Hubei Key Laboratory of Natural Products Research and Development, China Three Gorges University, Yichang, Hubei 443002
b.
Key Laboratory of Inorganic Nonmetallic Crystalline and Energy Conversion Materials, College of Materials and Chemical Engineering, China Three Gorges University, Yichang, hubei 443002
c.
Key Laboratory of Pesticide&Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079
Received Date:
30 June 2020 Revised Date:
04 September 2020 Available Online:
25 November 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21702121), the Open Fund from Hubei Key Laboratory of Natural Products Research and Development (China Three Gorges University) (No. NPRD-2018010), the Research Fund for Excellent Dissertation of China Three Gorges University and the Programme of Introducing Talents of Discipline to Universities (111 Project, No. D20015)
Abstract:
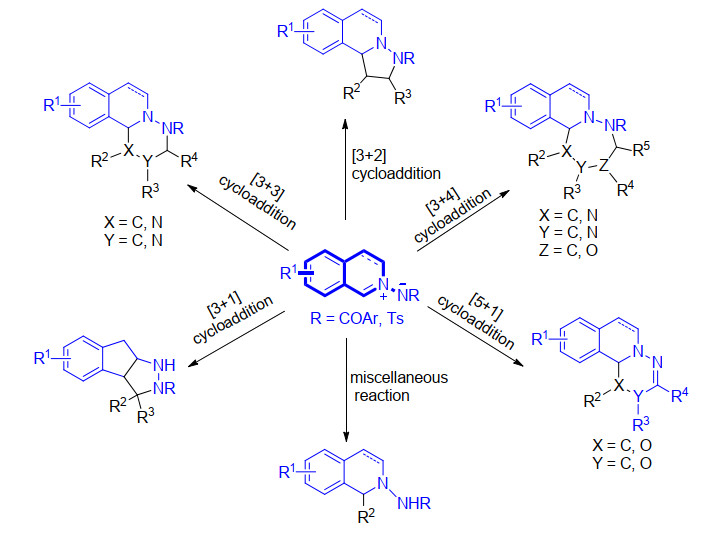
Among various 1, 3-dipoles of cyclic azomethine imines, C, N-cyclic azomethine imines are the most widely used reagents in the construction of diverse tetrahydroisoquinoline derivatives. The developments of reactions with C, N-cyclic azomethine imines including [3+2], [3+3], [3+4], [5+1], [3+1] cycloaddition reactions and miscellaneous reactions are summarized. The properties of reactions, reaction processes and synthetic applications are discussed. Finally, the prospects of the reaction with this reagent are also proposed.
Dedicated to the 40th anniversary of Chinese Journal of Organic Chemistry
[1]
(a) Liu, C. J.; Liu, D. Y.; Xiang, L. Acta Pharm. Sin.2010, 45, 9. (b) Blay, J.-Y. Eur. J. Clin. Med. Oncol.2010, 2, 1. (c) Vincenzi, B.; Napolitano, A.; Frezza, A. M.; Schiavon, G.; Santini, D.; Tonini, G. Pharmacogenomics2010, 11, 865. (d) Carter, N. J.; Keam, S. J. Drugs2010, 70, 355.
[2]
(a) Aune, G. J.; Furuta, T.; Pommier, Y. Anti-Cancer Drugs2002, 13, 545. (b) Scott, J. D.; Williams, R. M. Chem. Rev.2002, 102, 1669. (c) Mujahidin, D.; Doye, S. Eur. J. Org. Chem.2005, 2005, 2689. (d) Bentley, K. W. Nat. Prod. Rep.2006, 23, 444. (e) Werner, F.; Blank, N.; Opatz, T. Eur. J. Org. Chem.2007, 2007, 3911. (f) Reddy, R. J.; Kawai, N.; Uenishi, J. J. Org. Chem.2012, 77, 11101.
[3]
(a) Sekine Y.; Brossi, A. J. Nat. Prod.1990, 53, 533. (b) Luk, L. Y. P.; Bunn, S.; Liscombe, D. K.; Facchini, P. J.; Tanner, M. E. Biochemistry2007, 46, 10153. (c) Minami, H.; Dubouzet, E.; Iwasa, K.; Sato, F. J. Biol. Chem.2007, 282, 6274.
[4]
(a) Chang, F.-R.; Wu, Y.-C. J. Nat. Prod.2005, 68, 1056. (b) Kashiwada, Y.; Aoshima, A.; Ikeshiro, Y.; Chen, Y.-P.; Furukawa, H.; Itoigawa, M.; Fujioka, T.; Mihashi, K.; Cosentino, L. M.; Morris-Natschke, S. L.; Lee, K.-H. Bioorg. Med. Chem.2005, 13, 443.
[5]
(a) Padwa, A.; Danca, M. D. Org. Lett.2002, 4, 715. (b) Simpkins, N. S.; Gill, C. D. Org. Lett.2003, 5, 535 (c) Pérard-Viret, J.; Souquet, F.; Manisse, M.-L.; Royer, J. Tetrahedron Lett. 2010, 51, 96.
Afanaseva, K. K.; Efremova, M. M.; Kuznetsova, S. V.; Ivanov, A. V.; Starova, G. L.; Molchanov, A. P. Tetrahedron2018, 74, 5665. doi: 10.1016/j.tet.2018.07.040
[16]
Ling, L.; Chen, J. Q.; Song, J. H.; Zhang, Y. H.; Li, X. Q.; Song, L. J.; Shi, F.; Li, Y. X.; Wu, C. R. Org. Biomol. Chem.2013, 11, 3894. doi: 10.1039/c3ob40448j
[17]
Alcaide, B.; Almendros, P.; Quirs, M. T. Chem.-Eur. J.2014, 20, 3384. doi: 10.1002/chem.201304509
[18]
Li, Z.; Yu, H.; Liu, H. L.; Zhang, L.; Jiang, H.; Wang, B.; Guo, H. C. Chem.-Eur. J.2014, 20, 1731. doi: 10.1002/chem.201303625
[19]
Jia, Q. F.; Chen, L.; Yang, G. M.; Wang, J.; Wei, J.; Du, Z. Y. Tetrahedron Lett.2015, 56, 7150. doi: 10.1016/j.tetlet.2015.11.030
[20]
Jing, C. F.; Na, R. S.; Wang, B.; Liu, H. L.; Zhang, L.; Liu, J.; Wang, M.; Zhong, J. C.; Kwon, O.; Guo, H. C. Adv. Synth. Catal.2012, 354, 1023. doi: 10.1002/adsc.201100831
Li, W. J.; Jia, Q. F.; Du, Z. Y.; Zhang, K.; Wang, J. Chem.-Eur. J.2014, 20, 4559. doi: 10.1002/chem.201400333
[23]
Li, W. J.; Wei, J.; Jia, Q. F.; Du, Z. Y.; Zhang, K.; Wang, J. Chem.-Eur. J.2014, 20, 6592. doi: 10.1002/chem.201402089
[24]
Izquierdo, C.; Esteban, F.; Parra, A.; Alfaro, R.; Alemán, J.; Fraile, A.; García Ruano, J. L. J. Org. Chem.2014, 79, 10417. doi: 10.1021/jo5018519
[25]
Hesping, L.; Biswas, A.; Daniliuc, C. G.; Mück-Lichtenfeld, C.; Studer, A. Chem. Sci.2015, 6, 1252. doi: 10.1039/C4SC02612H
[26]
(a) Li, B.-S.; Wang, Y. H.; Jin, Z. C.; Chi, Y. R. Chem. Sci.2015, 6, 6008. (b) Kirmse, W.; Rondan, N. G.; Houk, K. N. J. Am. Chem. Soc. 1984, 106, 7989.
[27]
Gao, Z. H.; Chen, X. Y.; Cheng, J. T.; Liao, W. L.; Ye, S. Chem. Commun.2015, 51, 9328. doi: 10.1039/C5CC01238D
[28]
Guo, C.; Fleige, M.; Janssen-Müller, D.; Daniliuc, C. G.; Glorius, F. Nat. Chem.2015, 7, 842. doi: 10.1038/nchem.2337
[29]
Zhang, P. F.; Zhou, Y.; Han, X.; Xu, J. Y.; Liu, H. J. Org. Chem.2018, 83, 3879. doi: 10.1021/acs.joc.8b00227
Liu, X. H.; Wang, Y. J.; Yang, D. X.; Zhang, J. L.; Liu, D. S.; Su, W. Angew. Chem., Int. Ed.2016, 55, 8100. doi: 10.1002/anie.201602880
[32]
Hashimoto, T.; Omote, M.; Maruoka, K. Angew. Chem., Int. Ed.2011, 50, 3489. doi: 10.1002/anie.201100331
[33]
Wang, Y.; Wang, Q.; Zhu, J. P. Chem.-Eur. J. 2016, 22, 8084. doi: 10.1002/chem.201601548
[34]
Mousseau, J. J.; Fortier, A.; Charette, A. B. Org. Lett.2010, 12, 516. doi: 10.1021/ol902710f
[35]
Mousseau, J. J.; Bull, J. A.; Ladd, C. L.; Fortier, A.; Roman, D. S.; Charette, A. B. J. Org. Chem.2011, 76, 8243. doi: 10.1021/jo201303x
[36]
Zhou, W.; Li, X.-X.; Li, G.-H.; Wu, Y.; Chen, Z. L. Chem. Commun.2013, 49, 3552. doi: 10.1039/c3cc41258j
[37]
Mao, B. M.; Xu, Y.; Chen, Y. H.; Dong, J. P.; Zhang, J. Y.; Gu, K. J.; Zheng, B.; Guo, H. C. Org. Lett.2019, 21, 4424. doi: 10.1021/acs.orglett.9b01064
[38]
Mao, B. M.; Zhang, J. Y.; Xu, Y.; Yan, Z. Y.; Wang, W.; Wu, Y. J.; Sun, C. Q.; Zheng, B.; Guo, H. C. Chem. Commun.2019, 55, 12841. doi: 10.1039/C9CC06670E
[39]
Koptelov, Y. B.; Saik, S. P.; Molchanov. A. P. Russ. J. Org. Chem.2011, 47, 537. doi: 10.1134/S1070428011040129
[40]
Koptelov, Y. B.; Molchanov, A. P.; Kostikov, R. R. Russ. J. Org. Chem.2015, 51, 11343. doi: 10.1134/S1070428015080126
[41]
Kawai, H.; Yuan, Z.; Tokunaga, E.; Shibata, N. Org. Lett. 2012, 14, 5330. doi: 10.1021/ol3025154
Liu, X. H.; Yang, D. X.; Wang, K. Z.; Zhang, J. L.; Wang, R. Green Chem.2017, 19, 82. doi: 10.1039/C6GC02517J
[44]
Hu, F. Z.; Chen, H.; Zhang, M. M.; Yu, S. W.; Xu, X. Y.; Yuan, W. C.; Zhang, X. M. J. Heterocycl. Chem.2017, 54, 2922. doi: 10.1002/jhet.2903
[45]
(a) Hong, L.; Kai, M.; Wu, C.; Sun, W.; Zhu, G.; Li, G.; Yao, X.; Wang, R. Chem. Commun.2013, 49, 6713. (b) Yin, C.; Lin, L.; Zhang, D.; Feng, J.; Liu, X.; Feng, X. J. Org. Chem.2015, 80, 9691. (c) Lu, Y. L.; Sun, J.; Jiang, Y. H.; Yan, C. G. RSC Adv.2016, 6, 50471.
[46]
Wu, Y. F.; Tian, B.; Hu, C.; Sekine, K.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Org. Biomol. Chem.2019, 17, 5505. doi: 10.1039/C9OB00740G
[47]
Na, R. S.; Liu, H. L.; Li, Z.; Wang, B.; Liu, J.; Wang, M.-A.; Wang, M.; Zhong, J. C.; Guo, H. C. Tetrahedron2012, 68, 2349. doi: 10.1016/j.tet.2012.01.029
[48]
Zhang, L.; Jing, C. F.; Liu, H. L.; Wang, B.; Li, Z.; Jiang, H.; Yu, H.; Guo, H. C. Synthesis2013, 45, 0053.
[49]
Li, F. L.; Chen, J. F.; Hou, Y. D.; Li, Y. J.; Wu, X.-Y.; Tong, X. F. Org. Lett.2015, 17, 5376. doi: 10.1021/acs.orglett.5b02724
[50]
He, L. W. Z.; Liu, L.; Han, R. F.; Zhang, W. W.; Xie, X. G.; She, X. G. Org. Biomol. Chem.2016, 14, 6757. doi: 10.1039/C6OB01095D
[51]
Zhao, J. J.; Li, P.; Wu, C. R.; Chen, H. L.; Ai, W. Y.; Sun, R. H.; Ren, H. L.; Larockb, R. C.; Shi, F. Org. Biomol. Chem.2012, 10, 1922. doi: 10.1039/c2ob06611d
Xu, X. F.; Zavalij, P. Y.; Doyle, M. P. Angew. Chem., Int. Ed.2013, 52, 12664. doi: 10.1002/anie.201305539
[60]
Marichev, K. O.; Adly, F. G.; Carranco, A. M.; Garcia, E. C.; Arman, H.; Doyle, M. P. ACS Catal.2018, 8, 10392. doi: 10.1021/acscatal.8b03391
[61]
Wang, K. K.; Li, Y. L.; Wang, Z. Y.; Hu, M. W.; Qiua, T. T.; Zhu, B. K. Org. Biomol. Chem.2019, 17, 244. doi: 10.1039/C8OB02932F
[62]
Li, Z.; Yu, H.; Feng, Y. L.; Hou, Z. F.; Zhang, L.; Yang, W. J.; Wu, Y.; Xiao, Y. M.; Guo, H. C. RSC Adv.2015, 5, 34481. doi: 10.1039/C5RA04374C
[63]
Wang, Y. F.; Zhu, L. P.; Wang, M. R.; Xiong, J. L.; Chen, N. N.; Feng, X.; Xu, Z. Q.; Jiang, X. X. Org. Lett. 2018, 20, 6506. doi: 10.1021/acs.orglett.8b02828
Xu, J. F.; Yuan, S. R.; Peng, J. Y.; Miao, M. Z.; Chen, Z. K.; Ren, H. J. Org. Biomol. Chem.2017, 15, 7513. doi: 10.1039/C7OB01783A
[70]
Zheng, P. F.; Zeng, R.; Jiang, K.; Li, H. W.; Ye, Y.; Mu, C.; Shuai, L.; Ouyang, Q.; Chen, Y. C. Org. Lett. 2019, 21, 10052. doi: 10.1021/acs.orglett.9b03996
Lariveé, A.; Mousseau, J. J.; Charette, A. B. J. Am. Chem. Soc.2008, 130, 52. doi: 10.1021/ja710073n
[74]
Zhao, Z.-Q.; Zhao, X.-L.; Shi, M.; Zhao, M.-X. J. Org. Chem.2019, 84, 14487. doi: 10.1021/acs.joc.9b01955
[75]
Fang, L.; Chen, L. S.; Yu, J. J.; Wang, L. M. Eur. J. Org. Chem.2015, 2015, 1910. doi: 10.1002/ejoc.201403479
[76]
Hashimoto, T.; Omote, M.; Maruoka, K. Angew. Chem., Int. Ed.2011, 50, 8952. doi: 10.1002/anie.201104017
[77]
Hua, Z. R.; Fang, L.; Wu, S. Y.; Wang, L. M. Eur. J. Org. Chem.2016, 2016, 4953. doi: 10.1002/ejoc.201600905
[78]
Li, D.; Yang, D. X.; Wang, L. Q.; Liu, X. H.; Wang, K. Z.; Wang, J.; Wang, P. X.; Liu, Y. Y.; Zhu, H. Y.; Wang, R. Chem.-Eur. J.2017, 23, 6974. doi: 10.1002/chem.201700970
[79]
Zhang, D.; Liu, J. W.; Kang, Z. H.; Qiu, H.; Hu, W. H. Org. Biomol. Chem.2019, 17, 9844. doi: 10.1039/C9OB02303H


 下载:
下载:





































































































































 下载:
下载:






























































 下载:
下载:
