrath, N. A.; Brichacek, M.; Njardarson, J. T. J. Chem. Educ. 2010, 87, 1348.
(b) Barreiro, E. J.; Kmmerle, A. E.; Fraga, C. A. M. Chem. Rev. 2011, 111, 5215.
(a) Potthast, A.; Rosenau, T.; Chen, C.-L.; Gratzl, J. S. J. Org. Chem. 1995, 60, 4320.
(b) Friedman, L.; Fishel, D. L.; Shechter, H. J. Org. Chem. 1965, 30, 1453.
(c) Friedman, L. Org. Synth. 1963, 43, 80.
(d) Das, S.; Bhowmick, T.; Punnyamurthy, T.; Dey, D.; Nath, J.; Chaudhuri, M. K. Tetrahedron Lett. 2003, 44, 4915.
(e) Shimada, K.; Nanae, T.; Aoyagi, S.; Takikawa, Y.; Kabuto, C. Tetrahedron Lett. 2001, 42, 6167.
(a) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
(b) Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2011, 111, 1596.
(c) Xiao, Q.; Zhang, Y.; Wang, J. Acc. Chem. Res. 2013, 46, 236.
(d) Yan, G.-B.; Borah, A. J.; Wang, L.-G.; Yang, M.-H. Adv. Synth. Catal. 2015, 357, 1333.
(e) Chen, Y.-T. Chem.-Eur. J. 2019, 25, 3405.
(f) Hu, L.; Liu, Y.-A.; Liao, X. Synlett 2018, 29, 375.
(g) Feng, K.; Quevedo, R. E.; Kohrt, J. T.; Oderinde, M. S.; Reilly, U.; White, M. C. Nature 2020, 580, 621.
(h) Feng, B.; Yang, Y.; You, J. Chem. Sci. 2020, 11, 6031.
(i) Chen, X.-Y.; Sorensen, E. J. J. Am. Chem. Soc. 2018, 140, 2789.
(j) He, Z.-T.; Li, H.; Haydl, A.; Whiteker, G.; Hartwig, J. F. J. Am. Chem. Soc. 2018, 140, 17197.
(k) Serpier, F.; Pan, F.; Ham, W. S.; Jacq, J.; Genicot, C.; Ritter, T. Angew. Chem., Int. Ed. 2018, 57, 10697.
(l) Lv, W.; Wen, S.; Liu, J.; Cheng, G. J. Org. Chem. 2019, 84, 9786.
(a) Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102.
(b) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322.
(c) Schultz, D. M.; Yoon, T. P. Science 2014, 343, 1239176.
(d) Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075.
(e) Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828.
(f) Chen, Y.; Lu, L.-Q.; Yu, D.-G.; Zhu, C.-J.; Xiao, W.-J. Sci. China: Chem. 2019, 62, 24.
(a) Tasker, S. Z.; Standley, E. A.; Jamison, T. F. Nature 2014, 509, 299.
(b) Netherton, M. R.; Fu, G. C. Adv. Synth. Catal. 2004, 346, 1525.
(c) Rudolph, A.; Lautens, M. Angew. Chem., Int. Ed. 2009, 48, 2656.
ana, R.; Pathak, T. P.; Sigman, M. S. Chem. Rev. 2011, 111, 1417. doi: 10.1021/cr100327p
Zhang, P.; Le, C. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2016, 138, 8084. doi: 10.1021/jacs.6b04818
Biswas, S.; Weix, D. J. J. Am. Chem. Soc. 2013, 135, 16192. doi: 10.1021/ja407589e
Kariofillis, S. K.; Shields, B. J.; Tekle-Smith, M. A.; Zacuto, M. J.; Doyle, A. G. J. Am. Chem. Soc. 2013, 135, 16192. doi: 10.1021/ja407589e
Sato, Y.; Nakamura, K.; Sumida, Y.; Hashizume, D.; Hosoya, T.; Ohmiya, H. J. Am. Chem. Soc. 2020, 142, 9938. doi: 10.1021/jacs.0c04456
Schuster, G. B. Pure Appl. Chem. 1990, 62, 156.
(a) Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinummo, M. Tetrahedron 1971, 27, 3575.
(b) Proctor, R. S.; Phipps, R. J. Angew. Chem., Int. Ed. 2019, 58, 13666.
DiRocco, D. A.; Dykstra, K.; Krska, S.; Vachal, P.; Conway, D. V.; Tudge, M. Angew. Chem., Int. Ed. 2014, 53, 4802. doi: 10.1002/anie.201402023
Komai, T.; Matsuyama, K.; Matsushima, M. Bull. Chem. Soc. Jpn. 1988, 61, 1641. doi: 10.1246/bcsj.61.1641
MorletSavary, F.; Wieder, F.; Fouassier, J. P. J. Chem. Soc. Faraday Trans. 1997, 93, 3931. doi: 10.1039/a704951j
Tarantino, K. T.; Liu, P.; Knowles, R. R. J. Am. Chem. Soc. 2013, 135, 10022. doi: 10.1021/ja404342j
Jin, J.; MacMillan, D. W. C. Nature 2015, 525, 87. doi: 10.1038/nature14885
Wessig, P.; Muehling, O. Eur. J. Org. Chem. 2007, 2219. doi: 10.1002/chin.200732280
Li, G.-X.; Christian A.; Rivera, M.; Wang, Y.-X.; Gao, F.; He, G.; Liu, P.; Chen, G. Chem. Sci. 2016, 7, 6407. doi: 10.1039/C6SC02653B
(a) Huang, H.; Jia, K.; Chen, Y. Angew. Chem., Int. Ed. 2015, 54, 1881.
(b) Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 2280.
(c) Tellis, J. C.; Primer, D. N.; Molander, G. A. Science 2014, 345, 433.
(d) Khatib, M. E.; Seraphim, R. A. M.; Molander, G. A. Angew. Chem., Int. Ed. 2016, 55, 254.
Zhang, W.-M.; Dai, J.-J.; Xu, J.; Xu, H.-J. J. Org. Chem. 2017, 82, 2059. doi: 10.1021/acs.joc.6b02891
Liu, W.-B.; Yang, X.-B.; Zhou, Z.-Z.; Li, C.-J. Chemistry 2017, 2, 688. doi: 10.1016/j.chempr.2017.03.009
Sherwood, T. C.; Li, N.; Yazdani, A. N.; Dhar, T. G. M. J. Org. Chem. 2018, 83, 3000. doi: 10.1021/acs.joc.8b00205
Garza-Sanchez, R. A.; Patra, T.; Tlahuext-Aca, A.; Strieth-Kalthoff, F.; Glorius, F. Chem.-Eur. J. 2018, 24, 10064. doi: 10.1002/chem.201802352
Hu, A.-H.; Guo, J.-J.; Pan, H.; Zuo, Z.-W. Science 2018, 361, 668. doi: 10.1126/science.aat9750
Zidan, M.; Morris, A. O.; McCallum, T.; Barriault, L. Eur. J. Org. Chem. 2020, 1453.
Li, Z.-L.; Wang, X.-F.; Xia, S.-Q.; Jin, J. Org. Lett. 2019, 21, 4259. doi: 10.1021/acs.orglett.9b01439
Le, C.; Liang, Y.-F.; Evans, R. W.; Li, X.-M.; MacMillan, D. W. C. Nature 2017, 547, 79. doi: 10.1038/nature22813

图式 3 可见光促进的硅自由基介导的交叉偶联反应机理
Scheme 3 Proposed mechanistic cycle for silyl-mediated cross-coupling via photoredox

图式 5 可见光促进的芳基氯化物甲基化反应机理
Scheme 5 Proposed mechanistic cycle for the visible light mediated methylation of aryl chlorides

图式 6 可见光直接激发甲基硼酸酯络合物生成甲基自由基
Scheme 6 Generation of methyl radical through direct excitation of boracene-based methylborate

图式 7 可见光直接激发甲基硼酸酯络合物生成甲基自由基的芳甲基化反应机理
Scheme 7 Mechanism of generation of methyl radical through direct excitation of boracene-based methylborate

图式 8 通过光氧化还原催化作用的生物活性芳杂环的后期功能化
Scheme 8 Late-stage functionalization of biologically active heterocycles through photoredox catalysis

图式 9 光促芳杂环的甲基化反应的可能机理
Scheme 9 Proposed catalytic cycle for the photocatalyzed methylation of heterocycles with tBPA

图式 10 可见光促进的甲醇作为甲基化试剂杂芳环键官能团化反应
Scheme 10 Visible light mediated methanol as methylation agents in heteroarene functionalization

图式 11 可见光促进的甲醇作为甲基化试剂杂芳环键官能团化反应机理
Scheme 11 Proposed mechanism for methanol as methylation agents in heteroarene functionalization

图式 12 甲基硼酸参与的氮-杂芳环碳氢键甲基化反应
Scheme 12 Photoredox-mediated Minisci C—H methylation of N-heteroarenes using methylboronic acid

图式 13 吡啶氮氧化物的C2位键甲基化反应机理
Scheme 13 Mechanism of visible-light-induced C2 methylation of pyridine N-oxides

图式 14 可见光促进的甲醇做甲基化试剂的杂芳烃的甲基化反应
Scheme 14 Photo-induced methylation of heteroarenes with MeOH

图式 15 可见光促进的芳杂环甲基化反应
Scheme 15 Visible-light photoredox-mediated, methylation of heteroarenes

图式 16 可见光促进的DMSO作甲基化试剂杂芳烃的甲基化反应
Scheme 16 Visible-light photoredox-mediated, methylation of heteroarene using DMSO as the methylation agent

图式 17 可见光促进的DMSO作为甲基化试剂杂芳烃的甲基化反应机理
Scheme 17 Mechanism of methylation of heteroarene with DMSO

图式 18 可见光促进的甲烷参与的杂芳烃甲基化反应
Scheme 18 Visible-light photoredox-mediated, methylation of heteroarene using methane as the methylation agent

图式 19 可见光促进的甲醇作为甲基化试剂杂芳烃的甲基化反应
Scheme 19 Visible-light photoredox-mediated, methylation of heteroarene using methanol as the methylation agent

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:



 下载:
下载: