
Scheme 1.
Applications and syntheses of gem-difluoroalkenes

gem-Difluoroalkene was an important fluoro synthon in a variety of transformations and applications (Scheme 1, a). For example, α-aminometheneyl gem-difluorostyrene was the inhibitor of monoamine oxidase (MAO)[1] and pesticide building block.[2] gem-Difluoroalkenes are reactive substrate in a variety of transformation to build complicated molecules.[3] For example, Hu et al.[4] achieved the AgF-mediated fluorinative cross-coupling of gem-difluoroalkenes. Feng and Loh et al.[5] utilized the gem-difluoroalkenes as starting material in a series of transitionalmetal-catalyzed and photoredox catalysis transformations. The gem-difluorostyrenes were also applied in the thiolation by Dilman group[6] and fluorohalogenation by Jiang group.[7] With these divergent applications, the syntheses of gem-difluoroalkenes were extensively studied using a variety of protocols.[8] For example, by using bromodifluoroacetate as starting material, the gem-difluoroalkene ester was prepared in 4 steps by Matsuda and Kitazume (Scheme 1b).[9] With the same starting material, the preparation was realized via Julia-Kochienski olefination in 3 steps by the Liu group (Scheme 1b).[10] König group[11] reported an photoredox cross-coupling reaction between ketone-derived hydrazone and sodium trifluoromethane-sulfonate catalyzed by Ir complex (Scheme 1b). Xiao et al.[12] achieved the ZnO-promoted Wittig reaction. There has been one substrate reported by Zhou et al.[13] as an unexpected reaction in the Ni-catalyzed transfer hydrogenation reaction in which the gem-difluoror styrene 2a was obtained (Scheme 1b). Despite no further investigation, this example suggested a potential allylic-hydrodefluorination protocol to prepare the gem-difluoroalkenes.
Electrochemical synthesis recently emerged as a powerful strategy to prepare molecules with innovative pathways.[14] The electrochemical dechlorination and debromination reactions have been well-established.[15] However, the direct cleavage of C—F remains a challenging task due to strong bond energy. For example, activation of the Ar—F requires the synergistic catalysis involving photocatalyst and electrolysis in an oxidation process.[16] So far, the cathodic reduction of C—F bonds focused on the conversion of trifluoromethyl ketones to silyl gem-difluoroenol ether.[17] We reasoned that the cathodic reductive hydrodefluorination is hard to achieve due to the choice of hydrogen source. As the C—F activation on cathode requires the high reduction potential, the cathodic hydrogen evolution might be predominant at such conditions. Recently, we[18] reported the electrochemical hydrogenation reaction using gaseous ammonia as the hydrogen source. With the combination of graphite felt cathode and ammonia, the cathodic hydrogen evolution could be inhibited at -2.0 V (vs. SCE). Herein, we reported the electrochemical allylic hydrodefluorination reaction with gaseous ammonia as hydrogen source.
At onset of the study, compound 1a was used as the model substrate to screen various parameters of the allylic hydrodefluorination reaction (Table 1). With graphite felt as both anode and cathode, using gaseous NH3 at balloon pressure, the desired transformation took place readily at room temperature in MeCN with nBu4NBF4 as supporting electrolyte, giving product 2a in 78% NMR yield and 66% isolated yield (Entry 1). As expected, in the absence either electricity (Entry 2) or gaseous ammonia (Entry 3), no conversion was observed. Other supporting electrolytes instead of nBu4NBF4 were evaluated, and the NMR yield were inferior to that obtained in standard conditions (Entries 4~7). When the reaction was carried out in other solvents than MeCN, for example dimethyl sulfoxide (DMSO) and MeOH, the yields dropped dramatically. On the other hand, N, N-dimethylformamide (DMF) gave almost the identical yield to MeCN (Entry 10). Next, the constant current method was applied instead of controlled cell potential, and found constant current at a density of 15 mA/cm3 could offer 66% yield (Entry 11). It was noted as the graphite felt was porous material, the planar surface did not make sense. The volume of graphite felt was used in the expression of current density. Varied cell potentials were examined as well, the yield increased from 49% to 78% when the cell potential moved up from 2.8 to 3.2 V. Further elevation of cell potential resulted in the decrease of yields (Entry 12). This observation was in accord with the results from constant current experiments in Entry 11. Finally, the cathode material was comparaed. By changing the graphite felt to graphite rod or platinum, the reaction was inhibited (Entries 13, 14).
 下载:
导出CSV
下载:
导出CSV
 |
||
| Entry | Variation from standard condition | Yieldb/% |
| 1 | None | 78 (66c) |
| 2 | No voltage | 0 |
| 3 | No NH3 | 0 |
| 4 | LiClO4 instead of nBu4NBF4 | 62 |
| 5 | LiBF4 instead of nBu4NBF4 | 54 |
| 6 | nBu4NPF6 instead of nBu4NBF4 | 67 |
| 7 | nBu4NClO4 instead of nBu4NBF4 | 71 |
| 8 | DMSO as solvent | 48 |
| 9 | MeOH as solvent | 32 |
| 10 | DMF as solvent | 76 |
| 11 | Constant current is 55, 111, 167, 222 mA/gd | 23, 47, 66, 42 |
| 12 | Constant voltage is 2.8, 3.0, 3.4, 3.6 V | 49, 59, 68, 28 |
| 13 | Graphite rod instead of graphite felt | 14 |
| 14 | Platinum sheet as cathode graphite felt as anode trace | Trace |
| a Standard condition: 1a (0.2 mmol), nBu4NBF4 (0.3 equiv.), CH3CN (5 mL), graphite felt anode and cathode, undivided cell, 3.2 V cell voltage, r.t., NH3 atmosphere, 5 h. b Yields determined by 1H NMR with dichloromethane as the internal standard. c Isolated yield. d As graphite felt is the complex of fibre, the mA/g is more suitable to describe the constant current condition. | ||
With the optimal conditions in hand (Table 1, Entry 1), the substrate scope of electrochemical hydrodefluorination reaction was explored (Table 2). The methyl substitution in substrates 1b and 1c unexpectedly sabotaged the reaction yield substantially, due to overdefluorination reaction. It is gratifying to isolate the product 2d with p-methoxy substitution in 89% yield. Electron-withdrawing group, for example, CF3 group, impact the yield with a 59% yield. The product 2f with para-tBu group was obtained in moderate yield. It was glad to observe that the vinyl group in product 2g was intact and the corresponding yield was 70%. The good yield of 2h was also realized in 5 h. Next, a series of substrates 1i~1n bearing halogen atom were subject to the standard hydrodefluorination reaction. The yields were around 60%, except the products 2k and 2l containing Cl atom. The low yields of both products were majorly caused by the extensive purification. Especially, the para-bromo atom in product 2m survived the electrochemical process, demonstrating the functional group compatibility of this protocol. Products 2o~2q bearing fused rings were achieved in good yields. Gladly, product 2r containing thiophenyl group could be prepared in 84% isolated yield. Subsequently, the substrates other than ethyl ester were prepared and evaluated. Several esters 1s~1u, bearing n-butyl, t-butyl or allyl group, underwent the transformation readily, and the desired products 2s~2u were generated in yields from 52% to 87%. When trifluoroethyl ester 1v was employed as starting material, the desired product 2v was obtained in only 42% yield partially due to the in situ transamidation reaction. Finally, by using an amide as starting material, product 2w was obtained in good yield (81%).
Next, the electrochemistry analyses were conducted to gain further information of this transformation. The cyclic voltammetry experiments were performed to study the cathodic reduction process (Figure 1, a). At first, the ammonia solution showed totally inert during the whole scan. By deducting the signals of blank sample, the first reduction peak of 1a was observed at -2.0 V (vs. SCE). When the cyclic voltammetry (CV) analysis was carried out under gaseous ammonia, a second weak peak was detected at -2.2 V (vs. SCE). In order to separate these two peaks, square wave voltammetry (SWV) experiment was carried out (Figure 1, b). With the significant separation at a frequency of 10 Hz, a two-step electron transfer was supported.

(a) CV condition: glassy carbon working electrode, Pt wire counter electrode, SCE reference electrode, 50 mV/S. (b) 1a (0.04 mmol), nBu4BF4 (0.012 mol/L), MeCN (5 mL), pulse height 25 mV, step height 4 mV, frequency 10 Hz
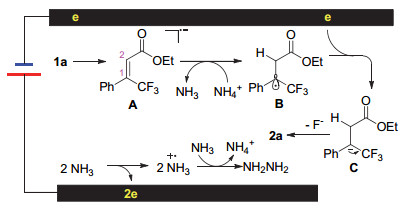
With the data achieved, a plausible reaction pathway was proposed in Scheme 2. At first the cathodic electron transfer converts substrate, for example 1a, to its' corresponding radical anion A, in which C(2) was supposed to hold more negative charge because of the carbonyl group. The protonation takes place at this position at first with ammonium cation generated via anodic oxidation of ammonia. A neutral radical B was produced. Consequently, a second anodic electron transfer gives rise to the benzylic anion C that undergoes an elimination to desired product 2a.
In summary, an electrochemical hydrodefluorination reaction with gem-difiluoroalkene esters as products was developed. The catalyst-free reaction employs inexpensive graphite felt as both anode and cathode, ammonia as a safe and environment-friendly hydrogen source. With this protocol, a series of α-trifluoromethyl cinnmates were converted to the gem-difluoro styrenes in moderate to good yields. The CV and SWV analyses supported a two-step electron transfer pathway. Especially, the reaction showed that the combination of ammonia and graphite felt cathode was a highly efficient approach to perform cathodic reduction with substrates requiring high reduction potential.
Column chromatography was generally performed on silica gel (300~400 mesh) and reactions were monitored by thin-layer chromatography (TLC) using 254 nm UV light to visualize the course of the reactions. NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer. High resolution mass spectra were recorded on Agilent G6500 Q-TOF instrument. Electrochemical analyses were carried out on CHI 730E electrochemistry workstation. 1H NMR (400 MHz), 13C NMR (101 MHz) and 19F (376 MHz) were recorded on Bruker Avance III 400 (400 MHz and 100 MHz). All 1H NMR and 13C NMR spectra are reported in parts per million (ppm) downfield of TMS. High-resolution mass spectra (HRMS) were equipped with an ESI source and a TOF detector mass spectrometer. The melting points were measured with digital melting point detector.
All reactions that required anhydrous conditions were carried by standard procedures under nitrogen atmosphere. Unless otherwise noted, materials (AR grade) were purchased from commercial suppliers, such as J & K, Innochem, and Energy chemicals, and used without further purification. The solvents were dried by distillation over the appropriate drying reagents.
A 10 mL two-necked heart-shaped flask was charged with the substrate 1a (0.2 mmol, 1.0 equiv.), nBu4NBF4 (0.06 mmol, 0.3 equiv.) and a magnetic stir bar. The flask was equipped with a rubber stopper. And graphite felt (2 cm×1 cm×0.5 cm) was used as anode and cathode. Two electrodes were separated with a Teflon film. The graphite felt anode was attached to a platinum wire and cathode was attached to a silver wire. A Teflon wire was tied around two electrodes. The flask was evacuated and backfilled with NH3, and then CH3CN (5 mL) was added by syringe. The mixture was stirred under room temperature and controlled cell potential electrolysis. After the reaction was completed by monitoring with TLC or GC-MS analysis, the mixture was diluted with water and extracted with EtOAc twice. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by chromatography on silica gel to afford the desire product 2a. Copounds 2b~2w were synthesized using the same method.
Ethyl 4, 4-difluoro-3-phenylbut-3-enoate (2a): 29 mg, yield 66%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.35 (d, J=6.1 Hz, 4H), 7.31~7.27 (m, 1H), 4.12 (q, J=7.1 Hz, 2H), 3.40 (t, J=2.2 Hz, 2H), 1.20 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 170.20~170.10 (m), 154.79 (dd, J=292.3, 289.0 Hz), 133.03 (t, J=3.8 Hz), 128.49, 127.84 (t, J=3.5 Hz), 87.16 (dd, J=21.6, 17.9 Hz), 127.53, 61.10, 33.89 (d, J=2.6 Hz), 14.05; 19F NMR (376 MHz, Chloroform-d) δ: -86.38 (d, J=34.8 Hz), -89.22 (d, J=34.8 Hz); HRMS (ESI) calcd for C12H13F2O2 [M+H]+ 227.0803, found 227.0873.
Ethyl 4, 4-difluoro-3-(p-tolyl)but-3-enoate (2b): 24 mg, yield 49%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.23 (d, J=7.9 Hz, 2H), 7.16 (d, J=8.0 Hz, 2H), 4.12 (q, J=7.2 Hz, 2H), 3.38 (s, 2H), 2.34 (s, 3H), 1.20 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 170.19 (dd, J=4.2, 2.9 Hz), 154.72 (dd, J=291.8, 288.7 Hz), 137.32, 130.03 (t, J=3.8 Hz), 129.19, 127.70 (t, J=3.5 Hz), 87.01 (dd, J=21.3, 18.0 Hz), 61.03, 33.92 (d, J=2.6 Hz), 21.09, 14.06; 19F NMR (376 MHz, Methanol-d4) δ: -91.09 (d, J=40.1 Hz), -92.25 (d, J=39.6 Hz); HRMS (ESI) calcd for C13H15F2O2 [M+H]+ 241.1040, found 241.1033.
Ethyl 3-(3, 5-dimethylphenyl)-4, 4-difluorobut-3-enoate (2c): 23 mg, yield: 46%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 6.95 (s, 2H), 6.92 (s, 1H), 4.13 (q, J=7.1 Hz, 2H), 3.36 (s, 2H), 2.31 (s, 6H), 1.21 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 170.22 (dd, J=4.2, 2.9 Hz), 154.77 (dd, J=291.9, 288.7 Hz), 137.96, 132.91 (t, J=3.8 Hz), 130.14, 129.27, 126.67, 125.68 (t, J=3.4 Hz), 87.19 (dd, J=21.0, 18.1 Hz), 61.01, 34.02 (d, J=2.7 Hz), 21.30, 14.08; 19F NMR (376 MHz, Methanol-d4) δ: -90.46 (d, J=39.1 Hz), -91.46 (d, J=39.1 Hz); HRMS (ESI) calcd for C14H17F2O2 [M+H]+ 255.1196, found 255.1187.
Ethyl 4, 4-difluoro-3-(4-methoxyphenyl)but-3-enoate (2d): 46 mg, yield: 89%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.26 (d, J=8.6 Hz, 2H), 6.88 (d, J=8.4 Hz, 2H), 4.12 (q, J=7.1 Hz, 2H), 3.80 (s, 3H), 3.36 (s, 2H), 1.20 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 170.22 (dd, J=4.2, 2.9 Hz), 158.89, 154.63 (dd, J=291.0, 288.5 Hz), 129.05 (t, J=3.5 Hz), 125.21 (t, J=3.8 Hz), 113.96, 86.69 (dd, J=21.6, 18.1 Hz), 61.03, 55.22, 34.03 (d, J=2.6 Hz), 14.07; 19F NMR (376 MHz, Methanol-d4) δ: -91.74 (d, J=41.5 Hz), -92.87 (d, J=41.9 Hz); HRMS (ESI) calcd for C13H15F2O3 [M+H]+ 257.0909, found 257.0981.
Ethyl 4, 4-difluoro-3-(4-(trifluoromethyl)phenyl)but- 3-enoate (2e): 35 mg, yield 59%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.61 (d, J=8.1 Hz, 2H), 7.47 (d, J=8.1 Hz, 2H), 4.13 (q, J=7.1 Hz, 2H), 3.42 (s, 2H), 1.20 (t, J=6.9 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 169.75 (dd, J=4.1, 2.7 Hz), 155.13 (dd, J=294.1, 290.8 Hz), 128.20 (t, J=3.7 Hz), 125.68 (q, J=3.7 Hz), 125.46 (q, J=3.8 Hz), 123.96 (q, J=272.1 Hz), 86.72 (dd, J=22.5, 17.1 Hz), 61.29, 33.55 (d, J=2.5 Hz), 14.01; 19F NMR (376 MHz, Methanol-d4) δ: -63.81, -88.29 (d, J=34.0 Hz), -89.85 (d, J=34.2 Hz); HRMS (ESI) calcd for C13H12F5O2 [M+H]+ 295.0757, found 295.0747.
Ethyl 3-(4-(tert-butyl)phenyl)-4, 4-difluorobut-3-enoate (2f): 25 mg, yield 44%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.30 (d, J=8.6 Hz, 2H), 7.20 (d, J=8.9 Hz, 2H), 4.06 (q, J=7.1 Hz, 2H), 3.30 (s, 2H), 1.24 (s, 9H), 1.13 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 170.27 (d, J=7.0 Hz), 154.83 (dd, J=292.3, 288.8 Hz), 150.47, 130.02 (t, J=3.9 Hz), 127.42 (t, J=3.6 Hz), 125.43, 86.91 (dd, J=21.3, 17.6 Hz), 61.05, 34.52, 33.88, 33.85, 31.22, 14.06; 19F NMR (376 MHz, Chloroform-d) δ: -87.15 (d, J=39.9 Hz), -88.31 (d, J=40.0 Hz); HRMS (ESI) calcd for C16H21F2O2 [M+H]+ 283.1509, found 283.1502.
Ethyl 4, 4-difluoro-3-(4-vinylphenyl) but-3-enoate (2g): 36 mg, yield 70%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.40 (d, J=8.2 Hz, 2H), 7.31 (d, J=7.8 Hz, 2H), 6.70 (dd, J=17.6, 10.9 Hz, 1H), 5.76 (d, J=17.6 Hz, 1H), 5.26 (d, J=10.8 Hz, 1H), 4.13 (q, J=7.1 Hz, 2H), 3.39 (s, 2H), 1.21 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 170.08 (dd, J=4.1, 2.8 Hz), 154.85 (dd, J=292.9, 289.4 Hz), 136.19, 132.40 (t, J=4.0 Hz), 127.92 (t, J=3.8 Hz), 126.29, 114.26, 87.04 (dd, J=21.7, 17.5 Hz), 61.09, 33.73 (d, J=2.8 Hz), 14.09; 19F NMR (376 MHz, Methanol-d4) δ: -90.63 (d, J=38.3 Hz), -91.76 (d, J=38.5 Hz); HRMS (ESI) calcd for C14H15F2O2 [M+H]+ 253.1040, found 253.1032.
Ethyl 3-([1, 1'-biphenyl]-4-yl)-4, 4-difluorobut-3-enoate (2h): 49mg, yield 81%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=95:5], colorless solid. 1H NMR (400 MHz, Chloroform-d) δ: 7.59 (d, J=7.8 Hz, 4H), 7.48~7.40 (m, 4H), 7.36 (t, J=7.4 Hz, 1H), 4.15 (q, J=7.1 Hz, 2H), 3.44 (s, 2H), 1.22 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 170.20 (d, J=2.9 Hz), 140.36 (d, J=11.7 Hz), 132.02~131.87 (m), 128.79, 128.15 (t, J=3.7 Hz), 127.44, 127.18, 127.00, 86.92 (dd, J=21.6, 17.3 Hz), 61.16, 33.77 (d, J=2.5 Hz), 14.08; 19F NMR (376 MHz, Chloroform-d) δ: -86.06 (d, J=38.1 Hz), -87.32 (d, J=37.5 Hz); HRMS (ESI) calcd for C18H17F2O2 [M+H]+ 303.1196, found 303.1188.
Ethyl 4, 4-difluoro-3-(4-fluorophenyl) but-3-enoate (2i): 30 mg, yield 62%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.33~7.28 (m, 2H), 7.08~7.01 (m, 2H), 4.12 (q, J=7.1 Hz, 2H), 3.36 (t, J=2.2 Hz, 2H), 1.20 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 169.97 (dd, J=4.1, 2.7 Hz), 162.01 (d, J=247.5 Hz), 158.06~151.53 (m), 129.69 (dt, J=7.8, 3.5 Hz), 128.96 (q, J=3.6 Hz), 115.60, 115.38, 86.48 (dd, J=22.2, 18.2 Hz), 61.13, 33.98 (d, J=2.6 Hz), 14.03; 19F NMR (376 MHz, Methanol-d4) δ: -90.13 (d, J=38.6 Hz), -91.39 (d, J=38.1 Hz), -115.41; HRMS (ESI) calcd for C12H12F3O2 [M+H]+ 245.0789, found 245.0782.
Ethyl 4, 4-difluoro-3-(3-fluorophenyl) but-3-enoate (2j): 30 mg, yield 62%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.32 (td, J=8.1, 6.0 Hz, 1H), 7.12 (d, J=7.8 Hz, 1H), 7.07 (d, J=8.8 Hz, 1H), 7.03~6.93 (m, 1H), 4.14 (q, J=7.1 Hz, 2H), 3.38 (t, J=2.2 Hz, 2H), 1.21 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 169.86 (dd, J=4.1, 2.8 Hz), 162.73 (d, J=245.8 Hz), 154.99 (dd, J=293.5, 289.9 Hz), 135.18 (dd, J=8.3, 4.2 Hz), 129.99 (d, J=8.5 Hz), 123.48 (q, J=3.2 Hz), 115.10 (dd, J=4.4, 3.3 Hz), 114.87 (dd, J=4.4, 3.3 Hz), 114.52 (d, J=21.0 Hz), 86.98~86.36 (m), 61.22, 33.68 (d, J=2.6 Hz), 14.05; 19F NMR (376 MHz, Chloroform-d) δ: -86.59 (d, J=32.6 Hz), -87.60 (d, J=32.0 Hz), -112.81; HRMS (ESI) calcd for C12H12F3O2 [M+H]+ 245.0789, found 245.0792.
Ethyl 3-(4-chlorophenyl)-4, 4-difluorobut-3-enoate (2k): 27 mg, yield 52%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.33 (d, J=8.6 Hz, 2H), 7.27 (d, J=8.8 Hz, 2H), 4.12 (q, J=7.1 Hz, 2H), 3.37 (s, 2H), 1.21 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 169.99~169.86 (m), 154.79 (dd, J=292.7, 289.9 Hz), 133.45, 131.48 (t, J=4.0 Hz), 130.25, 129.19 (t, J=3.7 Hz), 128.94, 128.73, 86.49 (dd, J=22.1, 17.6 Hz), 61.22, 33.71 (d, J=2.3 Hz), 14.06; 19F NMR (376 MHz, Chloroform-d) δ: -87.20 (d, J=33.8 Hz), -88.36 (d, J=33.8 Hz); HRMS (ESI) calcd for C12H11ClF2O2Na [M+Na]+ 283.0314, found 283.0306.
Ethyl 3-(3-chlorophenyl)-4, 4-difluorobut-3-enoate (2l): 21 mg, yield 41%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.26 (s, 1H), 7.23~7.12 (m, 3H), 4.06 (q, J=7.0 Hz, 2H), 3.30 (s, 2H), 1.14 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 169.79 (dd, J=4.2, 2.8 Hz), 154.97 (dd, J=293.4, 290.1 Hz), 134.90 (t, J=4.0 Hz), 134.43, 129.72, 128.05 (t, J=3.8 Hz), 127.74, 126.07 (t, J=3.5 Hz), 86.55 (dd, J=22.4, 17.5 Hz), 61.23, 33.68 (d, J=2.2 Hz), 14.05; 19F NMR (376 MHz, Chloroform-d) δ: -86.74 (d, J=32.6 Hz), -87.81 (d, J=32.5 Hz); HRMS (ESI) calcd for C12H11ClF2O2Na [M+Na]+ 283.0314, found 283.0310.
Ethyl 3-(4-bromophenyl)-4, 4-difluorobut-3-enoate (2m): 40 mg, yield 66%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.48 (d, J=8.5 Hz, 2H), 7.21 (d, J=8.4 Hz, 2H), 4.12 (q, J=7.1 Hz, 2H), 3.37 (s, 2H), 1.20 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 169.87 (dd, J=4.1, 2.8 Hz), 154.74 (dd, J=292.8, 289.9 Hz), 131.99 (t, J=4.0 Hz), 131.68, 129.50 (t, J=3.6 Hz), 121.58, 86.57 (dd, J=22.2, 17.6 Hz), 61.20, 33.65 (d, J=2.2 Hz), 14.05; 19F NMR (376 MHz, Chloroform-d) δ: -87.21 (d, J=33.7 Hz), -88.38 (d, J=33.8 Hz); HRMS (ESI) calcd for C12H11BrF2O2Na [M+Na]+ 326.9808, found 326.9798.
Ethyl 3-(3-bromophenyl)-4, 4-difluorobut-3-enoate (2n): 41 mg, yield 67%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.49 (s, 1H), 7.41 (d, J=7.6 Hz, 1H), 7.30~7.18 (m, 2H), 4.13 (q, J=7.1 Hz, 2H), 3.36 (s, 2H), 1.21 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 169.77 (dd, J=4.1, 2.8 Hz), 158.44~151.70 (m), 135.18 (t, J=4.1 Hz), 130.94 (t, J=3.7 Hz), 130.68, 129.99, 126.57 (t, J=3.5 Hz), 122.54, 86.48 (dd, J=22.5, 17.6 Hz), 61.24, 33.70 (d, J=2.4 Hz), 14.06; 19F NMR (376 MHz, Methanol-d4) δ: -90.05 (d, J=36.8 Hz), -91.16 (d, J=36.3 Hz); HRMS (ESI) calcd for C12H11BrF2O2Na [M+Na]+ 326.9808, found 326.9811.
Ethyl 3-(benzofuran-3-yl)-4, 4-difluorobut-3-enoate (2o): 28 mg, yield 78%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.67 (s, 1H), 7.59 (d, J=7.4 Hz, 1H), 7.50 (d, J=8.0 Hz, 1H), 7.30 (dt, J=20.8, 7.1 Hz, 2H), 4.14 (q, J=7.1 Hz, 2H), 3.42 (s, 2H), 1.20 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 169.94 (dd, J=4.1, 2.9 Hz), 154.66 (dd, J=292.1, 289.8 Hz), 143.32 (dd, J=5.2, 3.2 Hz), 124.68, 122.93, 120.49 (d, J=4.6 Hz), 113.69 (dd, J=4.2, 3.2 Hz), 111.66, 78.63 (dd, J=26.0, 20.9 Hz), 61.21, 33.88 (d, J=2.9 Hz), 14.03; 19F NMR (376 MHz, Chloroform-d) δ: -84.54 (d, J=32.1 Hz), -88.16 (d, J=32.6 Hz); HRMS (ESI) calcd for C14H13F2O3 [M+H]+ 267.0833, found 267.0828.
Ethyl 4, 4-difluoro-3-(phenanthren-9-yl) but-3-enoate (2p): 59 mg, yield 90%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless solid. 1H NMR (400 MHz, Chloroform-d) δ: 8.74 (d, J=7.8 Hz, 1H), 8.69 (d, J=8.2 Hz, 1H), 7.98 (d, J=7.9 Hz, 1H), 7.88 (d, J=7.3 Hz, 1H), 7.77 (s, 1H), 7.72~7.58 (m, 4H), 4.08 (q, J=7.1 Hz, 2H), 3.48 (s, 2H), 1.14 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 170.01~169.74 (m), 157.90~151.90 (m), 131.26, 130.75, 130.36, 130.10, 129.02 (d, J=3.8 Hz), 128.77, 127.15, 126.87 (d, J=3.7 Hz), 126.73, 125.45, 123.16, 122.53, 85.07 (t, J=22.4 Hz), 61.07, 35.19 (d, J=2.5 Hz), 14.02; 19F NMR (376 MHz, Chloroform-d) δ: -86.44 (d, J=34.8 Hz), -89.29 (d, J=34.8 Hz); HRMS (ESI) calcd for C20H17F2O2 [M+H]+ 327.1196, found 327.1188.
Ethyl 4, 4-difluoro-3-(naphthalen-1-yl) but-3-enoate (2q): 50 mg, yield 90%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless solid. 1H NMR (400 MHz, Chloroform-d) δ: 7.94 (d, J=8.1 Hz, 1H), 7.91~7.82 (m, 2H), 7.58~7.50 (m, 2H), 7.48 (d, J=5.6 Hz, 2H), 4.08 (q, J=7.1 Hz, 2H), 3.48 (s, 2H), 1.14 (d, J=14.3 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 171.98~167.34 (m), 154.52 (dd, J=291.4, 288.2 Hz), 133.77, 131.35 (d, J=2.5 Hz), 130.45 (dd, J=4.4, 1.8 Hz), 128.72, 127.70, 128.54, (dd, J=3.4, 1.5 Hz), 125.96, 126.43, 125.29, 124.76, 88.74~82.49 (m), 61.00, 35.37 (d, J=2.6 Hz), 13.96; 19F NMR (376 MHz, Chloroform-d) δ: -86.65 (d, J=34.9 Hz), -89.59 (d, J=34.9 Hz); HRMS (ESI) calcd for C16H14F2O2Na [M+Na]+ 299.0860, found 299.0868.
Ethyl 4, 4-difluoro-3-(thiophen-3-yl) but-3-enoate (2r): 39 mg, yield 84%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.32 (dd, J=5.2, 3.1 Hz, 1H), 7.23 (s, 1H), 7.18 (d, J=4.9 Hz, 1H), 4.15 (q, J=7.1 Hz, 2H), 3.38 (s, 2H), 1.23 (t, J=7.1 Hz, 3H).13C NMR (101 MHz, Chloroform-d) δ: 170.33~170.06 (m), 155.04 (dd, J=294.1, 288.4 Hz), 133.11 (dd, J=5.4, 3.4 Hz), 126.55 (dd, J=7.0, 2.3 Hz), 125.76, 121.91 (t, J=5.5 Hz), 83.81 (dd, J=23.5, 17.5 Hz), 61.17, 33.40 (d, J=3.1 Hz), 14.05; 19F NMR (376 MHz, Chloroform-d) δ: -85.32 (d, J=34.0 Hz), -89.32 (d, J=34.0 Hz); HRMS (ESI) calcd for C10H11F2O2S [M+H]+ 233.0448, found 233.0449.
Butyl 4, 4-difluoro-3-phenylbut-3-enoate (2s): 44 mg, yield 87%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.35 (d, J=5.5 Hz, 4H), 7.28 (dt, J=4.9, 2.5 Hz, 1H), 4.07 (t, J=6.6 Hz, 2H), 3.40 (t, J=2.1 Hz, 2H), 1.55 (dt, J=14.6, 6.7 Hz, 2H), 1.29 (dq, J=14.6, 7.4 Hz, 2H), 0.89 (t, J=7.4 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 170.19 (dd, J=4.1, 2.8 Hz), 154.80 (dd, J=292.4, 289.0 Hz), 133.04 (t, J=3.8 Hz), 128.48, 127.90~127.71 (m), 127.51, 87.23 (dd, J=21.5, 17.7 Hz), 64.97, 33.91, 33.89 (d, J=2.6 Hz), 30.51, 18.97, 13.58; 19F NMR (376 MHz, Methanol-d4) δ: -91.09 (d, J=40.1 Hz), -92.25 (d, J=39.5 Hz); HRMS (ESI) calcd for C14H17F2O2 [M+H]+ 255.1196, found 255.1195.
Tert-butyl 4, 4-difluoro-3-phenylbut-3-enoate (2t): 26 mg, yield 52%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.38~7.28 (m, 4H), 7.34~7.25 (m, 1H), 3.34 (t, J=2.3 Hz, 2H), 1.39 (s, 9H); 13C NMR (101 MHz, Chloroform-d) δ: 169.31 (dd, J=4.0, 2.8 Hz), 154.71 (dd, J=292.1, 288.7 Hz), 133.26 (t, J=3.9 Hz), 128.40, 127.87 (t, J=3.5 Hz), 127.40, 87.63 (dd, J=21.6, 17.3 Hz), 81.27, 35.09 (d, J=2.5 Hz), 27.85; 19F NMR (376 MHz, Methanol-d4) δ: -91.07 (d, J=40.2 Hz), -92.41 (d, J=39.6 Hz); HRMS (ESI) calcd for C14H17F2O2 [M+H]+ 255.1196, found 255.1198.
Allyl 4, 4-difluoro-3-phenylbut-3-enoate (2u): 32 mg, yield 68%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.35 (d, J=5.7 Hz, 4H), 7.30~7.27 (m, 1H), 5.85 (ddt, J=16.6, 11.0, 5.7 Hz, 1H), 5.29~5.13 (m, 2H), 4.57 (d, J=5.5 Hz, 2H), 3.44 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ: 169.93~169.66 (m), 154.86 (dd, J=292.5, 289.3 Hz), 132.95 (t, J=3.8 Hz), 131.73, 128.52, 127.86 (t, J=3.5 Hz), 127.59, 118.37, 87.08 (dd, J=21.4, 18.0 Hz), 65.68, 33.83 (d, J=2.6 Hz); 19F NMR (376 MHz, Chloroform-d) δ: -86.44 (d, J=34.8 Hz), -89.29 (d, J=34.7 Hz); HRMS (ESI) calcd for C13H13F2O2 [M+H]+ 239.0883, found 239.0880.
2, 2, 2-Trifluoroethyl 4, 4-difluoro-3-phenylbut-3-enoate (2v): 23 mg, yield 42%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.41~7.27 (m, 5H), 4.45 (q, J=8.4 Hz, 2H), 3.52 (t, J=2.1 Hz, 2H); 13C NMR (101 MHz, Chloroform-d) δ: 168.65 (dd, J=4.3, 2.9 Hz), 158.17~151.73 (m), 132.56~132.28 (m), 128.65, 128.10~127.55 (m), 122.70 (q, J=277.1 Hz), 86.48 (dd, J=21.2, 18.9 Hz), 60.76 (q, J=36.9 Hz), 33.35 (d, J=2.6 Hz); 19F NMR (376 MHz, Chloroform-d) δ: -70.30, -87.50 (d, J=34.5 Hz), -88.58 (d, J=34.6 Hz); HRMS (ESI) calcd for C12H10F5O2 [M+H]+ 281.0601, found 281.0606.
N-Benzyl-4, 4-difluoro-3-phenylbut-3-enamide (2w): 46 mg, yield 81%, chromatography on silica gel [V(petroleum ether):V(EtOAc)=98:2], colorless oil. 1H NMR (400 MHz, Chloroform-d) δ: 7.28 (d, J=4.4 Hz, 4H), 7.27~7.20 (m, 1H), 7.21~7.13 (m, 3H), 7.01~6.93 (m, 2H), 5.83 (s, 1H), 4.31 (d, J=5.7 Hz, 2H), 3.31 (t, J=2.2 Hz, 2H); 13C NMR (101 MHz, Chloroform-d) δ: 168.87~168.61 (m), 154.79 (dd, J=294.5, 289.4 Hz), 132.44 (t, J=3.8 Hz), 128.74, 128.60, 127.88~127.66 (m), 127.41, 126.18 (d, J=3.2 Hz), 87.71 (dd, J=21.1, 15.9 Hz), 43.62, 35.70 (d, J=1.9 Hz); 19F NMR (376 MHz, Chloroform-d) δ: -86.70 (d, J=33.5 Hz), -87.41 (d, J=33.8 Hz); HRMS (ESI) calcd for C17H16F2NO [M+H]+ 288.1200, found 288.1192.
Supporting Information The preparation and characterization of substrate 1a~1w. 1H NMR and 13C NMR spectra for all pure products 2a~2w, the CV and SWV experiments. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) McDonald, I. A.; Lacoste, J. M.; Bey, P.; Palfreyman, M. G.; Zreika, M. J. Med. Chem. 1985, 28, 186.
(b) Sayre, L. M. WO2007005737A2. 2007.
Okada, H.; Morita, M.; Ueda, T.; Takeo, H.; Kominami, H.; Kiriyama, K.; Nakamoto, K.; Yoshida, Y. WO2004052872A1, 2004.
Koley, S.; Altman, R. A. Isr. J. Chem. 2020, 60, 313.
(a) Gao, B.; Zhao, Y.; Ni, C.; Hu, J. Org. Lett. 2014, 16, 102.
(b) Gao, B.; Zhao, Y.; Hu, J. Angew. Chem., Int. Ed. 2015, 54, 638.
(a) Tian, P.; Feng, C.; Loh, T.-P. Nat. Commun. 2015, 6, 7472.
(b) Tian, P.; Wang, C.-Q.; Cai, S.-H.; Song, S.; Ye, L.; Feng, C.; Loh, T.-P. J. Am. Chem. Soc. 2016, 138, 15869.
(c) Cai, S.-H.; Ye, L.; Wang, D.-X.; Wang, Y.-Q.; Lai, L.-J.; Zhu, C.; Feng, C.; Loh, T.-P. Chem. Commun. 2017, 53, 8731.
(d) Tang, H.-J.; Lin, L.-Z.; Feng, C.; Loh, T.-P. Angew. Chem., Int. Ed. 2017, 56, 9872.
(e) Zhu, C.; Song, S.; Zhou, L.; Wang, D.-X.; Feng, C.; Loh, T.-P. Chem. Commun. 2017, 53, 9482.
(f) Tang, H.-J.; Zhang, Y.-F.; Jiang, Y.-W.; Feng, C. Org. Lett. 2018, 20, 5190.
(g) Zhou, L.; Zhu, C.; Loh, T.-P.; Feng, C. Chem. Commun. 2018, 54, 5618.
(h) Liu, H.; Ge, L.; Wang, D.-X.; Chen, N.; Feng, C. Angew. Chem., Int. Ed. 2019, 58, 3918.
(i) Zhou, L.; Zhu, C.; Bi, P.; Feng, C. Chem. Sci. 2019, 10, 1144.
(j) Zhu, C.; Zhang, Y.-F.; Liu, Z.-Y.; Zhou, L.; Liu, H.; Feng, C. Chem. Sci. 2019, 10, 6721.
(k) Cao, Z.-C.; Liu, J.-C.; Chu, Y.-Q.; Zhao, F.-M.; Zhu, Y.-H.; She, Y.-B. Chin. J. Org. Chem. 2019, 39, 2499(in Chinese).
(曹志成, 刘建超, 褚有群, 赵峰鸣, 朱英红, 佘远斌, 有机化学2019, 39, 2499.)
(l) Du, H.-W.; Sun, J.; Gao, Q.-S.; Wang, J.-Y.; Wang, H.; Xu, Z.; Zhou, M.-D. Org. Lett. 2020, 22, 1542.
Zubkov, M. O.; Kosobokov, M. D.; Levin, V. V.; Kokorekin, V. A.; Korlyukov, A. A.; Hu, J.; Dilman, A. D. Chem. Sci. 2020, 11, 737. https://pubs.rsc.org/en/content/articlelanding/2019/sc/c9sc04643g#!divAbstract
Liu, C.; Zhu, C.; Cai, Y.; Yang, Z.; Zeng, H.; Chen, F.; Jiang, H. Chem.-Eur. J. 2020, 26, 1953.
Chelucci, G. Chem. Rev. 2012, 112, 1344. https://pubmed.ncbi.nlm.nih.gov/22085400/
Nihei, T.; Iwai, N.; Matsuda, T.; Kitazume, T. J. Org. Chem. 2005, 70, 5912. https://www.researchgate.net/publication/312260011_Pharmacokineticpharmacodynamic_analysis_of_a_novel_anti-hyperuricemic_compound_derived_from_oxidized_porcine_hemin
Cao, C.-R.; Ou, S.; Jiang, M.; Liu, J.-T. Tetrahedron Lett. 2017, 58, 482. doi: 10.1002/ejoc.201301021#support-information-section
Wang, S.; Cheng, B.-Y.; Sršen, M.; König, B. J. Am. Chem. Soc. 2020, 142, 7524.
于蛟, 林锦鸿, 肖吉昌, 有机化学, 2019, 39, 265. https://www.zhangqiaokeyan.com/academic-journal-cn_chinese-journal-organic-chemistry_thesis/0201271521750.htmlYu, J.; Lin, J.-H.; Xiao, J.-C. Chin. J. Org. Chem. 2019, 39, 265(in Chinese). https://www.zhangqiaokeyan.com/academic-journal-cn_chinese-journal-organic-chemistry_thesis/0201271521750.html
Guo, S.; Yang, P.; Zhou, J. Chem. Commun. 2015, 51, 12115.
(a) Chen, J.; Lv, S.; Tian, S. ChemSusChem 2019, 12, 115.
(b) Gandeepan, P.; Kaplaneris, N.; Santoro, S.; Vaccaro, L.; Ackermann, L. ACS Sustainable Chem. Eng. 2019, 7, 8023.
(c) Mei, H.; Yin, Z.; Liu, J.; Sun, H.; Han, J. Chin. J. Chem. 2019, 37, 292.
(d) Meyer, T. H.; Finger, L. H.; Gandeepan, P.; Ackermann, L. Trends Chem. 2019, 1, 63.
(e) Qiu, Y.; Struwe, J.; Ackermann, L. Synlett 2019, 30, 1164.
(f) Song, C.; Liu, K.; Dong, X.; Chiang, C.-W.; Lei, A. Synlett 2019, 30, 1149.
(g) Wang, H.; Gao, X.; Lv, Z.; Abdelilah, T.; Lei, A. Chem. Rev. 2019, 119, 6769.
(h) Xiong, P.; Xu, H.-C. Acc. Chem. Res. 2019, 52, 3339.
(i) Ye, Z.; Zhang, F. Chin. J. Chem. 2019, 37, 513.
(j) Yuan, Y.; Lei, A. Acc. Chem. Res. 2019, 52, 3309.
(k) Zhang, H.-Y.; Tang, R.-P.; Shi, X.-L.; Jie, L.; Wu, J.-W. Chin. J. Org. Chem. 2019, 39, 1837(in Chinese).
(张怀远, 唐蓉萍, 石星丽, 颉林, 伍家卫, 有机化学, 2019, 39, 1837.)
(l) Ackermann, L. Acc. Chem. Res. 2020, 53, 84.
(m) Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Acc. Chem. Res. 2020, 53, 300.
(n) Li, M.; Hong, J.; Xiao, W.; Yang, Y.; Qiu, D.; Mo, F. ChemSusChem 2020, 13, 1661.
(o) Rockl, J. L.; Pollok, D.; Franke, R.; Waldvogel, S. R. Acc. Chem. Res. 2020, 53, 45.
(p) Wang, P.; Gao, X. L.; Huang, P. F.; Lei, A. W. ChemCatChem 2020, 12, 27.
(q) Wang, X.-Y.; Xu, X.-T.; Wang, Z.-H.; Fang, P.; Mei. T.-S. Chin. J. Org. Chem. 2020, 40, 3738(in Chinese).
(王向阳, 徐学涛, 王振华, 方萍, 梅天胜, 有机化学, 2020, 40, 3738.)
Peters, D. G.; McGuire, C. M.; Pasciak, E. M.; Peverly, A. A.; Strawsine, L. M.; Wagoner, E. R.; Barnes, J. T. Rev. Soc. Quim. Mex. 2017, 58, 287.
Huang, H.; Lambert, T. H. Angew. Chem., Int. Ed. 2020, 59, 658.
(a) Uneyama, K.; Kato, T. Tetrahedron Lett. 1998, 39, 587.
(b) Uneyama, K.; Maeda, K.; Kato, T.; Katagiri, T. Tetrahedron Lett. 1998, 39, 3741.
(c) Uneyama, K.; Mizutani, G. Chem. Commun. 1999, 613.
(d) Uneyama, K.; Mizutani, G.; Maeda, K.; Kato, T. J. Org. Chem. 1999, 64, 6717.
(a) Liu, X.; Liu, R.; Qiu, J.; Cheng, X.; Li, G. Angew. Chem., Int. Ed., 2020, 59, 13962.
(b) Li, J.; He, L.; Liu, X.; Cheng, X.; Li, G. Angew. Chem., Int. Ed. 2019, 58, 1759.

Figure 1 Electrochemistry analyses of 1a
(a) CV condition: glassy carbon working electrode, Pt wire counter electrode, SCE reference electrode, 50 mV/S. (b) 1a (0.04 mmol), nBu4BF4 (0.012 mol/L), MeCN (5 mL), pulse height 25 mV, step height 4 mV, frequency 10 Hz
Table 1. Reaction optimization of electrochemical hydrodefluorination reactiona
|
||
| Entry | Variation from standard condition | Yieldb/% |
| 1 | None | 78 (66c) |
| 2 | No voltage | 0 |
| 3 | No NH3 | 0 |
| 4 | LiClO4 instead of nBu4NBF4 | 62 |
| 5 | LiBF4 instead of nBu4NBF4 | 54 |
| 6 | nBu4NPF6 instead of nBu4NBF4 | 67 |
| 7 | nBu4NClO4 instead of nBu4NBF4 | 71 |
| 8 | DMSO as solvent | 48 |
| 9 | MeOH as solvent | 32 |
| 10 | DMF as solvent | 76 |
| 11 | Constant current is 55, 111, 167, 222 mA/gd | 23, 47, 66, 42 |
| 12 | Constant voltage is 2.8, 3.0, 3.4, 3.6 V | 49, 59, 68, 28 |
| 13 | Graphite rod instead of graphite felt | 14 |
| 14 | Platinum sheet as cathode graphite felt as anode trace | Trace |
| a Standard condition: 1a (0.2 mmol), nBu4NBF4 (0.3 equiv.), CH3CN (5 mL), graphite felt anode and cathode, undivided cell, 3.2 V cell voltage, r.t., NH3 atmosphere, 5 h. b Yields determined by 1H NMR with dichloromethane as the internal standard. c Isolated yield. d As graphite felt is the complex of fibre, the mA/g is more suitable to describe the constant current condition. | ||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: