
图 1.
人类与机器的学习模式
Figure 1.
Learning mode of human and machine

创造拥有人类智慧的机器是人类自古以来的梦想, 从中国周朝的偃师造人到古希腊神话中的黄金机器人, 人类一直在探索生命和智慧背后的秘密.伴随着计算机基础理论和技术的诞生和快速发展, 这一古老梦想正逐渐成为现实. 1956年, 麦卡锡[1]首次提出了人工智能(Artificial intelligence, AI)的概念, 开启了这一全新的研究领域.半个多世纪以来, 伴随着计算机计算能力的快速提升、大数据技术的发展以及学术界和工业界的通力合作, 人工智能领域取得了令人瞩目的进展[2]. 2016年, 阿尔法狗(AlphaGo)[3]打败围棋世界冠军李世石这一历史性时刻标志着人工智能走向了一个全新的高度, 其在各个领域的应用也开始进入公众视野.特别是最近十几年开始兴起的机器学习和深度学习等相关领域的爆发式进展, 在自然语言处理、图像识别、自动驾驶等领域, 已经大有赶超人类现有认知水平的势头[4].
机器学习(Machine learning)作为人工智能研究领域的重要分支, 近年来得到飞速发展和广泛应用. 1959年Samuel等[5]首次提出机器学习的概念, 将其定义为在不进行特定编程情况下给予计算机学习的能力(Give computers the ability to learn without being explicitly programmed).如图 1所示, 机器和人类的学习模式不乏相通之处[6].人类的大脑通过阅读、观察、探索和学习周围世界的规则并获得各种技能, 比如语言、决策、游戏等.机器则通过海量数据和程序的自我学习, 获得类似人类的语言、决策、游戏等各种能力.机器获得学习能力的前提是待解决问题存在基准真值(Ground truth), 通过建立一个假设空间, 程序学习数据中的通用模式, 建立模型并尝试预测基准真值.预测值与基准真值越接近, 模型越精确.程序通过这样一个不断学习与优化的过程, 试图获得人类大脑具有的学习和解决问题的能力.
随着化学信息学的发展, 机器学习在化学领域已展现出巨大的应用潜力, 在量子化学[7]、密度泛函理论(DFT)计算[8]、药物发现[9]、分子从头设计[10]、反应预测与逆合成分析[11]以及自动化合成[12]等领域均已有重要应用.传统的有机化学研究过程中, 物理有机和量子化学提供了强大的理论工具, 但面对复杂体系预测性有限, 大多数新发现仍高度依赖于“试错(Trio-errors)”和“偶缘(Serendipity)”.而人工智能和机器学习以其强大的学习能力、迭代能力、容易实现和易于部署的特性, 为有机化学研究范式的革新带来了曙光.本文主要综述机器学习在有机化学研究中的应用, 简要介绍机器学习的研究范式和在有机化学领域的应用模式, 结合最新的研究进展展示具体的应用实例, 并总结和展望当前研究的挑战和难题.本文面向有机化学多个研究领域, 兼顾基础性和前沿研究进展, 对涉及机器学习的诸多方面比如数据、算法、描述符以及工作流程做基本介绍.
机器学习应用于有机化学一般遵循以下步骤(图 2): (1)数据集:通过收集公开发表的文献、数据库、实验室原始数据等方式汇总特定任务的数据集; (2)分子描述:将化学分子和反应式转化为算法能够识别的形式; (3)模型建立:选择针对特定问题的模型, 选择合适的算法, 使用训练集对模型进行训练, 并用验证集对模型表现进行评价; (4)模型应用:使用训练好的模型预测未知结果; (5)讨论与分析:对预测结果(如物理化学特性或者反应活性等)进行归因与解释.本文重点介绍数据集、分子描述符和算法, 并结合具体实例介绍模型训练、应用和分析.

公开发表在杂志、专利中的数据需要通过提取、汇总、整理才能成为可用的数据集(比如SciFinder和Reaxys等)[13].但通常来说, 一般使用者并不能直接批量获取这些数据.最近, 一些课题组与Reaxys合作, 通过其拥有的海量数据开展化学反应预测和逆合成分析工作[11].当前也有一些公开的化学数据库, 如GDB-13[14]及其子库QM7/QM7b、GDB-17[15]及其子库QM8、QM9等, 收集了大量小分子化合物的量化信息. ESOL[16]和FreeSolv[17]则收集了大量小分子化合物的物理化学信息, 如水溶性和自由能信息. ZINC数据库[18]收录了大量市售化合物的3D结构、供应商等信息, 可用于虚拟筛选.美国专利及商标局(United States Patent and Trademark Office, USPTO)提供了数十年间专利反应数据, 并由Lowe等[19]整理成了专门的反应数据集可供下载使用.
数据的另一种获取来源是电子实验记录本.许多药物和化学相关公司运用电子实验记录本记录数据, 但相关数据主要用于数据存储和知识产权目的, 通常难以直接进行数据挖掘, 需要开放访问并且提供专门的化学信息学环境[20]. NextMove Software开发的HazELNut工具包可以从电子实验记录本系统中提取、格式转换和储存实验数据, 并添加反应分类等附加的标注[21]. 2013年, 罗氏与Elsevier、NextMove合作, 整合公司内部的实验数据和Elsevier的文献数据, 罗氏研究人员可以使用Reaxys的定制界面搜索和浏览所有的反应数据. 2014年, 阿斯利康提取了电子实验记录本的数据并将其加载到数据中心, 支持网页搜索和其他外部应用程序.除了商业软件工具, 还有用于基本反应分析的开源软件, 例如反应解码器(Reaction decoder tool, RDT)工具包, 能够快速从化学反应中提取特征关系[22].
另一方面, 发表的文献和专利大多为最优条件下的最好结果, 而失败的反应和被证明行不通的反应路线都没有发表, 这将造成严重的数据偏误.而电子实验记录本不仅记录了反应中较优的结果, 也记录了较差的甚至失败的实验结果, 不存在负面样本偏误.因此, 电子实验记录本记录的结果更加适用于实验数据挖掘以及预测模型的建立和训练.但即使已有数百万高质量的实验结果收录在案, 面对巨大的化学反应空间(反应物、产物、反应条件等参数), 这些数据在空间中的分布相对来说仍然十分稀疏[23].近年来, 随着微流控和自动化技术的不断进步, 高通量筛选装置能在较短时间里产生大量优质的反应数据, 使系统探索更大范围反应空间成为可能[24], 这些研究推动了有机合成的大数据化, 有望为智能化学合成提供数据基础[25].
数据收集完成后, 另一个关键的问题就是如何把分子式转化为计算机能够识别的形式.通过何种方式描述化合物分子以及描述符是否包含了化合物分子中暗含的关键信息, 将直接决定模型的预测效果.化合物分子描述方式种类繁多, 比较常见的有以下几种方式(图 3).
这种方法通过提取分子中各种物理化学参数, 比如log P、pKa和分子量(Molecular weight)等, 这些参数最终汇总为一组特征向量并作为输入训练机器学习模型.当前常用的化学信息学软件, 如ChemAxon[26]、RDKit[27]和CDK[28]等都可以快速方便地提取分子的特征.此外, 通过半经验或DFT理论计算获得高精度分子结构特征和理化参数也是较为常用的描述符生成方法.
这种方法将化合物分子转化为一串二进制向量, 这些二进制向量特征性地表示了某类分子碎片是否存在, 如同指纹一样描述了某一分子与其他分子的相似与不同之处.这里列举三种常见的分子指纹: (1)基于亚结构的分子描述子MACCS[29]和PubChem fingerprints[30]; (2)基于拓扑和路径的指纹, 如Daylight fingerprint和Tree fingerprint; (3)环形分子指纹, 如MolPrint2D[31]、Morgan fingerprint[32]以及类似的Extended-Connectivity Fingerprints (ECFP)[33]、Functional-Class Fingerprints(FCFP))等.一些免费的开源化学信息学工具包支持对分子指纹的提取, 例如RDKit、Open Babel[34]、CDK和Indigo[35]等.
最常用的分子简写式是简化分子线性输入规范[36] (Simplified molecular input line entry specification, SMILES)和国际化学标识符[37]((International chemical Identifier, InChI), 标准化的化学标识格式可以解决数据库中分子的非标准化命名问题. SMILES利用ASCII字符串, 通过简单的原子符号、键符号和语言规则可以描述分子的三维结构和化学反应. SMILES的一种扩展格式SMARTS[38] (SMILES arbitrary target specification)是一种精确的子结构特征识别和原子分类的符号. InChI能够唯一性地描述化学分子[39], 而SMILES不是唯一性的.
分子图法是将分子抽象为一张向量图或者无向图, 其中的原子和化学键抽象表达为图中的节点和边.库伦矩阵(Coulomb matrix[7, 40])是分子图的另一种表示方法, 包含原子核排斥和自由原子的势能信息, 并且对分子的平移和旋转保持不变, Rupp等[7a]将这一方法用于分子原子化能预测, 精度能达到12.54 kJ/mol.
格点表示法是将分子放置于二维或三维网格中, 通过每个网格的位置和包含的分子信息进行编码.其中三维格点方法可以提取分子的空间结构特征, 这些策略已经被用于蛋白质配体打分模型[41]和立体选择性预测模型[42]等工作中.
算法是人工智能的核心, 目前常用的机器学习算法种类繁多.从学习方式来分, 可以分为监督学习(Supervised learning)、无监督学习(Unsupervised learning)、半监督学习(Semi-supervised learning)和强化学习(Reinforcement learning[43])等.从机器学习的算法基础来分, 则可以分为基于统计学的传统机器学习算法和神经网络算法.目前常用的传统机器学习算法主要包括贝叶斯方法(Bayesian[44])、决策树(Decision tree[45])及其衍生方法、朴素贝叶斯(Naive Bayes[46])、支持向量机(Support vector machine[47])、聚类方法(Cluster analysis[48])、集成学习算法(Ensemble methods, 如著名的基于树的随机森林方法, Random forest[49])等, 这类方法被广泛应用于包括有机化学在内的各个领域.相关算法原理可参考相关综述及统计学相关书籍[50], 在此不做过多介绍. Scikit-learn[51] Python工具库囊括了大部分的经典机器学习算法, 可以结合RDkit等化学信息学工具直接调用, 使用非常方便.
另一类则是基于大脑神经元(Brain-inspired)的工作原理发展起来的神经网络模型.与创造“人工大脑”不同, 这些程序试图理解大脑是如何工作的.这类模型主要有两个分支.一类被称为脉冲神经网络(Spike neural network[52]), 该网络基于拟合生物神经元发射和接收脉冲信号的机制.另一类则是现在极为流行的神经网络(Neural network)[53].通过神经元的多层级连接, 神经网络能够提取输入中的关键特征, 并提取出这些特征与输出之间复杂的关系.随着计算机运算能力的爆炸性增长, 越来越复杂的神经网络, 即深度神经网络也开始被应用在化学研究的各个领域, 并展现出巨大的潜力.深度神经网络可以使用更多神经元节点和更深的网络层, 因此其特征抽象能力也大大提高.同时, 神经网络结构和算法也飞速发展, 比如通过Dropout[54]和DropConnect[55]方法解决过拟合问题, 通过Rectified linear unit (ReLU)[56]解决梯度消失问题, 通过图神经网络[57]解决分子拓扑结构表示的问题[58]等.目前流行的深度神经网络主要包含全连接神经网络(Full connected neural network, FCNN)、卷积神经网络(Convolutional neural network, CNN)、循环神经网络(Recurrent Neural Network, RNN)、自动编码器(Auto encoder, AE)、生成对抗网络(Generative adversarial network, GAN)及其相关变种.关于神经网络的工作原理目前已有大量书籍文献, 在此不做过多介绍.大部分机器学习框架都是开源的, 如TensorFlow、PyTorch、GLUON、Caffe、Theano等框架已经被广泛应用于各个方面.
对于有机化学工作者来说, 最关心的问题是如何选择已有的算法来解决面临的有机化学问题.从学习方式来看, 目前有机化学领域主要采用监督学习算法, 即利用已标记的有限化学数据集, 通过某种学习策略/方法建立模型, 实现对新数据/实例的标记(分类)或映射(回归).监督学习要求训练样本的标签(如物化性质、药理活性、反应选择性等)已知, 标签的精确度越高, 样本越具有代表性, 学习模型的准确度越高.此外, 很多有机化学问题并不符合相似性原则, 结构的微小变化就可能造成药物活性、催化活性等性质的巨大变化, 非基于距离的算法(Non-distance-based algorithms)比如随机森林、神经网络等对这类问题更加合适[59].在具体的研究中, 往往可通过对已有算法进行筛选和验证来确定最适用的机器学习算法.需要注意的是, 基于训练集和验证集得到的最优模型并不一定是最适用的模型, 它可能存在过拟合或者泛化能力差等问题, 一般还需要经过外部测试集的进一步检验来确定最优的预测模型.
机器学习在有机化学领域最早的工作可追溯到20世纪60年代, Corey等[60]发展了基于规则的计算机辅助合成设计程序(Computer-aided synthetic planning, CASP). CASP的目标是辅助化学家快速高效地实现小分子化合物的合成.其以分子结构作为输入, 输出一系列详细的反应方案, 每个方案通过一系列可行的反应步骤将目标连接到可购买的起始原料. 20世纪60年代至90年代, 受到计算资源的限制, 早期工作主要依赖于专家编码的反应规则和启发式策略, 通过给定的判断规则给出可能的化学键断裂和生成的位置.在过去的20年间, 随着大规模化学数据库的发展, 数以百万计的反应数据可以应用于机器学习模型的训练和验证.同时, 神经网络与深度学习算法的快速发展也推动了机器学习在化合物性质预测、分子从头设计、化学反应预测、逆合成分析以及智能合成机器等领域的应用[61].
机器学习很早就已应用于化合物性质预测.近年来, 深度神经网络在该领域的应用展现出了巨大的优势, 并频繁出现在各种化合物性质与活性预测挑战赛中. 2015年, Dahl等[62]使用2D拓扑描述符作为输入, 将深度神经网络方法应用于Merck Kaggle挑战赛.在15个化合物活性预测中有13个目标都显著高于当时最优的预测结果.这一方法有几个显著特点: (1)深度神经网络可以同时处理数千个描述符而无需特征选择; (2) Dropout可以避免传统神经网络过拟合的问题; (3)超参优化可以最大限度地提升深度神经网络的预测效果; (4)多任务深度神经网络的预测效果明显好于单任务神经网络. 2016年, Mayr等[63]运用多任务神经网络赢得了Tox21挑战赛, 他们选用多种描述符(3D/2D描述符、预先定义的药效基团)和ECFP分子指纹输入模型对12000种化合物的12种毒性指标进行了建模, 与Dahl等的神经网络类似, 他们也将Dropout方法和ReLu激活函数应用于深度神经网络, 并使用GPU并行计算加速模型训练.研究结果表明深度神经网络具备很强的特征提取能力, 模型可以有效识别并提取出各种可能的毒性基团特征.随后的多项对比研究均发现, 相较于传统机器学习算法, 神经网络算法在化合物生物活性预测方面表现出较优的预测能力[64].目前, 神经网络算法在药物生理活性、毒性、溶解度等性质预测方面均实现了较好的应用[65].需要指出的是, 深度神经网络的表现很大程度上取决于训练数据规模和质量, 数据不足或质量不高可能很难得到稳健且迁移能力较好的模型.最近, Kim等[66]利用贝叶斯推理进行定量的不确定性分析来提高预测的可靠性, 他们选择分子图作为模型输入, 采用贝叶斯图卷积网络(Bayesian graph convolutional network)对化合物生物活性和毒性进行分类建模和预测, 并可同时定量给出引起不确定性的因素, 有效提高了ChEMBL数据集中分子表皮生长因子受体(Epidermal growth factor receptor, EGFR)抑制活性预测的准确性[67].
机器学习在有机化合物物理化学性质预测方面也表现出不俗的潜力, 以有机物pKa预测为例, 著名的ADMET商业软件中的pKa预测模块就是通过神经网络集成实现的[68]. 2019年, Grzybowski等[69]通过图卷积神经网络(Graph convolutional neural network, GCNN)实现了C—H酸pKa在毫秒级时间的准确预测, 对大约13000个去质子反应的预测正确率大于90%, 平均绝对误差MAE≈2.1.最近, 罗三中和张龙等[70]基于机器学习方法建立了pKa的全局预测模型(图 4).他们从iBonD[71]数据库中收集了39种溶剂的pKa实验数据, 清洗和整理后得到了包含15338种化合物的数据集.在描述符方面, 他们开发了将分子指纹和物理有机参数相结合的SPOC (Structure and physical organic chemistry)描述符.在建模方面, 他们选择5重交叉验证方法对常用机器学习方法进行了筛选, 发现运用神经网络或XGBoost算法训练的全局模型具有最佳预测表现, MAE仅为0.87个pK单位.并可以实现多溶剂体系pKa的快速精准预测, 该研究还表明, 全局模型的预测结果优于所有的单一溶剂模型, 对不同溶剂中pKa预测值的相关度分析也验证了迁移学习的特征.此外, 对样本外药物分子, 二甲基亚砜(DMSO)中氢键催化剂以及乙腈中氨基催化剂pKa预测进一步验证了该模型的稳健性.

最近, 神经网络算法也被应用于化学键均裂能(BDE)预测中.江俊等[73]以原子种类和电荷分布为描述符, 采用3个隐藏层的神经网络对8000个有机化合物的量化计算BDE数据进行了建模和预测, 其预测的平均误差为4.05 kJ/mol, 相关系数R2可达0.97, 与DFT计算结果相当.同样, 针对其他量化计算所得的化合物性质如频率、零点能、最高已占分子轨道(HOMO)或最低空分子轨道(LUMO)能量等也可以通过机器学习预测快速获得. 2017年, 谷歌的研究人员[74]从几种流行的处理图结构数据的神经网络模型中抽象出共性, 提出了一种应用于图结构的监督学习框架, 该框架称为消息传递神经网络(Message passing neural networks, MPNNs).通过对QM9数据库中的DFT计算数据进行建模, 成功实现了包括原子化能、红外振动频率、零点振动能和HOMOLUMO能量等有机分子量子化学性质的预测.
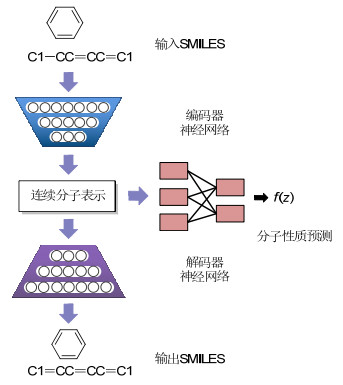
分子从头设计(De novo design)利用算法虚拟设计和评估一系列符合特定性质的分子, 可用于药物、材料等功能分子的发现[10, 75]. Gómez-Bombarelli等[10b]发展了使用变分自编码器(Variational autoencoder, VAE)生成分子结构的方法.他们从ZINC数据库获取分子并以SMILES格式输入, 编码器将分子的这种离散表示转换为隐含空间的连续向量, 解码器再将这些连续向量还原成分子SMILES(图 5).此方法的隐含空间中的分子表示是连续的, 因此可以通过随机解码、扰乱或插入等方法产生新的分子, 并通过一些优化算法产生特定性质的分子[76]. 2017年, Kadurin等[77]使用VAE结合生成对抗网络[78], 生成了具有特定抗癌活性的分子. 2019年, Zhavoronkov等[79]基于VAE实现了盘状结构域受体1 (Discoidin domain receptor 1, DDR1)有效抑制剂的快速设计.
近年来, 使用RNN模型进行分子从头设计受到了越来越多的关注. 2017年, Segler等[10c, 80]报道了使用RNN生成新型分子结构.通过使用大量SMILES训练RNN网络并学习SMILES的概率分布, 该网络可以高效生成训练集之外的各种分子结构(图 6). Segler等尝试预先在一个通用集上训练神经网络模型, 随后通过迁移学习将预训练的模型应用于特定数据集, 以提高小数据集的预测性能.通过这一策略, 该模型能生成14%从未出现过的抗金色葡萄球菌和28%新型的抗疟疾的活性分子.
2016年, Jaques等[81]使用一种强化学习算法Deep Q-learning结合预先训练的RNN模型, 生成了具有特定分子性质(比如cLog P[82]和QED drug-likeness[83])的新型分子.然而, 该方法依赖于包含手写规则的奖励函数, 也可能生成没有真实意义的简单分子结构.为解决此问题, Olivecrona等[84]提出了一种基于策略的强化学习方法, 通过预训练RNN, 可以生成目标结构的类似物和对生物靶标有预期活性的分子.此模型被用于训练生成药物塞来昔布的类似物, 在另一项训练任务中, 该模型能够以95%以上的准确率生成对多巴胺II型受体有效的分子.
从上述工作中可以看出, 机器学习算法, 特别是深度神经网络算法, 可以根据实际需要来定义分子特性并生成具有较高多样性和特异性的分子.然而, 对抗神经网络和增强学习方法容易陷入局域分子空间, 可能导致生成的分子仅仅是某类分子骨架的简单变化[85], 最近越来越多的工作开始关注并尝试解决这些问题[86].
关于反应预测和逆合成分析的研究起步较早, 自20世纪60年代开始就陆续出现了若干计算机辅助合成设计系统, 例如LHASA[87]和SYNGEN[88]等.然而, 传统的方法使用大量专家制定的规则来判断某条路径的可行性, 并没有得到完全令人满意的结果[89].最近, 基于机器学习方法的反应预测研究进展迅速[90].下面将简要介绍机器学习算法在前向反应预测、反应活性与选择性预测, 以及反应条件预测方面的应用进展.
2011年, Baldi课题组[91]使用自定义的分子轨道概念(Unfilled and filled molecular orbitals)和物理化学描述符作为输入, 通过对反应数据进行训练, 最终能以89.05%的精度预测极性反应.如果综合考虑前4种可能, 预测精度能进一步提升到99.86%(图 7).该策略考虑了具体的反应条件, 因而能得出更加真实可信的结果.同时, 该策略在一定程度上从机理层面阐释了化学反应中电子转移的基元过程, 并能识别和预测多步反应过程.随后, Baldi课题组[92]将这一方法进一步推广到自由基反应和周环反应中.
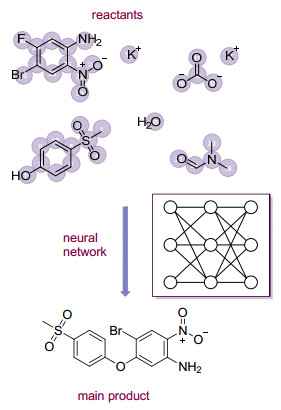
利用图卷积神经网络预测反应物到产物的原子和化学键变化也是一种反应预测的机器学习策略. 2019年, Jensen等[58b]使用分子图表示反应物分子.分子图的节点和边分别描述原子和化学键, 通过图卷积神经网络计算了每个原子对之间化学键变化的可能性, 可能性大的候选产物被组合列举出来并通过另一个图卷积网络重新预测出主要产物的概率分布.他们对来自专利文献中数十万个反应进行训练, 最终准确预测了85%以上的主要产物(图 8).
除了上述的方法, 预测化合物SMILES的序列到序列模型(Sequence-to-sequence model)也被应用于反应预测.基于序列的技术的关键思想是使用反应物、试剂和产物的文本表示(通常是SMILES), 并将反应预测视为从一种语言(反应物-试剂)到另一种语言(产物)的机器翻译问题. 2019年, Schwaller等[93]使用多头注意力(Multihead attention)的Transformer模型, 不需要手动创建规则, 只是通过推断数据集中的反应物、试剂、产物中化学结构之间的相关性来进行预测.他们将SMILES作为输入, 模型在一个通用的基准数据集上排名第一的预测结果精度达到了90.4%.
给定反应物和反应条件, 机器学习策略也能实现反应活性与选择性的预测.早期工作大多使用Hammet-Taft等自由能参数来描述反应特征[94], 成功实现了对α, β-不饱和化合物迈克尔加成反应活性的预测[95].后续也有使用分子拓扑描述符[96]和量子化学描述符[97], 或者综合两种方法[8]研究反应物、反应条件与反应活性之间的关系的报道; 基于分子描述符和位阻参数的多元线性回归模型可以预测反应活性[99]; 也有报道利用反应位点电子转移模式预测化学反应速率[100].采用氢键、位阻以及理论计算得到的振动频率、NPA电荷等作为描述符非常适用于反应选择性预测, 这方面工作已有相关总结[101], 在此不再赘述.由于反应条件对反应活性的影响是一个复杂的问题, 不同的反应在不同条件下的反应机理可能完全不同, 因此很难找到一类通用的方法描述该过程[102].机器学习方法能够从实验人员无法理解的大量数据集里发现反应模式, 这些构效关系的发现有助于发现高效催化剂, 从而快速优化催化反应[59].
近年来, 传统物理有机线性自由能关系被成功应用于手性催化反应定量构效关系(Quantitative structure-reactivity relationships, QSRR)模型, 实现了对一系列手性金属配合物催化反应活性和对映选择性的精准预测, 指导了催化剂的结构优化[103].但这些研究都只针对单一催化体系, 模型的普适性和拓展能力有限. 2019年, Denmark等[42]运用支持向量机和深度前馈神经网络预测了手性磷酸催化的硫醇与酰亚胺的加成反应的选择性.他们选择了5种硫醇底物、5种酰亚胺底物和43种磷酸催化剂生成的1075个实验数据作为训练和验证集.使用基于3D-格点的ASO描述符(Average steric occupancy, 平均空间占有率)来描述分子的空间特征, 并加入取代基的静电势能面最大值(Electrostatic potential energy maximum, ESPMAX)作为电子描述符.在训练集选取方面, 他们通过Kennard-Stone算法抽样选出具有代表性的23种催化剂子集作为训练集UTS (Universal training set), 该方法可以保证训练集特征在整个催化空间的均匀分布.最终模型以较高的精度预测了产物的ee值, MAE在0.995 kJ/mol以内, 并且成功预测了一种最优的大位阻磷酸催化剂(图 9), 显示了该策略在预测新颖催化剂方面的潜力.

尽管随机森林算法已有20多年的发展历史, 但其在当前的深度神经网络时代仍然具有竞争力.近期, 随机森林算法在催化反应预测方面表现不俗. 2018年, Doyle等[104]报道了随机森林模型对Buchwald-Hartwig胺化反应产率的预测.通过高通量实验得到了23种异噁唑添加剂、15种芳基和杂芳基卤化物、4种钯催化剂和3种碱所组成的4608个反应数据(图 10), 并提取了120种原子、分子和振动描述符作为输入.该模型以均方根误差(RMSE)=11.3%, R2=0.83的精度预测了产率.

Sunoj等[105]同样利用随机森林算法实现了不对称催化氢化反应对映选择性的预测.模型数据集包含了58种轴手性联萘配体、190种烯烃和亚胺共368个反应数据, 9种分子参数(如键长、键角、二面角)和22种代表整体分子特性的描述符(如HOMO和LUMO能量、偶极矩)作为模型的参数.与实验值相比, 随机森林算法能准确预测对映选择性, 其RMSE在(8.4±1.8)%范围内.
洪鑫等[106]对杂环自由基C—H官能团化的区域选择性进行了预测.模型的训练中使用了分子特征库, 包含C—C键形成过程的局部原子特性、自由基的整体分子特性以及物理有机化学描述符.通过物理有机特性与随机森林算法相结合, 建立了一个将DFT计算的孤立反应物性质与过渡态能垒联系起来的回归模型, 样本外测试集的预测结果显示位点预测精度为94.2%, 选择性预测精度为89.9%.
不同于上述采用物理化学意义明确的参数或描述符的工作, Glorius等[107]最近发展了一种基于多指纹特征(Multiple fingerprint features, MFFs)的分子表示方法.同样采用随机森林算法, 使用上文Denmark数据集实现了对映选择性的预测, MAE在1.179 kJ/mol以内.该方法还能够预测反应产率和反应性.采用分子指纹方法构建的模型计算成本低、具有一定的普适性, 但在模型的可解释性方面可能需要进一步分析.
传统的初始反应条件选择基本是经验性的或直接参考文献信息.然而, 化学信息的指数增长使人工分析和总结变得极其困难, 需要专门的方法和工具来有效地从原始数据中提取这些知识[108].因此, 发展一种高效、快速的反应条件筛选策略具有重要的科学意义.由于化合物的反应性很大程度上是由反应条件决定的, 因此反应条件的理论评估尤为重要.到目前为止, 已经报道了几种不同的反应条件建模方法. 2015年, Marcou等[109]使用反应压缩图(CGR)衍生片段描述符, 构建了支持向量机、随机森林和朴素贝叶斯分类模型来预测共轭加成反应的溶剂和催化剂的最佳组合.随后, Varnek等[108]基于相似的反应在相似的条件下进行的原理, 运用CGR表示化学反应, 预测的保护基团脱保护反应最佳实验条件具有较高的准确性(约90%).为准确预测适用于大型反应体系的完整反应条件, 2018年, Jensen等[110]发展了一种分层设计的神经网络模型来预测化学环境(催化剂、溶剂、试剂)和反应温度(图 11).该模型对约1000万个来自Reaxys的反应进行了训练, 在训练集以外的100万个反应中进行了测试, 以69.6%的准确率预测了排名前十的反应试剂, 以60%~70%的准确率预测了反应温度(±20 ℃).

未经优化的化学反应在反应时间、试剂方面经常面临低效和成本的问题.优化反应的一种常用方法是一次改变一个实验条件, 同时固定所有其他条件, 该方法常会错过最佳条件; 另一种方法是通过组合化学筛选反应条件的所有组合, 虽然这种方法有更大可能找到全局最优条件, 但是费时费力.因此, 通过机器学习方法构建有效的反应条件优化体系, 对学术研究和工业生产都具有重要意义.溶剂选择作为一个独立的问题在早期得到了广泛的研究[110]. 2013年, Struebing等[111]将基于DFT的速率常数计算与线性回归模型相结合, 对反应溶剂进行优化, 在1341种溶剂的搜索空间内测试了9种溶剂, 将Menschutkin反应的速率常数提高了40%. 2018年, Aspuru-Guzik等[112]开发了名为Phoenics的主动学习算法, 其将贝叶斯优化与贝叶斯核密度估计(Bayesian kernel density estimation)相结合, 实验过程可以按照Phoenics所提出的反应条件执行, 再将实验结果重新输入程序.通过此种反馈机制, Phoenics可以提出一系列实验条件集并最终确定最佳条件集.
计算机辅助合成设计的目的是确定一系列可行的反应步骤, 从而由可获取的原料合成目标产物. Corey等[60]开发的逆合成方法考虑了逆向策略, 即从产物出发, 搜索可能的前体.一些综述介绍了各种逆合成方法和工具的历史[89, 113], 在此不再详细介绍.本文只对当前常用的逆合成策略:基于模板(Template-based)和无模板(Template-free)两种逆合成策略的发展做一简介.
基于模板的逆合成方法将反应规则与目标分子匹配以产生一个或多个候选前体[114].模板可以由专家整理或者从反应数据库中自动提取[115]. CHEMATICA软件[86]整合了超过一万条有机化学家手动编辑的经验反应规则, 该反应规则与启发式策略一起使用, 以确定合成路径.从数据库中自动提取反应规则是当前更主流的方式. 2019年, Law等[116]按照“处理反应数据-建立原子/原子映射-识别反应中心及其外围环境”的流程建立了ARChem合成路线设计系统. 2017年, Coley等[117]表明, 模板可以通过目标分子与数据库中反应产物之间的分子相似性(Molecular similarity)进行排序.
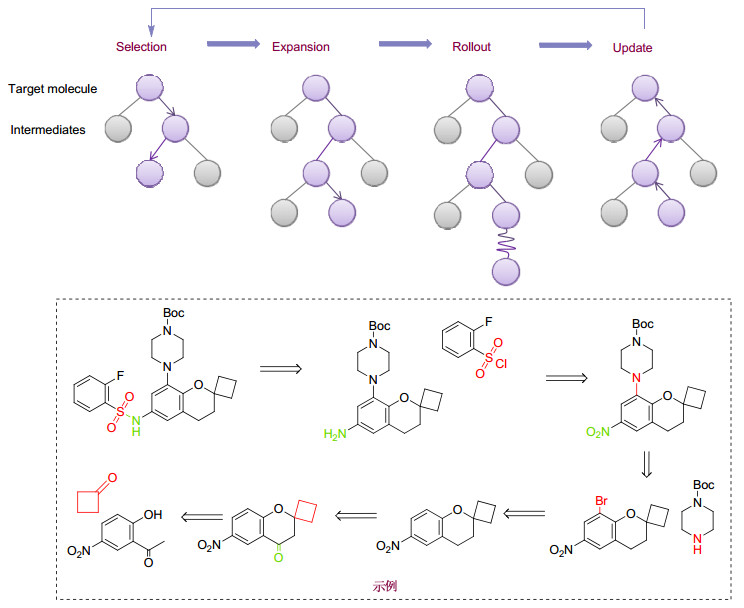
2017年, Waller等[11]运用深度神经网络模型对收集的350万个反应数据进行训练.该模型可以自动提取反应规则, 在近100万个反应的验证集中预测的前十个目标反应的命中率达到了97%.随后, Waller课题组尝试使用蒙特卡洛树搜索和深度神经网络策略, 搜索40种药物类似分子的合成路线.与之前发展的方法相比, 该策略能以更高的精度推测可行的合成路线(与基准预测的87.5%的准确率相比, 该策略的准确率达95%).随后, Waller课题组[11]结合策略网络和蒙特卡洛树搜索对1200万反应数据进行训练(图 12), 结果表明该策略显著加速了逆合成路线预测的速度和准确率.盲测结果表明受试者很难分辨出模型给出的逆合成路线与人类专家设计的逆合成路线.当使用USPTO的5万组反应数据作为测试集时, 模型也能取得较高的预测精度.
无模板的逆合成方法中, 化学结构的所有转变是在没有任何反应规则的情况下进行的, 其中基于深度学习的策略越来越流行. 2017年, Liu等[118]建立了将反应产物SMILES转换为反应物SMILES的序列到序列模型, 该模型以美国专利文献中的50000个实验反应实例为训练样本, 与基于规则的专家系统基准模型的性能相当.最近, 来鲁华和裴剑锋等[119]提出了一种无模板方法, 使用Transformer框架构建了单步逆合成分析模型, 并结合蒙特卡洛树搜索(MCTS)和启发式打分函数建立了逆合成路线规划系统AutoSynRoute(图 13), 该系统成功实现了4种分子的逆合成路线设计.
当前, 前向反应预测主要用于预测反应产物、潜在的副产物和杂质, 而将其与逆合成分析相结合用于反应路径的工作较少.最近, Schwaller等[120]使用前向预测模型[121]和反应分类模型[122]来评估逆合成预测的质量, 提升了单步逆合成模型的性能.
智能合成机器是合成化学研究发展的长期目标, 旨在从根本上改善合成化学的效率和成本, 能够辅助化学家进行重复性、高成本或危险性的实验.随着自动化化学[123]、在线分析[124]和实时优化[125]的发展, 有机合成机器已经初露锋芒, 通过建立机器学习算法控制的反应系统, 可以开发出能够自主探索化学反应性的智能合成装置[12].
2018年, Perera等[126]的工作表明流动装置可以在药物发现过程的早期加速反应优化, 并提供了可靠的数据帮助其他实验室建立机器学习算法.随后, Felpin等[127]提出了一种将监测技术与反馈算法相结合的模块化自动流动反应器.该自动化反应装置通过在线高效液相色谱(HPLC)和在线NMR检测产物, 并根据结果自动优化条件, 通过尝试66种实验组合, 最终通过四步反应, 以67%的产率合成了天然产物Carpanone.
同年, Cronin等[12]发展了一种结合了机器学习、在线核磁、红外、质谱的自动化反应筛选装置.他们的工作表明, 由机器学习算法控制的自动反应处理系统可能比人工处理反应快一个数量级.这种方法能够以结构化的方式寻找失败或无反应性的实验信息.机器学习算法能够对来自辉瑞数据集的1000个钯催化的Suzuki-Miyaura反应组合的反应性进行预测, 在考虑了略多于10%的数据集的结果后, 精度超过80%.为了实现反应装置的模块化和自动化, 他们之后设计了一套基于化学编程语言的自动化合成系统Chemputer, 该系统通过标准的化学语言控制和协调各反应模块, 最终实现化合物的多步合成[128].这一策略是自动化合成与化学反应模块化和标准化的一个很好范例.
2019年, Jamison和Jensen等[129]开发了一种基于人工智能的自动化合成机器人平台(图 14).他们首先开发了合成规划软件ASKCOS, 该软件采用基于模板的逆合成方法, 从USPTO和Reaxys数据库收集的反应数据中提取转换规则, 用于训练前馈神经网络模型, 最终的合成路线选择包括逆向综合规划、反应条件推荐和路径评估, 部分路线包含化学配方文件(Chemical recipe files, CRFs), 需要用户提供额外的信息来确定停留时间、化学计量和浓度.之后, 他们设计了一个模块化的连续流动平台来执行这些文件, 根据CRF中确定的合成路线, 机械臂将模块化过程单元(反应器和分离器)组装成连续流动路径, 实现了15种药物或类药物分子的自动化合成.该平台将CASP与机器流动化学平台相结合, 加速了小分子的自动合成, 但合成路线仍需要人工输入.最近, Gilmore等[130]报道了一种有机分子的自动化径向合成(Radial synthesis)仪器, 该仪器由四部分组成:溶剂和试剂输送系统(Reagent delivery system, RDS)、中央交换站(Central switching station, CSS)、备用模块(Standby module, SM)以及收集容器(Collection vessel, CV), 一系列连续流动模块被径向布置在CSS周围.这种全自动仪器能够进行线性和循环合成, 不同过程之间无需重新手动配置.

(a) Workflow of synthetic planning software. (b) mobile chemical robot platform[129]. Copyright 2019 American Association for the Advancement of Science
当前, 虽然有一些免费开放的化学信息学数据库[131], 但这类数据库数量仍然较少, 数据也不够完善.机器学习在化学领域的发展仍然受数据集规模和质量的限制.同时也需要建立更多标准化学数据库, 方便不同机器学习方法的验证和比较.在图像识别领域, ImageNet数据库[132]的建立推动了该领域快速发展.受ImageNet和WordNet[133]启发, Wu等[134]建立了MoleculeNet数据集, MoleculeNet基于DeepChem开源软件包并提供相关接口, 收集汇总了大量包含量子化学、物理化学、生物物理和生理学信息的数据集, 并发展了处理分子表示和机器学习算法的全套流程, 该数据集将有助于新算法的开发与横向比较.
数据的共享也是当前面临的一个挑战.研究工作的代码和数据集应尽可能公开, 这不仅能方便基准测试, 更能够促进机器学习算法的开发.为推动数据的开放与共享, 开发化学语言标准是十分必要的.化学标记语言(Chemical markup language, CML)是一种代表性的通用化学语言标记格式, 可以表示分子、反应、光谱、分析数据和实验操作流程等信息, 目前在化学专利数据库的标注和数据挖掘领域已有广泛应用[19].
另一个挑战是如何通过文献报告机器学习工作是如何完成的, 以便其他人可以重复[135].机器学习模型参数众多, 程序结构复杂, 数据庞大, 很难通过简单的流程图或文本描述所有细节, 文献和补充材料无法实现有效的参数和代码共享.因此, 有必要开发和采用新的数据和代码共享方法来确保机器学习方法在此领域的可用性.
如果Reaxys和SciFinder等数据库能够开放接口给化学工作者们使用, 同时化学工作者共享最新实验数据, 结合数据的标准化, 将极大推动有机化学领域机器学习的发展.未来, 化学界应该努力打破不同研究机构、高校和组织之间的数据壁垒, 以更加开放的姿态共同推动化学领域的理性化进程.
Norquist等[136]指出, 机器学习程序可以从失败的实验数据中学习并成功用于反应预测, 因而失败的反应数据对于机器学习理解反应非常重要.然而, 记录文献报道的数据库通常只包括较为成功的实验数据.大多数反应预测都是基于成功的反应(例如USPTO和Reaxys数据集)进行训练的, 因而模型很难从这些数据集中获取更多有效信息.此外, 由于识别所有化学物质的时间和成本都很高, 因此反应混合物中副产物的全部表征并不经常公开, 这限制了预测反应性模型的构建[137].
电子实验记录本作为一种有效的实验记录手段, 记录了包括大量成功和失败的反应等的实验室第一手数据.近年来, 越来越多的电子实验记录本被开发出来, 但是由于实验人员的传统思维和对新技术的谨慎, 电子实验记录本除了在有规定性要求的公司中使用以外, 其普及率仍然较低.电子实验记录本的普及使用将有助于改善数据集中失败数据匮乏的问题, 从而推动机器学习整体预测能力的提高.
除此之外, 实验数据的质量与可重复性也是一个挑战.近期发展的反应自动化是一种获取可重复性和标准化数据的有效方法[12, 138], 通过模块化实验装置、标准化操作和标准化学语言, 该策略排除了个体和环境差异, 实验结果的可靠性和重复性更高.此外, 通过将实验和分析数据整合到一个统一的数据平台, 研究者可以方便获取更多高质量和多维度的实验数据, 这将大大促进使用机器学习方法进行数据挖掘和反应预测的发展[139].
如何有效地表示分子结构和化学反应也是机器学习面临的挑战之一.大多数应用于化学反应或性质的机器学习方法使用分子或原子描述符来建立模型, 模型的成功与否取决于这些描述符的有效性和相关性.利用神经网络为反应中的分子创建指纹等进展推动了反应预测的改进[140].提高机器学习方法的性能需要开发新的化学描述符.
机器学习算法面临的一个挑战是如何从较小的数据集中获得更多知识.机器学习方法通常需要大量的数据才能有效地学习.虽然这在图像识别等领域很少是一个问题, 但在化学领域通常仅限于数百或数千个高质量的数据.有限数据集问题的一个有希望的解决方案是元学习, 即在问题内部和问题之间学习[141].诸如神经图灵机[142]和模仿学习[143]等新的发展使这一过程得以实现.最近有报道称, 贝叶斯程序学习(Bayesian program learning, BPL)能够在数据有限的情况下对one-shot分类任务表现出接近人类水平的性能[144].
匹配网络(Matching networks)[145]可以使用辅助数据预先训练, 并引入记忆机制和注意力机制, 从而提高小数据集下的训练表现.类似的方法通常用于药物化学领域, 因为新型靶标的信息和数据较少, 使用传统的机器学习算法很难通过少量数据训练出稳定可靠的模型. 2017年, Altae-Tran等[146]使用长短期记忆(long short-term memory, LSTM)方法实现了小数据集下的模型训练.最近, 记忆增强神经网络(Memory augmented neural networks)通过在传统的神经网络模型里增加记忆模块, 使模型获得外部记忆, 因此可以利用特定领域的先验知识提高学习效率和实现小规模数据集的快速学习[147].
可解释的机器学习模型能够为有机化学领域机制与规律的认识提供启发与补充.如何构建机制明晰的“白箱”模型将是未来一个重要的研究方向.通过机器学习模型的建立再反馈于有机化学的认识, 未来机器学习与有机化学研究将是互相促进、互相依存的关系.
总的来说, 机器学习(特别是深度学习技术)已在有机化学研究中获得了初步的应用, 可以进行分子从头设计、提出切合实际的合成路线、预测给定反应的产物和产率、开发新的选择性催化剂、优化反应条件, 并应用于自动化平台中.现阶段, 为有效地使用机器学习来促进研究进展, 在有机化学领域开展机器学习的使用教学工作是十分必要的.此外, 如何开发开放的大型数据库、获取高质量和标准化的数据、更有效的表示分子和反应、运用和开发适合研究有机化学领域的机器学习算法以及建立有效、通用的算法评价基准, 将是未来机器学习在有机化学领域应用的重要议题.未来机器学习在有机化学研究中的应用会持续增加, 有机化学工作者有必要了解模型背后的理论框架, 在机器学习中引入物理有机的策略和理念, 发展有化学意义的分子描述, 用物理有机的语言连接量子力学和数学算法, 变黑箱模型为白箱模型.可以预期, 以深度机器学习为代表的人工智能技术的引入和贯通应用, 将为有机化学研究的范式变革提供诸多可能.
McCarthy, J.; Minsky, M. L.; Rochester, N.; Shannon, C. E. A Proposal for the Dartmouth Summer Research Project on Artificial Intelligence, August 31, 1955, AI Magazine, 2006, 27, 12.
Jordan, M. I.; Mitchell, T. M. Science 2015, 349, 255. doi: 10.1126/science.aaa8415
Silver, D.; Huang, A.; Maddison, C.; Guez, A.; Sifre, L.; van den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M.; Dieleman, S.; Grewe, D.; Nham, J.; Kalchbrenner, N.; Sutskever, I.; Lillicrap, T.; Leach, M.; Kavukcuoglu, K.; Graepel, T.; Hassabis, D. Nature 2016, 529, 484. doi: 10.1038/nature16961
Skoraczyński, G.; Dittwald, P.; Miasojedow, B.; Szymkuć, S.; Gajewska, E. P.; Grzybowski, B. A.; Gambin, A. Sci. Rep. 2017, 7, 3582. doi: 10.1038/s41598-017-02303-0
Samuel, A. L. IBM J. Res. Dev. 1959, 3, 210. doi: 10.1147/rd.33.0210
Tenenbaum, J. B.; Kemp, C.; Griffiths, T. L.; Goodman, N. D. Science 2011, 331, 1279. doi: 10.1126/science.1192788
(a) Rupp, M. Phys. Rev. Lett. 2012, 108, 058301.
(b) Müller, K.-R. J. Chem. Theory Comput. 2013, 9, 3404.
Brockherde, F.; Vogt, L.; Li, L.; Tuckerman, M. K.; Burke, K.; Müller, K.-R. Nat. Commun. 2017, 8, 872. doi: 10.1038/s41467-017-00839-3
(a) Stokes, J. M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N. M.; MacNair, C. R.; French, S.; Carfrae, L. A.; Bloom-Ackerman, Z.; Tran, V. M.; Chiappino-Pepe, A.; Badran, A. H.; Andrews, L. W.; Chory, E. J.; Church, G. M.; Brown, E. D.; Jaakkola, T. S.; Barzilay, R.; Collins, J. J. Cell 2020, 180, 688.
(b) Li, W.; Yang, J. C.; Huang, N. Acta Pharm. Sin. 2019, 54, 761(in Chinese).
(李伟, 杨金才, 黄牛, 药学学报, 2019, 54, 761.)
(a) Sun, T. L.; Pei, J. F. Chin. Sci. Bull. 2015, 60, 689(in Chinese).
(孙潭霖, 裴剑锋, 科学通报, 2015, 60, 689.)
(b) Gómez-Bombarelli, R.; Wei, J. N.; Duvenaud, D.; Hernández-Lobato, J. M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T. D.; Adams, R. P.; Aspuru-Guzik, A. ACS Cent. Sci. 2018, 4, 268.
(c) Segler, M. H. S.; Kogej, T.; Tyrchan, C.; Waller, M. P. ACS Cent. Sci. 2018, 4, 120.
(a) Segler, M. H. S.; Preuss, M.; Waller, M. P. Nature 2018, 555, 604.
(b) Segler, M. H. S.; Waller, M. P. Chem.-Eur. J. 2017, 23, 5966.
Granda, J. M.; Donina, L.; Dragone, V.; Long, D.-L.; Cronin, L. Nature 2018, 559, 377. doi: 10.1038/s41586-018-0307-8
Warr, W. A. Mol. Inf. 2014, 33, 469. doi: 10.1002/minf.201400052
Blum, L. C.; Reymond, J.-L. J. Am. Chem. Soc. 2009, 131, 8732. doi: 10.1021/ja902302h
Ruddigkeit, L.; van Deursen, R.; Blum, L. C.; Reymond, J.-L. J. Chem. Inf. Model. 2012, 52, 2864. doi: 10.1021/ci300415d
Delaney, J. S. J. Chem. Inf. Comput. Sci. 2004, 44, 1000. doi: 10.1021/ci034243x
Mobley, D. L.; Guthrie, J. P. J. Comput.-Aided Mol. Des. 2014, 28, 711. doi: 10.1007/s10822-014-9747-x
Sterling, T.; Irwin, J. J. J. Chem. Inf. Model. 2015, 55, 2324. doi: 10.1021/acs.jcim.5b00559
(a) Akhondi, S. A.; Klenner, A. G.; Tyrchan, C.; Manchala, A. K.; Boppana, K.; Lowe, D.; Zimmermann, M.; Jagarlapudi, S. A. R. P.; Sayle, R.; Kors, J. A.; Muresan, S. PloS One 2014, 9, el07477.
(b) Southan, C. Drug Discovery Today: Technol. 2015, 14, 3.
(c) Lowe, D. M. PhD. Dissertation, University of Cambridge, Cambridge, 2012.
Manickam, Y.; Chaturvedi, R.; Babbar, P.; Malhotra, N.; Jain, V.; Sharma, A. Drug Discovery Today 2018, 23, 6.
(a) Schneider, N.; Lowe, D. M.; Sayle, R. A.; Landrum, G. A. J. Chem. Inf. Model. 2015, 55, 39.
(b) Schneider, N.; Lowe, D. M.; Sayle, R. A.; Tarselli, M. A.; Landrum, G. A. J. Med. Chem. 2016, 59, 4385.
Rahman, S. A.; Torrance, G.; Baldacci, L.; Cuesta, S. M.; Fenninger, F.; Gopal, N.; Choudhary, S.; May, J. W.; Holliday, G. L.; Steinbeck, C.; Thornton, J. M. Bioinformatics 2016, 32, 2065. doi: 10.1093/bioinformatics/btw096
Cooper, T. W. J.; Campbell, I. B.; Macdonald, S. J. F. Angew. Chem., Int. Ed. 2010, 49, 8082. doi: 10.1002/anie.201002238
Buitrago Santanilla, A.; Regalado, E. L.; Pereira, T.; Shevlin, M.; Bateman, K.; Campeau, L.-C.; Schneeweis, J.; Berritt, S.; Shi, Z.-C.; Nantermet, P.; Liu, Y.; Helmy, R.; Welch, C. J.; Vachal, P.; Davies, I. W.; Cernak, T.; Dreher, S. D. Science 2015, 347, 49. doi: 10.1126/science.1259203
Tetko, I. V.; Engkvist, O.; Chen, H. Future Med. Chem. 2016, 8, 1801. doi: 10.4155/fmc-2016-0163
ChemAxon http://chemaxon.com.
Landrum, G. RDKit: Open-source Cheminformatics, 2014, http://www.rdkit.org.
(a) Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. J. Chem. Inf. Comput. Sci. 2003, 43, 493.
(b) Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E. L. Curr. Pharm. Des. 2006, 12, 2111.
(c) Chemistry Development Kit, 2014, https://cdk.github.io/.
Durant, J. L.; Leland, B. A.; Henry, D. R.; Nourse, J. G. J. Chem. Inf. Comput. Sci. 2002, 42, 1273. doi: 10.1021/ci010132r
(a) Cereto-Massague, A.; Jose Ojeda, M.; Valls, C.; Mulero, M.; Garcia-Vallve, S.; Pujadas, G. Methods 2015, 71, 58.
(b) Muegge, I.; Mukherjee, P. Expert Opin. Drug Discovery 2016, 11, 137.
(a) Bender, A.; Mussa, H. Y.; Glen, R. C.; Reiling, S. J. Chem. Inf. Comput. Sci. 2004, 44, 170.
(b) Bender, A.; Mussa, H. Y.; Glen, R. C.; Reiling, S. J. Chem. Inf. Comput. Sci. 2004, 44, 1708.
Morgan, H. L. J. Chem. Doc. 1965, 5, 107. doi: 10.1021/c160017a018
Rogers, D.; Hahn, M. J. Chem. Inf. Model. 2010, 50, 742. doi: 10.1021/ci100050t
O'Boyle, N. M.; Banck, M.; James, C. A.; Morley, C.; Vandermeersch, T.; Hutchison, G. R. J. Cheminf. 2011, 3, 33. doi: 10.1186/1758-2946-3-33
Indigo-GGA Software Services 2014. https://github.com/ggasoft-ware/indigo.
Weininger, D. J. Chem. Inf. Comput. Sci. 1988, 28, 31. doi: 10.1021/ci00057a005
Heller, S.; McNaught, A.; Stein, S.; Tchekhovskoi, D.; Pletnev, I. J. Cheminf. 2013, 5, 7. doi: 10.1186/1758-2946-5-7
Jeliazkova, N.; Kochev, N. Mol. Inf. 2011, 30, 707. doi: 10.1002/minf.201100028
Raymond, J. W.; Willett, P. J. Comput.-Aided Mol. Des. 2002, 16, 521. doi: 10.1023/A:1021271615909
Rupp, M.; Tkatchenko, A.; Muller, K. R.; von Lilienfeld, O. A. Phys. Rev. Lett. 2012, 108, 058301. doi: 10.1103/PhysRevLett.108.058301
(a) Hochuli, J.; Helbling, A.; Skaist, T.; Ragoza, M.; Koes, D. R. J. Mol. Graphics Modell. 2018, 84, 96.
(b) Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D. R. J. Chem. Inf. Model. 2017, 57, 942.
Zahrt, A. F.; Henle, J. J.; Rose, B. T.; Wang, Y.; Darrow, W. T.; Denmark, S. E. Science 2019, 363, eaau5631. doi: 10.1126/science.aau5631
(a) Sutton, R. S. Mach. Learn. 1988, 3, 9.
(b) François-Lavet, V.; Henderson, P.; Islam, R.; Bellemare, M. G.; Pineau, J. Found. Trends Mach. Learn. 2018, 11, 219.
(a) Koski, T.; Noble, J. Mathematica Applicanda (Matematyka Stosowana) 2012, 40, 51.
(b) Spiegelhalter, D. J. J. R. Statist. Soc. C 1998, 47, 115.
Quinlan, J. R. Mach. Learn. 1986, 1, 81.
(a) Domingos, P.; Pazzani, M. Mach. Learn. 1997, 29, 103.
(b) Webb, G. I.; Boughton, J. R.; Wang, Z. Mach. Learn. 2005, 58, 5.
(c) Maron, M. E. J. ACM 1961, 8, 404.
Cortes, C.; Vapnik, V. Mach. Learn. 1995, 20, 273.
(a) Achtert, E.; Böhm, C.; Kriegel, H.-P.; Kröger, P.; Müller-Gorman, I.; Zimek, A. In Finding Hierarchies of Subspace Clusters, Knowledge Discovery in Databases: PKDD 2006 Series 4213, Springer Berlin Heidelberg, Heidelberg, 2006, pp. 446~453.
(b) Kriegel, H.-P.; Kröger, P.; Zimek, A. WIREs Data Mining Knowl. Discov. 2012, 2, 351.
(c) Sibson, R. Comput. J. 1973, 16, 30.
(d) Banerjee, A.; Dave, R. N. In Validating Clusters using the Hopkins Statistic, 2004 IEEE International Conference on Fuzzy Systems (IEEE Cat. No. 04CH37542), IEEE, Budapest, 2004, pp. 149~153.
(e) Estivill-Castro, V. SIGKDD Explor. Newsl. 2002, 4, 65.
(a) Breiman, L. Mach. Learn. 2001, 45, 5.
(b) Ho, T. K. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 832.
周志华, 机器学习, 清华大学出版社, 北京, 2016.Zhou, Z. H. Machine Learning, Tsinghua University Press, Beijing, 2016 (in Chinese).
Pedregosa, F.; Varoquaux, G.; Gramfort, V.; Michel, B.; Thirion, O.; Grisel, M.; Blondel, P.; Prettenhofer, R.; Weiss, V.; Dubourg, V.; Vanderplas, J.; Passos, A.; Cournapeau, D.; Brucher, M.; Perrot, M.; Duchesnay, E. J. Mach. Learn. Res. 2011, 12, 2825.
(a) Maass, W. Neural Net. 1997, 10, 1659.
(b) Wang, W.; Pedretti, G.; Milo, V.; Carboni, R.; Calderoni, A.; Ramaswamy, N.; Spinelli, A. S.; Ielmini, D. Sci. Adv. 2018, 4, eaat4752.
(c) Tavanaei, A.; Ghodrati, M.; Kheradpisheh, S. R.; Masquelier, T.; Maida, A. Neural Netw. 2019, 111, 47.
McCulloch, W. S.; Pitts, W. Bull Math. Biophys. 1943, 5, 115. doi: 10.1007/BF02478259
Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. J. Mach. Learn. Res. 2014, 15, 1929.
Li, W.; Matthew, Z.; Sixin, Z.; Yann Le, C.; Rob, F. In Proceedings of the 30th International Conference on Machine Learning, Proceedings of Machine Learning Research Series 28, Eds.: Dasgupta, S.; McAllester, D., Proceedings of Machine Learning Research, Atlanta, 2013, pp. 1058~1066.
Nair, V.; Hinton, G. E. In International Conference on Machine Learning, Proceedings of the 27th International Conference on Machine Learning, International Conference on Machine Learning Series 27, International Conference on Machine Learning, Haifa, 2010.
(a) Zhou, J.; Cui, G.; Zhang, Z. Y.; Yang, C.; Liu, Z. Y.; Wang, L. F.; Li, C. C.; Sun, M. arXiv e-prints 2018, arXiv: 1812.08434.
(b) Zhang, Z. W.; Cui, P.; Zhu, W. W. IEEE Trans. Knowl. Data Eng. 2020, doi: 10.1109/TKDE.2020.2981333.
(a) Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gómez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R. P. In Advances in Neural Information Processing Systems 28, Neural Information Processing Systems 2015, Eds.: Cortes, C.; Lawrence, N.; Lee D.; Sugiyama, M.; Garnett, R., Neural Information Processing Systems, 2015, pp. 2215~2223.
(b) Coley, C. W.; Jin, W.; Rogers, L.; Jamison, T. F.; Jaakkola, T. S.; Green, W. H.; Barzilay, R.; Jensen, K. F. Chem. Sci. 2019, 10, 370.
Mitchell, J. B. O. Wires Comput. Mol. Sci. 2014, 4, 468. doi: 10.1002/wcms.1183
Corey, E. J.; Wipke, W. T. Science 1969, 166, 178. doi: 10.1126/science.166.3902.178
Muratov, E. N.; Bajorath, J.; Sheridan, R. P.; Tetko, I. V.; Filimonov, D.; Poroikov, V.; Oprea, T. I.; Baskin, I. I.; Varnek, A.; Roitberg, A.; Isayev, O.; Curtalolo, S.; Fourches, D.; Cohen, Y.; Aspuru-Guzik, A.; Winkler, D. A.; Agrafiotis, D.; Cherkasov, A.; Tropsha, A. Chem. Soc. Rev. 2020, 49, 3525. doi: 10.1039/D0CS00098A
Ma, J.; Sheridan, R. P.; Liaw, A.; Dahl, G. E.; Svetnik, V. J. Chem. Inf. Model. 2015, 55, 263. doi: 10.1021/ci500747n
Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. Front. Environ. Sci. 2016, 3, 80.
(a) Ramsundar, B.; Liu, B.; Wu, Z.; Verras, A.; Tudor, M.; Sheridan, R. P.; Pande, V. J. Chem. Inf. Model. 2017, 57, 2068.
(b) Koutsoukas, A.; Monaghan, K. J.; Li, X.; Huan, J. J. Cheminf. 2017, 9, 42.
(c) Lenselink, E. B.; ten Dijke, N.; Bongers, B.; Papadatos, G.; van Vlijmen, H. W. T.; Kowalczyk, W.; Ijzerman, A. P.; van Westen, G. J. P. J. Cheminf. 2017, 9, 45.
(a) Subramanian, G.; Ramsundar, B.; Pande, V.; Denny, R. A. J. Chem. Inf. Model. 2016, 56, 1936.
(b) Aliper, A.; Plis, S.; Artemov, A.; Ulloa, A.; Mamoshina, P.; Zhavoronkov, A. Mol. Pharm. 2016, 13, 2524.
(c) Lusci, A.; Pollastri, G.; Baldi, P. J. Chem. Inf. Model. 2013, 53, 1563.
(d) Xu, Y.; Dai, Z.; Chen, F.; Gao, S.; Pei, J.; Lai, L. J. Chem. Inf. Model. 2015, 55, 208.
Ryu, S.; Kwon, Y.; Kim, W. Y. Chem. Sci. 2019, 10, 8438. doi: 10.1039/C9SC01992H
Gaulton, A.; Hersey, A.; Nowotka, M.; Patrícia Bento, A.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L. J.; Cibrián-Uhalte, E.; Davies, M.; Dedman, N.; Karlsson, A.; Magariños, M. P.; Overington, J. P.; Papadatos, G.; Smit, I.; Leach, A. L. Nucleic Acids Res. 2017, 45, 945. doi: 10.1093/nar/gkw1074
(a) Fraczkiewicz, R.; Lobell, M.; Göller, A. H.; Krenz, U.; Schoenneis, R.; Clark, R. D.; Hillisch. A. J. Chem. Inf. Model. 2015, 55, 389.
(b) Fraczkiewicz, R.; Lobell, M.; Göller, A. H.; Krenz, U.; Schoenneis, R.; Clark, R. D.; Hillisch, A. J. Chem. Inf. Model. 2015, 55, 389.
Roszak, R.; Beker, W.; Molga, K.; Grzybowski, B. A. J. Am. Chem. Soc. 2019, 141, 17142. doi: 10.1021/jacs.9b05895
Yang, Q.; Li, Y.; Yang, J.-D.; Liu, Y. D.; Zhang, L.; Luo, S. Z.; Cheng, J.-P. Angew. Chem., Int. Ed. 2020, 59, 19282. doi: 10.1002/anie.202008528
Yang, J.-D.; Xue, X.-S.; Ji, P.; Li, X.; Cheng, J.-P. Internet Bond-energy Databank (pKa and BDE): iBonD Home Page, http://ibond.chem.tsinghua.edu.cn or http://ibond.nankai.edu.cn.
Hall, L. H. J. Chem. Inf. Comput. Sci. 1995, 35, 1039. doi: 10.1021/ci00028a014
Feng, C.; Sharman, E.; Ye, S.; Luo, Y.; Jiang, J. Sci. China:Chem. 2019, 62, 1698. doi: 10.1007/s11426-019-9619-8
Gilmer, J.; Schoenholz, S. S.; Riley, P. F.; Vinyals, O.; Dahl, G. E. Proceedings of the 34th International Conference on Machine Learning, In Proceedings of Machine Learning Research Series 70, Eds.: Precup, D.; Teh, Y. W., Proceedings of Machine Learning Research, Sydney, 2017, pp. 1263~1272.
Ertl, P.; Lewis, R.; Martin, E.; Polyakov, V. arXiv e-prints 2017, arXiv: 1712.07449.
梁礼, 邓成龙, 张艳敏, 滑艺, 刘海春, 陆涛, 陈亚东, 药学进展, 2020, 44, 18.Liang, L.; Deng, C. L.; Zhang, Y. M.; Hua, Y.; Liu, H. C.; Lu, T.; Chen, Y. D. Prog. Pharm. Sci. 2020, 44, 18(in Chinese).
Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. Mol. Pharm. 2017, 14, 3098. doi: 10.1021/acs.molpharmaceut.7b00346
Goodfellow, I. arXiv e-prints 2016, arXiv: 1701.00160.
Zhavoronkov, A.; Ivanenkov, Y. A.; Aliper, A.; Veselov, M. S.; Aladinskiy, V. A.; Aladinskaya, A. V.; Terentiev, V. A.; Polykovskiy, D. A.; Kuznetsov, M. D.; Asadulaev, A.; Volkov, Y.; Zholus, A.; Shayakhmetov, R. R.; Zhebrak, A.; Minaeva, L. I.; Zagribelnyy, B. A.; Lee, L. H.; Soll, R.; Madge, D.; Xing, L.; Guo, T.; Aspuru-Guzik, A. Nat Biotechnol. 2019, 37, 1038. doi: 10.1038/s41587-019-0224-x
Yu, L. T.; Zhang, W. N.; Wang, J.; Yu, Y. arXiv e-prints 2016, arXiv: 1609.05473.
Jaques, N.; Gu, S.; Bahdanau, D.; Hernández-Lobato, J. M.; Turner, R. E.; Eck, D. arXiv e-prints 2016, arXiv: 1611.02796.
Leo, A.; Hansch, C.; Elkins, D. Chem. Rev. 1971, 71, 525. doi: 10.1021/cr60274a001
Bickerton, G. R.; Paolini, G. V.; Besnard, J.; Muresan, S.; Hopkins, A. L. Nat. Chem. 2012, 4, 90. doi: 10.1038/nchem.1243
Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. J. Cheminf. 2017, 9, 48. doi: 10.1186/s13321-017-0235-x
Benhenda, M. arXiv e-prints 2017, arXiv: 1708.08227.
(a) Metz, L.; Poole, B.; Pfau, D.; Sohl-Dickstein, J. arXiv e-prints 2016, arXiv: 1611.02163.
(b) Unterthiner, T.; Nessler, B.; Seward, C.; Klambauer, G.; Heusel, M.; Ramsauer, H.; Hochreiter, S. arXiv e-prints 2017, arXiv: 1708.08819.
Corey, E. J.; Wipke, W. T.; Cramer, R. D.; Howe, W. J. J. Am. Chem. Soc. 1972, 94, 431. doi: 10.1021/ja00757a021
Hendrickson, J. B. Recl. Trav. Chim. Pays-Bas. 1992, 111, 323. doi: 10.1002/recl.19921110611
(a) Todd, M. H. Chem. Soc. Rev. 2005, 34, 247.
(b) Szymkuć, S.; Gajewska, E. P.; Klucznik, T.; Molga, K.; Dittwald, P.; Startek, M.; Bajczyk, M.; Grzybowski, B. A. Angew. Chem., Int. Ed. 2016, 55, 5904.
de Almeida, A. F.; Moreira, R.; Rodrigues, T. Nat. Rev. Chem. 2019, 3, 589. doi: 10.1038/s41570-019-0124-0
Kayala, M. A.; Azencott, C.-A.; Chen, J. H.; Baldi, P. J. Chem. Inf. Model. 2011, 51, 2209. doi: 10.1021/ci200207y
Kayala, M. A.; Baldi, P. J. Chem. Inf. Model. 2012, 52, 2526. doi: 10.1021/ci3003039
Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C. A.; Bekas, C.; Lee, A. A. ACS Cent. Sci. 2019, 5, 1572. doi: 10.1021/acscentsci.9b00576
McDaniel, D. H.; Brown, H. C. J. Org. Chem. 1958, 23, 420. doi: 10.1021/jo01097a026
(a) Friedman, M.; Wall, J. S. J. Am. Chem. Soc. 1964, 86, 3735.
(b) Friedman, M.; Cavins, J. F.; Wall, J. S. J. Am. Chem. Soc. 1965, 87, 3672.
(c) Friedman, M.; Wall, J. S. J. Org. Chem. 1966, 31, 2888.
Toropov, A. A.; Kudyshkin, V. O.; Voropaeva, N. L.; Ruban, I. N.; Rashidova, S. S. J. Struct. Chem. 2004, 45, 945. doi: 10.1007/s10947-005-0084-8
Yu, X.; Yi, B.; Wang, X. Eur. Polym. J. 2008, 44, 3997. doi: 10.1016/j.eurpolymj.2008.09.028
Morrill, J. A.; Biggs, J. H.; Bowman, C. N.; Stansbury, J. W. J. Mol. Graphics Modell. 2011, 29, 763. doi: 10.1016/j.jmgm.2010.12.009
Schwöbel, J. A. H.; Wondrousch, D.; Koleva, Y. K.; Madden, J. C.; Cronin, M. T. D.; Schüürmann, G. Chem. Res. Toxicol. 2010, 23, 1576. doi: 10.1021/tx100172x
Wondrousch, D.; Böhme, A.; Thaens, D.; Ost, N.; Schüürmann, G. J. Phys. Chem. Lett. 2010, 1, 1605. doi: 10.1021/jz100247x
(a) Zhang, L.; Li, X.; Luo, S. Z.; Cheng, J.-P. Sci. Sin.: Chim. 2016, 46, 535(in Chinese).
(张龙, 李鑫, 罗三中, 程津培, 中国科学: 化学, 2016, 46, 535.)
(b) Li, Y.; Luo, S. Z. Chin. J. Org. Chem. 2018, 38, 2363(in Chinese).
(李遥, 罗三中, 有机化学, 2018, 38, 2363.)
Halberstam, N. M.; Baskin, I. I.; Palyulin, V. A.; Zefirov, N. S. Mendeleev Commun. 2002, 12, 185. doi: 10.1070/MC2002v012n05ABEH001620
Harper, K. C.; Bess, E. N.; Sigman, M. S. Nat. Chem. 2012, 4, 366. doi: 10.1038/nchem.1297
Ahneman, D. T.; Estrada, J. G.; Lin, S.; Dreher, S. D.; Doyle, A. G. Science 2018, 360, 186. doi: 10.1126/science.aar5169
Singh, S.; Pareek, M.; Changotra, A.; Banerjee, S.; Bhaskararao, B.; Balamurugan, P.; Sunoj, R. B. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 1339. doi: 10.1073/pnas.1916392117
Li, X.; Zhang, S.-Q.; Xu, L.-C.; Hong. X. Angew. Chem., Int. Ed. 2020, 59, 13253. doi: 10.1002/anie.202000959
Sandfort, F.; Strieth-Kalthoff, F.; Kuhnemund, M.; Beecks, C.; Glorius, F. Chem 2020, 6, 1. doi: 10.1016/j.chempr.2019.12.023
Lin, A. I.; Madzhidov, T. I.; Klimchuk, O.; Nugmanov, R. I.; Antipin, I. S.; Varnek, A. J. Chem. Inf. Model. 2016, 56, 2140. doi: 10.1021/acs.jcim.6b00319
Marcou, G.; Aires, de Sousa. J.; Latino, D. A. R. S.; de Luca, A.; Horvath, D.; Rietsch, V.; Varnek, A. J. Chem Inf. Model. 2015, 55, 239. doi: 10.1021/ci500698a
Gao, H.; Struble, T. J.; Coley, C. W.; Wang, Y.; Green, W. H.; Jensen, K. F. ACS Cent. Sci. 2018, 4, 1465. doi: 10.1021/acscentsci.8b00357
Struebing, H.; Ganase, Z.; Karamertzanis, P.; Siougkrou, E.; Haycock, P.; Piccione, P. M.; Armstrong, A.; Galindo, A.; Adjiman, C. S. Nat. Chem. 2013, 5, 952. doi: 10.1038/nchem.1755
Häse, F.; Roch, L. M.; Kreisbeck, C.; Aspuru-Guzik, A. ACS Cent. Sci. 2018, 4, 1134. doi: 10.1021/acscentsci.8b00307
(a) Baskin, I. I.; Madzhidov, T. I.; Antipin, I. S.; Varnek, A. A. Russ. Chem. Rev. 2017, 86, 1127.
(b) Cook, A.; Johnson, A. P.; Law, J.; Mirzazadeh, M.; Ravitz, O.; Simon, A. Science 2012, 2, 79.
(c) Zefirov, N. S.; Gordeeva, E. V. Russ. Chem. Rev. 1987, 56, 1002.
Coley, C. W.; Green, W. H.; Jensen, K. F. Acc. Chem. Res. 2018, 51, 1281. doi: 10.1021/acs.accounts.8b00087
Varnek, V.; Baskin, I. I. In Systems Medicine, Vol. 2, Eds.: Wolkenhauer, O., Academic Press, Oxford, 2021, pp. 190~197.
Law, J.; Zsoldos, Z.; Simon, A.; Reid, D.; Liu, Y.; Khew, S. Y.; Johnson, A. P.; Major, S.; Wade, R. A.; Ando, H. Y. J. Chem. Inf. Model. 2019, 49, 593.
Coley, C. W.; Rogers, L.; Green, W. H.; Jensen, K. F. ACS Cent. Sci. 2017, 3, 1237. doi: 10.1021/acscentsci.7b00355
Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Luu Nguyen, Q.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. ACS Cent. Sci. 2017, 3, 1103. doi: 10.1021/acscentsci.7b00303
Lin, L. J.; Xu, Y. J.; Pei, J. F.; Lai, L, H. Chem. Sci. 2020, 11, 3355. doi: 10.1039/C9SC03666K
Schwaller, P.; Petraglia, R.; Zullo, V.; Nair, V. H.; Haeuselmann, R. A.; Pisoni, R.; Bekas, C.; Iuliano, A.; Laino, T. Chem. Sci. 2020, 11, 3316. doi: 10.1039/C9SC05704H
(a) Segler, M. H. S.; Preuss, M.; Waller, M. P. Nature 2018, 555, 604.
(b) Satoh, H.; Funatsu, K. J. Chem. Inf. Comput. Sci. 1995, 35, 34.
Schwaller, P.; Vaucher, A.; Nair, V. H.; Laino, T.; Reymond, J.-L. ChemRxiv Preprint 2019, https://doi.org/10.26434/chemrxiv.9897365.v2. doi: 10.26434/chemrxiv.9897365.v2
(a) Trobe, M.; Burke, M. D. Angew. Chem., Int. Ed. 2018, 57, 4192.
(b) Ley, S. V.; Fitzpatrick, D. E.; Ingham, R. J.; Myers, R. M. Angew. Chem., Int. Ed. 2015, 54, 3449.
Sans, V.; Cronin, L. Chem. Soc. Rev. 2016, 45, 2032. doi: 10.1039/C5CS00793C
Houben, C.; Lapkin, A. A. Curr. Opin. Chem. Eng. 2015, 9, 1. doi: 10.1016/j.coche.2015.07.001
Perera, D.; Tucker, J. W.; Brahmbhatt, S.; Helal, C. J.; Chong, A.; Farrell, W.; Richardson, P.; Sach, N. W. Science 2018, 359, 429. doi: 10.1126/science.aap9112
Cortés-Borda, D.; Wimmer, E.; Gouilleux, B.; Barré, E.; Oger, N.; Goulamaly, L.; Peault, L.; Charrier, B.; Truchet, C.; Giraudeau, P.; Rodriguez-Zubiri, M.; Le Grognec, E.; Felpin, F.-X. J. Org. Chem. 2018, 83, 14286. doi: 10.1021/acs.joc.8b01821
Steiner, S.; Wolf, J.; Glatze, S.; Andreou, A.; Granda, J. M.; Keenan, G.; Hinkley, T.; Aragon-Camarasa, G.; Kitson, P. J.; Angelone, D.; Cronin, L. Science 2019, 363, eaav2211. doi: 10.1126/science.aav2211
Coley, C. W.; Thomas D. A.; Lummiss, J. A. M.; Jaworski, J. N.; Breen, C. P.; Schultz, V.; Hart, T.; Fishman, J. S.; Rogers, L.; Gao, H.; Hicklin, R. W.; Plehiers, P. P.; Byington, J.; Piotti, J. S.; Green, W. H.; Hart, A. J.; Jamison, T. F.; Jensen, K, F. Science 2019, 365, eaax1566. doi: 10.1126/science.aax1566
Chatterjee, S.; Guidi, M.; Seeberger, P. H.; Gilmore. K. Nature 2020, 579, 379. doi: 10.1038/s41586-020-2083-5
(a) Mysinger, M. M.; Carchia, M.; Irwin, J. J.; Shoichet, B. K. J. Med. Chem. 2012, 55, 6582.
(b) Sun, J.; Jeliazkova, N.; Chupakhin, V.; Golib-Dzib, J.-F.; Engkvist, O.; Carlsson, L.; Wegner, J.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V.; Kochev, N.; Ashby, T. J.; Chen, H. J. Cheminf. 2017, 9, 17.
Russakovsky, O.; Deng, J.; Su, H.; Krause, J.; Satheesh, S.; Ma, S.; Huang, Z.; Karpathy, A.; Khosla, A.; Bernstein, M.; Berg, A. C.; Li, F.-F. Int. J. Comput. Vision 2015, 115, 211. doi: 10.1007/s11263-015-0816-y
Miller, G. A. Commun. ACM 1995, 38, 39.
Wu, Z.; Ramsundar, B.; Feinberg, Evan N.; Gomes, J.; Geniesse, C.; Pappu, A. S.; Leswing, K.; Pande, V. Chem. Sci. 2018, 9, 513. doi: 10.1039/C7SC02664A
Kitchin, J. R. Nat. Cat. 2018, 1, 230. doi: 10.1038/s41929-018-0056-y
Raccuglia, P.; Elbert, K. C.; Adler, P. D. F.; Falk, C.; Wenny, M. B.; Mollo, A.; Zeller, M.; Friedler, S. A.; Schrier, J.; Norquist, A. J. Nature 2016, 533, 73. doi: 10.1038/nature17439
Struble, T. J.; Alvarez, J. C.; Brown, S. P.; Chytil, M.; Cisar, J.; DesJarlais, R. L.; Engkvist, O, ; Frank, S. A.; Greve, D. R.; Griffin, D. J.; Hou, X. J.; Johannes, J. W.; Kreatsoulas, C.; Lahue, B.; Mathea, M.; Mogk, G.; Nicolaou, C. A.; Palmer, A. D.; Price, D. J.; Robinson, R. I.; Salentin, S.; Xing, L.; Jaakkola, T.; Green, W. H.; Barzilay, R.; Coley, C. W.; Jensen, K. F. J. Med. Chem. 2020, 63, 8667. doi: 10.1021/acs.jmedchem.9b02120
Caramelli, D.; Salley, D.; Henson, A.; Camarasa, G. A.; Sharabi, S.; Keenan, G.; Cronin, L. Nat. Commun. 2018, 9, 3406. doi: 10.1038/s41467-018-05828-8
(a) Goodell, J. R.; McMullen, J. P.; Zaborenko, N.; Maloney, J. R.; Ho, C.-X.; Jensen, K. F.; Porco, J. A. Jr.; Beeler, A. B. J. Org. Chem. 2009, 74, 6169.
(b) Heublein, N.; Moore, J. S.; Smith, C. D.; Jensen, K. F. RSV Adv. 2014, 4, 63627.
(c) Weber, A.; von Roedern, E.; Stilz, H. U. J. Comb. Chem. 2005, 7, 178.
Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gómez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, A. P. In Advances in Neural Information Processing Systems 28, Neural Information Processing Systems 2015, Eds.: Cortes, C.; Lawrence, N.; Lee D.; Sugiyama, M.; Garnett, R., Neural Information Processing Systems, 2015, pp. 2224~2232.
Jankowski, N., Duch, W.; Gra̧bczewski, K. Meta-Learning in Computational Intelligence, Springer, Berlin, 2011.
Graves, A.; Wayne, G.; Danihelka, I. arXiv e-prints 2014, arXiv: 1410.5401.
Duan, Y.; Andrychowicz, M.; Stadie, B. C.; Ho, J.; Schneider, J.; Sutskever, I.; Abbeel, P.; Zaremba W. In Advances in Neural Information Processing Systems 30, Neural Information Processing Systems 2017, Eds.: Guyon, I.; Luxburg, U. V.; Bengio, S.; Wallach, H.; Fergus, R.; Vishwanathan, S.; Garnett, R., Neural Information Processing Systems, 2017, pp. 1087~1098.
Lake, B. M.; Salakhutdinov, R.; Tenenbaum, J. B. Science 2015, 350, 1332. doi: 10.1126/science.aab3050
Vinyals, O.; Blundell, C.; Lillicrap, T.; Kavukcuoglu, K.; Wierstra, D. In Advances in Neural Information Processing Systems 29, Neural Information Processing Systems 2016, Eds.: Lee, D.; Sugiyama, M.; Luxburg, U.; Guyon, I.; Garnett, R., Neural Information Processing Systems, 2016, pp. 3630~3638.
Altae-Tran, H.; Ramsundar, B.; Pappu, A. S.; Pande, V. ACS Cent. Sci. 2017, 3, 283. doi: 10.1021/acscentsci.6b00367
(a) Santoro, A.; Bartunov, S.; Botvinick, M.; Wierstra, D.; Lillicrap, T. In Proceedings of The 33rd International Conference on Machine Learning, Proceedings of Machine Learning Research Series 48, Eds.: Balcan, M. F.; Weinberger, K. Q., Proceedings of Machine Learning Research, New York, 2016, pp. 1842~1850.
(b) Ha, H.; Hwang, U.; Hong, Y.; Yoon, S. arXiv e-prints 2018. arXiv: 1805.10768.

图 2 机器学习应用于有机化学的工作流程
Figure 2 Workflow of applying machine learning in organic chemistry

图 9 预测高选择性催化剂的工作流程
Figure 9 Workflow of the prediction of higher-selectivity catalysts

图 10 钯催化的Buchwald-Hartwig反应产率预测
Figure 10 Prediction of yield in palladium catalyzed Buchwald-Hartwig reaction

图 11 分层设计的神经网络模型预测反应条件
Figure 11 Neural network model of layered design for predicting reaction conditions

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





 下载:
下载:









 下载:
下载: