
图 1.
铁载体介导的铁转运系统及“特洛伊木马”策略[8]
Figure 1.
Siderophore-mediated iron acquisition and its exploitation by "Trojan horses"

20世纪40年代, 抗生素被视为“神奇药物”[1-5], 它优异的杀菌功效和较强的选择性一度使人们认为感染性疾病将成为过去.但是, 随着抗生素的广泛使用, 由于细菌产生耐药性而导致的感染治疗失败, 对人类健康和生存构成了重大威胁[5], 因此临床上迫切需要新型抗生素和抗菌策略来解决这一问题.
20世纪80年代, Miller团队提出“特洛伊木马”分子抗生素策略, 将抗生素与天然铁载体或合成铁载体分子偶联, 利用细菌自身的铁载体转运系统靶向将抗生素递送到抗菌作用靶点[6].这种策略的目的是促进抗生素进入细菌细胞, 从而增加其抗菌活性或扩大其抗菌谱.铁是几乎所有生命形式的重要营养素, 在所有生物中都起着重要作用.大多数的生物都需要铁, 它不仅是氧化还原酶等重要生物酶的辅助因子, 也是参与氧气运输、电子传递、能量产生和其它重要代谢过程的生物必需元素[7].在生理pH (7.35~7.40)条件下, 人血清中的铁以Fe3+形式存在, 约为10-24 mol/L, 而每个细菌细胞分裂需要约10-5~10-6 mol/L Fe3+(细胞内至少维持10-6 mol/L)浓度.为了在感染期间获得足够的铁来维持自身生长并完成各种生理活动, 细菌进化出了一系列能够摄取铁的系统, 通过合成一系列小的有机化合物铁载体(分子量在200~2000之间), 并将其分泌到胞外, 利用主动转运系统从胞外获取各种形式的铁来维持生命活动[8](图 1).
已经发现的天然铁载体-抗生素偶联物有链霉菌和放线菌产生的阿波霉素、铁霉素和沙霉素等[9].阿波霉素和沙霉素是研究较多的两类天然铁载体-抗生素偶联物, 已经证明它们可使用特定的铁转运系统进入目标细菌[10].阿波霉素(1)主要由具有铁螯合能力的三异羟肟酸部分和抗菌的硫代核糖嘧啶部分(图 2)组成, 在阴性菌大肠杆菌内, 阿波霉素通过TonB依赖性转运蛋白FhuA[11](外膜识别)、FhuD[12] (周质)、FhuB(细胞质膜)和FhuC(一种ATPase, 为跨内膜转运提供能量)主动转运至细胞质中.一旦阿波霉素进入细胞质, 就会被一种肽酶裂解, 在细胞质中释放抗菌成分硫代核糖嘧啶, 通过抑制氨酰基-tRNA合成酶以阻断蛋白质合成发挥抗菌作用.沙霉素(2)(图 2)由三异羟肟酸酯和氨基糖苷两个部分组成, 沙霉素可以通过铁还原来引发分子内环化作用释放其氨基糖苷部分来抑制蛋白质合成发挥抗菌作用[13].这些天然铁载体-抗生素偶联物, 通过借助铁离子吸收系统进入到细菌体内, 进而将药物分子传递到胞内作用靶点来发挥抗菌作用.这些天然铁载体-抗生素偶联物的发现鼓舞科研人员利用“特洛伊木马”分子抗生素策略来设计新型具有抗菌作用的化合物. 2019年11月14日, 美国食品药品监督管理局(FDA)批准了一个铁载体分子偶联抗生素药物头孢地尔(Cefiderocol)上市, 进一步验证了“特洛伊木马”分子抗生素策略可以作为一种靶向传递抗生素分子来解决细菌耐药问题的可行性[14].但在已有的文献中, 偶联物发挥抗菌作用有成功的例子也有失败的例子, 什么样的抗生素与什么样的铁载体分子偶联可以达到“特洛伊木马”策略的效果?还没有此类药物分子设计的指引.在本文中, 我们将详细总结分析已有文献中不同作用机制抗生素与铁载体分子偶联对抗菌活性的影响, 试图找出一些规律, 为新型铁载体分子偶联抗生素药物研发提供参考.
迄今为止, 已经有超过500种铁载体在细菌、真菌、植物中被发现[18].铁载体通常被细菌合成并释放到环境中, 可以极高的亲和力螯合Fe3+, Fe3+-铁载体螯合物可被细菌的特定膜蛋白受体识别, 从而将螯合物转运至细菌内部.一旦螯合物进入到细菌内的还原性细胞质中, Fe3+就会被还原为Fe2+, 导致铁载体失去亲和力, 随后释放的Fe2+被细菌利用[19].铁载体根据来源主要分为两大类, 天然铁载体和人工合成类铁载体.天然铁载体是由细菌、真菌或植物分泌出的铁载体[9, 20], 例如革兰氏阳性菌金黄色葡萄球菌(Staphylococcus aureus)分泌的葡萄铁蛋白[21](Staphyloferrin, 3)(图 3)、革兰氏阴性菌的大肠杆菌(E. coli)分泌的肠杆菌素[22](Enterobactin, 4)(图 3)、铜绿假单胞菌(Pseudomonas aeruginosa)分泌的荧光嗜铁素(Pyoverdine, 5)和螯铁蛋白[23](Pyochelin, 6)(图 3), 还有从果蝇红酵母菌[24] (Rhodotorula pilimanae)中提取出来的红酵母酸(Rhodotorulic acid, 7)(图 3)和镰刀菌(Fusarium roseum)产生的一种线型Fusarinine B (8)[25]铁载体(图 3)等.这些天然铁载体以极高的亲和力与三价铁离子结合形成螯合物, 被相应的受体识别并转运进入胞内, 为生物提供铁离子, 从而提供竞争性的生长优势.
天然铁载体具有高度多样化的结构, 根据铁螯合功能基团的化学性质, 天然铁载体可以分为三大类:异羟肟酸型、儿茶酚型和α-羟基羧酸型[26-27].异羟肟酸型(9)(图 4)是自然界中最普遍存在的铁载体, 大部分由细菌真菌等微生物产生.其N-羟基和羰基氧原子作为Fe3+的配体, 与Fe3+的结合常数大约在1022~1032之间, 这种高亲和力的结合可以保护螯合物免受水解和酶促的降解.儿茶酚型(10)(图 4)则一般由一些细菌产生, 因为它们具有邻苯二酚结构, 所以苯环上相邻的两个羟基能够作为Fe3+的配体[28]; α-羟基羧酸型(11)铁载体(图 4)则通常由细菌和真菌(毛霉菌)产生, 通过羧基和羟基与Fe3+结合.合成类铁载体的最常见的结构主要也是以异羟肟酸、儿茶酚和α-羟基羧酸, 以及类儿茶酚结构的羟基吡啶酮(12)为主要骨架[29-31], 这些基团均可以与Fe3+形成六齿配位的稳定结构.这些天然及合成类铁载体借助铁特异性摄取系统, 可以促使与其连接的抗生素靶向输送至病原体特定作用靶点, 有助于抗生素跨过细菌细胞膜.对于具有复杂结构细胞壁的革兰氏阴性病原体, 抗生素可以借助铁载体的转运, 克服外膜通透性降低而产生的细菌耐药性.
作用于细菌细胞壁的抗生素-铁载体类偶联物的药物研发可追溯到20世纪80年代, Benz和同事们[32]发现阿波霉素中的N5-乙酰基-N5-羟基-L-鸟氨酸基团是细菌参与铁转运系统铁载体的必要基团. 1990年, Miller课题组[32]以N5-乙酰基-N5-羟基-L-鸟氨酸作为铁载体与第一代β-内酰胺类头孢菌素氯碳头孢(loracarbef)偶联得到偶联物13和14(图 5), 来探究β-内酰胺类抗生素偶联物的抗菌活性.初步活性结果表明, 偶联物13和14在1和10 μmol/L时均有效地抑制了大肠杆菌的生长, 进一步证实β-内酰胺类抗生素有望作为“特洛伊木马”分子策略中的抗生素进行深入研究.遗憾的是, 偶联物13、14相比母体抗生素氯碳头孢的抗菌活性没有显著提升.随后, Miller课题组[33-34]进一步探究了不同类型铁载体, 异羟肟酸和儿茶酚铁载体与氯碳头孢偶联物(15, 16)(图 5)的抗菌活性.初步活性结果表明, 异羟肟酸酯铁载体偶联物15和儿茶酚铁载体偶联物16可以分别利用不同的外膜受体蛋白Fhu和Cir进入周质释放氯碳头孢抗生素, 并与细菌内青霉素结合蛋白(PBPs)直接相互作用[35], 发挥优秀的大肠杆菌抗菌活性.但遗憾的是, 这些偶联物分别只能作用于具有Fhu和Cir膜蛋白受体的大肠杆菌来发挥抗菌活性, 对于Fhu和Cir膜蛋白缺陷型受体的细菌菌株无效, 其无法借助铁载体铁转运系统进入到细菌细胞提高杀菌活性.所以, Miller课题组[36]进一步设计并合成一种以氯碳头孢作为抗菌成分, 结构中具有儿茶酚和单异羟肟酸酯的混合型铁载体偶联物17(图 5)来提升偶联物的抗菌活性.初步活性探究发现, 17可以利用大肠杆菌中多种铁载体的运输途径来发挥抗菌活性, 不仅对大肠杆菌株有效, 对几种膜受体突变类型的菌株也均有显著的抗菌活性.此外, 偶联物17对耐甲氧西林金黄色葡萄球菌(MRSA)[37]的最小抑菌浓度(Minimum Inhibitory Concentration, MIC)值仅为2~8 μg/mL, 其抗菌活性更优于母体抗生素氯碳头孢(MIC值>128 μg/mL).这些研究表明相较于天然铁载体中的N5-乙酰基-N5-羟基-L-鸟氨酸基团和单一合成类铁载体, 混合类铁载体儿茶酚和异羟肟酸酯, 在规避常见的抗生素耐药机制, 如外膜通透性障碍和外排机制等方面更为有效, 具有更优秀的抗菌活性.所以, 合成类混合型铁载体与抗生素偶联物展现的优秀抗菌活性为未来“特洛伊木马”分子抗生素策略的铁载体选择提供了新发展方向.
为了进一步探究作用于细菌细胞壁的其它类抗生素是否也可以作为“特洛伊木马”分子的抗生素, 1996年Miller团队[37]分别将万古霉素(Vancomycin)与亚精胺基邻苯二酚、儿茶酚和异羟肟酸酯的混合型铁载体偶联, 得到偶联物18和19(图 6).初步活性结果表明, 亚精胺基邻苯二酚18和混合型铁载体偶联物19相比于母体抗生素万古霉素, 丧失了对革兰氏阳性菌的抗菌活性.所以, 后续的“特洛伊木马”分子抗生素策略研究没有再使用万古霉素作为抗生素. 1998年, Budzikiewicz等[38]合成了两种天然铁载体Ⅰ型荧光嗜铁素与氨苄青霉素的偶联物20和Ⅱ型荧光嗜铁素与氨苄青霉素的偶联物21(图 6).该系列偶联物选择二羧酸作为Ⅰ型荧光嗜铁素和Ⅱ型荧光嗜铁素中的Lys残基与抗生素之间的连接基团, 这种长而灵活的连接基团可以避免氨苄青霉素与其靶向的细菌转肽酶之间产生空间位阻, 使药物与铁载体紧密结合而不影响外膜识别.初步活性数据显示, 偶联物20和21具有显著的铜绿假单胞菌抗菌活性, 偶联物20和21的MIC值分别为0.39和0.24 μmol/L.这表明偶联物可以通过铁摄取途径进入细菌细胞, 在细菌内释放氨苄青霉素, 从而发挥抗菌活性.遗憾的是该类型天然铁载体仅对利用荧光嗜铁素类菌株有效, 具有很大的局限性.
2002年, 为了启动基于铁载体的“特洛伊木马”策略抗生素最全面的研究, 德国汉诺尔研究所(HKI)的Möllmann研究小组[39]合成了大量的青霉素与铁载体的偶联物, 主要是利用完全合成的仿生铁载体类似物与抗生素偶联来探究偶联物的抗菌活性.他们将人工合成双儿茶酚类铁载体与氨苄青霉素偶联得到偶联物22(图 7), 初步抗菌活性测试表明, 与母体抗生素氨苄青霉素相比, 偶联物22的铜绿假单胞菌菌株的抗菌能力提升1000倍以上(22的MIC<0.05 μg/mL; 氨苄青霉素>100 μg/mL). 2003年, Möllmann研究小组[40]进一步探究双儿茶酚和异羟肟酸酯混合的铁载体与氨苄青霉素偶联后偶联物23(图 7)的抗菌活性, 发现23不仅对革兰氏阴性菌铜绿假单胞菌的抗菌活性比母体氨苄青霉素提升了2000倍, 对革兰氏阳性菌的抗菌活性也有所提升(23对金黄色葡萄球菌MIC值为0.4 μg/mL; 氨苄青霉素MIC值为25 μg/mL).综上, 双儿茶酚和异羟肟酸酯类混合型结构对于识别细菌铁载体至关重要.由于混合型铁载体具有可被细菌膜受体识别的优势, 2012年, Miller团队[41-42]设计并合成具有简化型的三脚骨架连接基的三儿茶酚类铁载体与抗生素氨苄青霉素和阿莫西林偶联, 得到相应的偶联物24和25(图 7), 进一步探究三脚骨架连接基的混合型铁载体对偶联物抗菌活性影响.初步抗菌活性发现, 24和25对革兰氏阴性菌铜绿假单胞菌呈现抗菌活性, 已有研究表明氨苄青霉素与阿莫西林本身对野生型铜绿假单胞菌不具备抗菌活性(MIC>200 μmol/L), 但氨苄青霉素与阿莫西林与铁载体偶联后却显现出优秀的抗菌活性, 尤其在缺铁培养基中, 其MIC值为0.05~0.39 μmol/L.早在2009年, Möllmann研究小组[43]就阐述了合成类铁载体双儿茶酚的氨苄青霉素偶联物的作用机制(22), 利用细菌铁转运系统, 绕过外排泵进入到革兰氏阴性菌体内发挥抗菌活性.更令人兴奋的是, 该偶联物对小鼠具有良好的耐受性, 且没有诱变作用.这些结果表明, 与天然铁载体相比, 人工合成类铁载体提高了以氨苄青霉素或阿莫西林作为抗生素的偶联物抗菌活性, 尤其是混合型人工合成类铁载体的偶联物具有更优的抗菌活性, 混合类合成铁载体可能是未来的“特洛伊木马”分子抗生素策略中铁载体发展的主要方向.
通过铁载体偶联物来增强β-内酰胺类抗生素的尝试, 不仅仅停留在青霉素和头孢菌素类抗生素.氨曲南26(图 8)也可以作为“特洛伊木马”分子策略中的抗生素, 且是临床上唯一使用的[44], 对β-内酰胺酶更具抗性的单环β-内酰胺类抗生素[45]. 1985年, Squibb医学研究所[46-47]报道了以氨曲南(26)作为抗菌成分, 咪唑啉酮为连接部分, 羟基吡啶酮基团作为铁载体的偶联物27(图 8)的合成与研究.虽然羟基吡啶酮不是天然的铁载体的组成部分, 但它们通常被认为是儿茶酚类铁载体的模拟结构, 并且具有高亲和性的铁螯合能力.初步抗菌活性显示, 与母体抗生素氨曲南的抗菌活性一样, 偶联物27对革兰氏阴性菌也具有显著的抗菌活性, 尤其对铜绿假单胞菌菌株的抗菌活性有所提高(27的MIC为0.5 μg/ mL, 氨曲南MIC值为4 μg/mL).随后, Tuominen小组[47]扩大了对该系列化合物连接部分的结构探究, 确定了结构中以三唑酮作为连接部分, 具有强大的抗革兰氏阴性菌的U-78608 (28)先导化合物.随着大型制药公司合并, 有关抗生素的研究工作也被削减.直到2011年[48-49], 随着抗生素的耐药性越来越普遍, 一些研究人员或制药公司(辉瑞公司)重新考虑了三唑酮作为连接部分的羟基吡啶酮衍生物类“特洛伊木马”分子抗生素的研究, 设计并合成偶联物MC-1(图 8, 29)[50], 主要是在U-78608 (28)结构基础上将三唑酮甲基替换为丙二醇.初步体外活性表明, MC-1 (29)对革兰氏阴性菌具有显著的抗菌活性, 与临床使用的氨曲南相比其抗菌活性显著提升, 对铜绿假单胞菌的MIC值为0.5 μg/mL.进一步的机制研究表明, 在铜绿假单胞菌的菌株中, MC-1 (29)利用了PiuA膜蛋白, 一种依赖TonB的铁载体外膜蛋白, 进入细菌内发挥抗菌活性.该小组还证明, MC-1 (29)对传统的低通透性突变体也具有活性, 可绕过细菌外排泵也不会被大量β-内酰胺酶水解进入细菌发挥抗菌活性.作者得出的结论是: “MC-1可能是该系列中最具代表性的分子, 它可以承受多种抗生素耐药机制的作用, 为临床医生与铜绿假单胞菌等革兰氏阴性病原体的持续斗争提供了武器”.在此基础上, 辉瑞药业也报导了单酰胺羟基吡啶酮MB-1 (30)[51](图 8), 为了模拟MC-1中铁螯合剂基团的位置, MB-1结构中羟基吡啶酮连接在C-4官能化的单环β内酰胺上.初步活性数据显示, 与MC-1一样, 新的单环β内酰胺MB-1偶联物也表现出优秀的革兰氏阴性抗菌活性, 且不易产生耐药性.综上, 单环β内酰胺类“特洛伊木马”分子抗生素也呈现优秀的抗菌活性, 为未来铁载体类抗生素的研发提供了另一个全新的方向.

20世纪80年代至今, 已有cefetecol (32)、BAL30072 (31)、GSK3342830和头孢地尔(33)(图 9)等4种“特洛伊木马”分子抗生素进入临床试验[52]. BAL30072是一种新型羟基吡啶酮与单环β-内酰胺抗生素的偶联物. BAL30072对铜绿假单胞菌和鲍曼不动杆菌均具有优秀的抗菌活性(MIC90分别为4和8 μg/mL), 但由于BAL30072严重肝毒性仅到Ⅰ期临床.头孢菌素cefetecol[53]主要由于哺乳动物体内的儿茶酚O-甲基转移酶可使儿茶酚类铁载体的邻苯二酚上的1个酚基甲基化, 从而导致其体内活性下降也停止于Ⅰ期临床. 2019年11月14日[54], 美国食品药品监督管理局(FDA)批准日本盐野义公司研发的新型铁载体头孢菌素类抗生素头孢地尔(Cefiderocol, Fetroja)上市.该药物主要用于治疗易感革兰氏阴性微生物引起的复杂性尿路感染的18岁及以上成人患者.首个新型铁载体头孢菌素类抗生素的成功, 为复杂性尿路感染患者带来了新的希望.

历时30多年, 日本盐野义制药有限公司完成了从铁载体类抗生素研发到头孢地尔铁载体类抗生素进入市场的历程.在20世界90年代初期, 盐野义制药有限公司发现铁载体头孢菌素S-9096 (34)[55](图 9), 其对革兰阴性菌(绿脓杆菌)具有强效的抗菌活性.该化合物C-3侧链上具有儿茶酚铁载体的结构, 可利用三价铁转运系统对革兰氏阴性细菌的外膜具有良好的渗透性, 从而发挥优秀的抗菌活性.但是, 由于心血管毒性和化合物的稳定性差, S-9096尚未到临床开发.之后的15年里, 盐野义制药有限公司终止了对“特洛伊木马”策略抗生素的研发.随着抗生素的耐药性越来越普遍, 多家制药公司和课题组对β-内酰胺类“特洛伊木马”抗生素展开深入的研究. 1992年, 法国ICI制药公司通过对BAL30072引入氯原子得到37(图 10)偶联物来降低邻苯二酚部分的pKa值, 防止哺乳动物体内的儿茶酚O-甲基转移酶使其甲基化, 保留了铁载体的螯合特性, 除了具有出色的抗菌活性, 37更具有较长的药代动力学半衰期.遗憾的是, 由于缺乏针对β-内酰胺酶的水解的稳定性, 37化合物未进入市场[57]. 2008年, 盐野义制药有限公司重启探寻“特洛伊木马”分子抗生素之路并聚焦于β-内酰胺类抗生素与铁载体偶联物的研究.十年之后, 盐野义制药有限公司的Aoki等[56]对头孢地尔抗生素药物的设计与合成进行了报道, 详细描述了头孢地尔的构效关系.新型铁载体类头孢菌素头孢地尔是由氯儿茶酚铁载体的部分、C-7侧链引入了头孢他啶(35)(图 10)酰氨基的部分和C-3侧链结合了头孢吡肟(36)(图 10)季铵盐的部分组成.头孢地尔抗生素中氯儿茶酚基团的铁载体部分以偶联物37为基础设计, 可以避免铁载体不被甲基转移酶甲基化, 保留铁载体螯合活性. C-3侧链上引入与头孢吡肟相似的吡咯烷基团, 提高了头孢地尔对β-内酰胺酶的稳定性发挥抗菌活性.头孢地尔在C-7侧链上引入头孢他啶中羧基丙氧基亚氨基基团部分, 可改善偶联物的跨外膜运输, 将抗生素转运至细胞周质发挥抗菌活性.铁载体、抗生素、连接基团三个部分的绝对优化, 让他们发现了新型“特洛伊木马”分子抗生素头孢地尔.头孢地尔的作用机制是与三价铁离子结合, 并通过细菌铁转运蛋白, 从细胞膜外膜被主动运输至细菌外周胞质内通过抑制细胞壁生物合成所必需的青霉素结合蛋白(PBPs), 导致细菌细胞死亡.这种“特洛伊木马”策略允许头孢地尔在细菌细胞外周胞质中达到更高的浓度, 在外周胞质空间中与青霉素结合蛋白结合并抑制细菌细胞壁的合成, 发挥抗菌活性.

综上, 可以发现天然铁载体与作用于细菌细胞壁类抗生素偶联, 其抗菌活性大都很难优于母体抗生素, 而且也容易出现其他挑战, 如偶联物的溶解性低、穿过细菌胞质膜的通道不足, 以及活性抗生素的释放困难等问题; 与天然铁载体相比, 合成类铁载体偶联作用于细菌细胞壁类抗生素的抗菌活性显著增强, 其结构简单、靶向特异性强和活性抗生素释放充足, 也更深受科学家和制药公司的喜爱; 相较于其它作用机制类的抗生素, 作用靶标位于外周胞质空间内的细胞壁生物合成的青霉素结合蛋白(PBPs)的β-内酰胺类抗生素是“特洛伊木马”分子策略最成功的抗生素类别.这可能是因为“特洛伊木马”策略使抗生素更容易穿过细菌外膜, 在细菌周质中达到更高的浓度, 高浓度下抗生素更容易与周质空间中的青霉素结合蛋白结合, 发挥更佳的抗菌活性.所以, 作用于细菌细胞壁类抗生素-铁载体类偶联物的药物研发一直是科学家和制药企业的研究热点.首个新型铁载体头孢菌素类抗生素头孢地尔的发现, 为“特洛伊木马”分子抗生素的研发带来了曙光.
早在1995年, 为了确定作用于核糖体的抗生素是否可以作为偶联物的药物部分, Miller课题组[58]率先合成三异羟肟酸酯铁载体-红霉素胺偶联物38和39(图 11).初步活性结果表明, 这些偶联物的抗菌活性不及红霉素胺本身抗菌活性. 1998年, Poras等[59]随后合成了一系列儿茶酚铁载体-螺旋霉素偶联物(40~46)(图 11)来探究该系列偶联物的抗菌活性.遗憾的是, 在铁离子缺乏和铁离子充足的两种琼脂培养基条件下的活性测定中, 也没有发现优于母体螺旋霉素抗菌活性的偶联物.大多数作用于细菌核糖体类抗生素-铁载体偶联物的“特洛伊木马”策略均未取得成功, 它们的抗菌活性和抗菌谱几乎没有优于母体抗生素.这些大环内酯类抗生素与铁载体偶联物的抗菌活性比原抗生素没有显著提升, 可能是由于偶联物无法正常转运或释放到达细菌内核糖体作用靶点所致.
由于大环内酯类抗生素与铁载体偶联物的抗菌局限性, 我们分析作用于细菌核糖体类抗生素-铁载体偶联物的一个限制因素, 可能是偶联物无法实现高效细菌内膜转运, 其铁载体部分可能是影响抗生素跨过细菌内膜的转运效率的原因; 此外, 即使部分偶联物可顺利穿过细菌内膜, 偶联物也可能由于到达核糖体后缺乏相应的抗生素释放机制, 从而在一定程度上降低抗生素与其作用靶点的结合能力, 导致抗菌活性降低.因此, 对于此类偶联物来说, 连接基团的选择至关重要, 既要保证偶联物在细菌外环境中不会提前水解, 又不能过于稳定, 必须保证偶联物在细胞质内水解释放抗菌成分以发挥抗菌作用. Schalk课题组[60]合成了噁唑烷酮类抗生素与儿茶酚型铁载体连接得到的偶联物, 进一步探究“特洛伊木马”分子抗生素策略对噁唑烷酮类抗生素的抗菌谱的影响.噁唑烷酮类抗生素是继磺胺和喹诺酮类抗生素新发展起来的合成类抗菌药, 其利奈唑胺已经被批准用于临床治疗革兰氏阳性菌感染, 特别是多重耐药的革兰氏阳性菌感染, 都具有优秀的抗菌活性[61-62], 但对铜绿假单胞菌等革兰氏阴性菌效果不佳[63].初步活性结果表明, 偶联物47(图 12)对铜绿假单胞菌PAO1菌株的MIC值为128 μmol/L, 其抗菌活性优于对照的利奈唑胺(MIC>1024 μmol/L).这是一个良好的开端, 表明在缺铁条件下, 噁唑烷酮类抗生素的“特洛伊木马”策略具有可行性.随后, 在另一项研究中, Schalk课题组[64]将噁唑烷酮类抗生素与铜绿假单胞菌的铁载体pyochelin结合, 得到偶联物48(图 12)及其类似物.遗憾的是, 这项研究中所有的偶联物几乎没有抗菌活性.作者认为这可能是因为噁唑烷酮类抗生素难以从偶联物上释放, 导致偶联物溶解度过低失活[65].近期, Miller课题组[66-67]设计并合成了一种独特的铁载体与头孢菌素和噁唑烷酮双药偶联物49(图 12), 其中头孢菌素充当连接基团的主要成分, 作者设想头孢菌素作为连接基团可以被细菌的青霉素结合蛋白或β-内酰胺酶裂解释放噁唑烷酮类抗生素, 以提高偶联物的抗菌活性.偶联物49的抗菌活性比头孢菌素或缺乏β-内酰胺连接的铁载体-噁唑烷酮偶联物要高出125倍以上, 尤其是在β-内酰胺酶高表达的鲍曼不动杆菌株仍可保持一个较高的抗菌活性(MIC值为6 μmol/L).进一步以鲍曼不动杆菌为参考菌株, 通过实验验证49和头孢菌素-铁载体偶联物对正常的鲍曼不动杆菌ATCC 17978的MIC均为0.4 μmol/L, 表明这两种偶联物对正常菌株的抗菌活性均归因于头孢菌素的抗菌活性.之后, 作者通过进一步构建质粒编码的β-内酰胺酶(ADC-1:一种鲍曼不动杆菌固有的内酰胺酶, 能够水解头孢菌素类抗生素)过表达的鲍曼不动杆菌ATCC 17978菌株进行测试, 发现头孢菌素-铁载体偶联物表现出了明显的耐药性(MIC>50 μmol/L), 而偶联物49仍保持良好的抗菌活性(MIC=6 μmol/L), 表明在β-内酰胺酶高表达菌株中噁唑烷酮类化合物主要发挥抗菌作用. Miller课题组关于铁载体-噁唑烷酮类偶联物的最新研究进展向我们提供了一个成功的例子, β-内酰胺类化合物可以作为一个理想的连接体, 将其应用于其它以胞质为靶点的铁载体-抗生素偶联物中, 为“特洛伊木马”分子抗生素策略提供了新的方向.
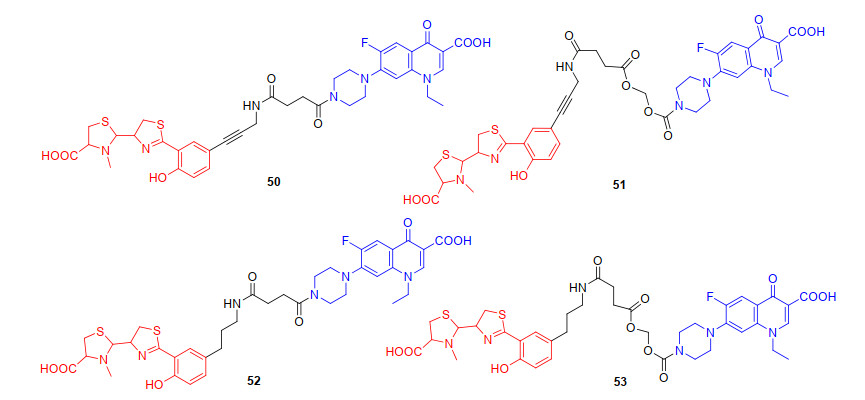
为了探索作用于细菌DNA螺旋酶和拓扑异构酶类抗生素-铁载体偶联物作为“特洛伊木马”分子抗生素策略的可行性, 2006年, Rivault等[68]合成以螯铁蛋白为铁载体的诺氟沙星喹诺酮类抗生素偶联物50~53(图 13).初步抗菌活性评估发现, 不稳定连接基团(亚甲基二氧基酯)的偶联物51和53的抗菌活性与母体抗生素诺氟沙星相比, 显示出相同的铜绿假单胞菌的抗菌活性, 作者猜测造成这种结果是由于51和53偶联物连接基团不稳定, 未到达细胞质之前就被非特异性酯酶部分水解, 而未被水解的部分偶联物进入细胞内并释放游离的诺氟沙星, 从而发挥抗菌活性[69].遗憾的是, 稳定的连接基团(酰胺键)偶联物50和52, 对细菌生长曲线没有明显的抑制作用.对于偶联物50、52的失活, 作者猜测偶联物可以借助荧光嗜铁素(pyoverdine)被细菌受体识别[70], 转运进入细菌细胞膜, 但由于连接基团太过稳定, 使得抗生素难以释放, 造成偶联物50、52失活. 这说明铁载体-抗生素偶联物中, 连接基团可能是喹诺酮类偶联物发挥抗菌作用的关键因素, 当连接基团不易水解时, 诺氟沙星可能无法从偶联物中释放作用于细胞质中的DNA螺旋酶, 或者偶联物中铁载体部分在空间上阻碍了诺氟沙星与DNA螺旋酶的相互作用, 导致抗菌活性下降.
2013年, Wencewicz等[71]合成了三异羟肟酸酯铁载体与环丙沙星连接的偶联物54(图 14), 初步活性数据显示, 54对革兰氏阳性菌金黄色葡萄球菌(MIC值为1 μmol/L)具有与环丙沙星(MIC值为0.5 μmol/L)等同的抗菌活性, 但对革兰氏阴性菌无效.这说明完整的三羟肟酸铁载体骨架参与了细菌膜转运, 这种类型的转运铁载体对具有羟肟酸铁载体转运蛋白的革兰氏阳性细菌具有选择性. 2014年, Fardeau等[72-73]合成了三儿茶酚基团作为铁载体, 可水解的酯键作为连接基团, 环丙沙星作为抗生素的偶联物55~58(图 14), 探究该系列偶联物的抗菌活性.初步活性结果表明, 偶联物55和57对铜绿假单胞菌DSM 1117呈现中等的抗菌活性(MIC值分别为8和 64 μg/mL), 而58在128 μg/mL依然没有呈现抗菌活性, 所有偶联物的抗菌活性均低于母体抗生素环丙沙星.这可能是因为偶联物在水性介质中的溶解度低或在细胞质中连接基团酯键未发生水解或部分水解导致抗菌成分未得到完全释放.
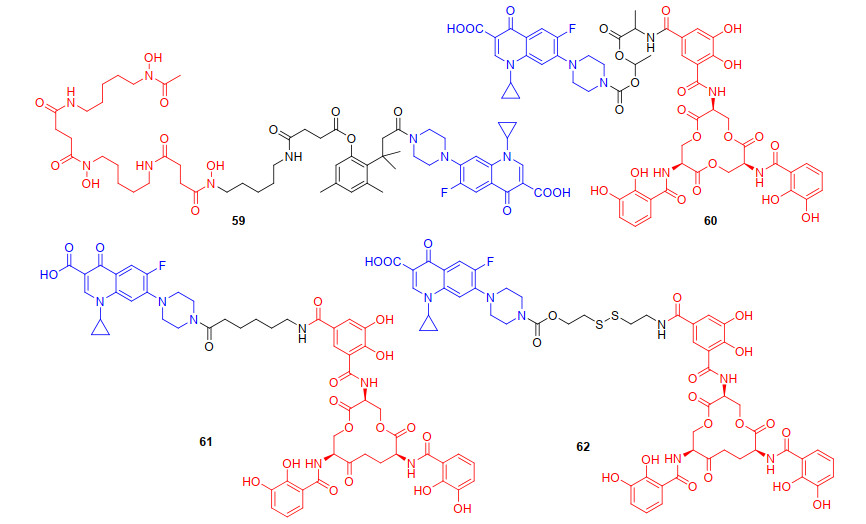
综上, 我们可以发现“特洛伊木马”分子以喹诺酮类抗生素来探究时, 连接基团对抗菌活性有至关重要的影响, 为了更深入探究铁载体-抗生素偶联物中连接基团对抗菌活性的影响. 2012年, Ji等[74]设计并合成了以邻羟基肉桂酸衍生物为连接基团的环丙沙星偶联物59 (图 15).邻羟基肉桂酸衍生物作为连接基团, 易被细菌和真菌分泌的酯酶或磷酸酶内酯化, 迅速形成氢化香豆素, 并伴随抗生素的释放.初步抗菌活性评估59对革兰氏阳性菌株(枯草芽孢杆菌、金黄色葡萄球菌)以及铜绿假单胞菌呈现中等抗菌活性(MIC90为1~32 μg/mL), 而对粪肠球菌无抗菌活性.遗憾的是, 59抗菌活性低于母体抗生素环丙沙星, 可能是酯酶水解导致抗生素释放不完全[75]. 2015年, Zheng等[76]设计并合成了带有不稳定(酰氧基)烷基酯为连接基团与肠杆菌素铁载体和环丙沙星偶联物60(图 15), 进一步探究连接基团对抗菌活性的影响.先前的研究表明, (酰氧基)烷基酯连接基对空间环境敏感[77], 通过调节(酰氧基)烷基酯附近的取代基, 增大位阻, 可增强偶联物的稳定性, 避免药物的过早释放. 2018年, Neumann等[78]报道了以烷基链为连接基团, 肠杆菌素铁载体与环丙沙星偶联得到的偶联物61(图 15), 探究了烷基类连接基团对抗菌活性的影响, 并评估了偶联物药物释放作用机制.烷基连接的偶联物61对两种大肠杆菌UTI89和大肠杆菌CFT073都具有很高的抗菌活性, 其MIC值与环丙沙星(MIC值为0.1~1 μmol/L)接近.已有研究[79]和当前研究的结果表明, 连接基团可能会阻止偶联物的跨膜转运和药物释放.随后Neumann等[80]合成了不稳定二硫键作为连接基团的偶联物62(图 15), 探讨了连接基团的稳定性对偶联物抗菌活性的影响, 遗憾的是, 62只对部分大肠杆菌株有生长抑制作用.作者猜测62可能在未进入细菌细胞膜时提前还原裂解释放抗生素, 所以二硫键不能广泛应用于铁载体-抗生素偶联物的设计中.偶联物主要是通过连接基团使铁载体与喹诺酮类抗生素偶联, 我们发现连接基团的选择对喹诺酮类偶联物抗菌活性的提升至关重要, 大部分可水解的连接基团, 由于其不稳定性容易导致抗生素过早释放, 而不可水解的连接基团相比可水解的连接基团, 可使偶联物在体外和转运过程中更加稳定, 然后在细菌内使偶联物释放, 提高偶联物的抗菌活性.但过于稳定的连接基团, 又增加了抗生素释放的难度, 比如, 偶联物62与母体抗生素相比, 抗菌活性并没有提高.
喹诺酮类抗生素作为“特洛伊木马”分子的抗生素时, 偶联物的抗菌活性大都没有提高, 主要原因可能有以下几种: (1)偶联物自身溶解性的问题; (2)铁载体结构较大, 阻碍了偶联物转运至细胞质与喹诺酮类抗生素的作用靶标结合; (3)铁载体与抗生素之间的连接基团不可水解, 不能释放抗生素; (4)连接基团具有非特异性水解的特性, 可能还未到达药物作用靶标过早释放等.
达托霉素是由玫瑰孢链霉菌(Streptomyces roseosporus)发酵得到的含有十个碳脂肪侧链的环脂肽. 2003年, 达托霉素(Cubicin)获得美国食品与药物管理局(FDA)批准, 用于治疗革兰氏阳性菌引起的并发性皮肤感染, 并在美国等多个国家上市.达托霉素可以与细菌细胞膜结合并破坏其功能, 使内容物外泄而发挥杀菌的作用[81].但由于达托霉素分子量大, 具有阴电性的特点, 导致其无法穿过革兰氏阴性菌外膜.因此, 临床上达托霉素主要用于治疗革兰氏阳性菌引起的感染. 2017年, Gosh等[82]将天然鲍曼不动杆菌(Acinetobacter baumannii)的fimsbactin A铁载体进行改造, 人工合成了结构中包含两个儿茶酚和一个异羟肟酸酯的fimsbactin A铁载体类似物, 再与达托霉素通过酰胺键连接得到偶联物63(图 16).初步活性研究发现, 偶联物63对革兰氏阳性菌金黄色葡萄球菌菌株的MIC大约为6~12.5 µmol/L, 其抗菌活性低于达托霉素(MIC=0.05~0.4 µmol/L).但是, 与对革兰氏阴性菌生长无抑制作用的达托霉素相比, 63对鲍曼不动杆菌及其耐药菌株的MIC为0.4~0.8 µmol/L, 表明人工合成的fimsbactin A铁载体类似物可以被革兰氏阴性菌外膜的转运蛋白识别, 偶联物可以通过主动转运进入细胞周质中, 与细菌细胞膜结合发挥抗菌活性.达托霉素作用靶点位于细胞膜而不在细胞质中, 所以连接部分是否水解并不影响偶联物的抗菌活性. 2018年, Miller课题组[83]对偶联物63(图 16)进行结构改造, 将fimsbactin A铁载体类似物替换为双儿茶酚类(bis-catechol)和三儿茶酚类(tri-catechol)结构的铁载体, 分别合成偶联物64和65(图 16).初步活性检测发现, 偶联物64和65对鲍曼不动杆菌及其耐药菌株抗菌活性均弱于偶联物63, 可能是因为人工改造的铁载体fimsbactin A类似物与人工合成儿茶酚类铁载体相比, 更易于被鲍曼不动杆菌细胞外膜上的转运蛋白识别, 使得偶联物更高效地被转运到细菌周质内.
此外, 三种达托霉素与铁载体连接得到的偶联物对鲍曼不动杆菌外的其它革兰氏阴性菌大肠杆菌以及铜绿假单胞菌均未显现出抗菌活性, 这与达托霉素抗菌作用靶点在大肠杆菌以及铜绿假单胞菌分布较少有关[84].综上, 偶联物63能够通过铁载体介导的铁离子吸收系统, 跨过鲍曼不动杆菌的外膜到达细胞膜发挥抗菌活性, 是“特洛伊木马”策略拓宽达托霉素抗菌谱成功的典型例子, 为后续抗菌药物研发提供了全新的可能.
Lantibiotic是一类由革兰氏阳性菌产生, 结构中含有羊毛硫氨酸(lanthionine), 作用于其它革兰氏阳性菌的抗菌肽.根据化学结构和抗菌活性, lantibiotic类抗菌肽分为A型和B型. A型lantibiotic类抗菌肽是正电荷、结构较为延伸的肽. B型是一些较少量正电荷、结构为球形小分子的肽[85]. Lantibiotic类抗菌肽与细胞膜结合后, 以表面结合的方式插入到细胞膜的双分子层, 进一步导致细胞渗漏和死亡[86].但是, lantibbotic类抗菌肽缺乏穿透革兰氏阴性菌细胞外膜的能力, 限制了此类抗菌肽的使用范围.为了改善这类抗菌肽的缺陷, 2011年Yoganathan等[87]对gallidermin (A型lantibiotic类抗菌肽)进行结构改造, 在位于13号位的赖氨酸上引入人工合成的agrobactin铁载体类似物, 连接部分为方酸二甲胺(diamino squarate)得到偶联物66(图 17).初步活性数据显示, 与母体抗菌肽相比, 66保留了对阳性菌的抗菌活性, 但没有产生革兰氏阴性菌的抗菌活性.有趣的是, 偶联物66对铜绿假单胞菌的生长有轻微的促进作用.推测这种反常现象是偶联物为细菌提供了少量的铁离子导致的, 说明铁载体可以促使偶联物跨过阴性菌外膜.但是, 可能由于lantibiotic类抗菌肽分子量过大, 导致穿过外膜的抗菌肽的分子数极少.偶联物在较低浓度下无法发挥抗菌活性, 其螯合的可溶性铁离子被交换到细菌天然铁载体中, 为细菌生长提供了少量铁离子, 从而轻微促进了铜绿假单胞菌的生长.
综上, 达托霉素-铁载体偶联物的成功验证了“特洛伊木马”分子抗生素策略, 可以提升作用于细菌细胞膜的抗生素的抗菌活性, 并扩大已有抗生素的抗菌谱, 为后续扩大抗菌谱的相关抗菌药物研发提供了全新的策略和方向.
2009年, Wencewicz等[88]报道了以三氯生为抗生素, 去铁胺类似物为铁载体, 酯键作为连接片段的偶联物67(图 18).三氯生是一种广谱的抗菌药物, 其作用靶标为烯脂酰载体蛋白还原酶(FabI). FabI在细菌体内起到重要作用, 它是脂肪酸合成途径中的限速酶[89].三氯生通过与FabI以及烟酰胺辅因子形成稳定的三元配合物来影响FabI发挥作用, 抑制细菌体内的脂肪酸的合成[90].偶联物的生物活性实验表明, 67对革兰氏阳性菌金黄色葡萄球菌(Staphylococcus aureus)、革兰氏阴性菌大肠杆菌(E. coli)、铜绿假单胞菌(Pseudomonas aeruginosa)的生长都具有抑制作用, 对大肠杆菌菌株E. coli DC2的MIC值小于0.1 µmol/L.偶联物67的抗菌活性与母体三氯生的作用效果相似.其机制研究表明, 所用的大肠杆菌菌株均无法利用去铁胺来摄取培养基中的铁离子, 表明偶联物中铁载体并没有发挥将三氯生转运到细菌胞内的作用.因此, 三氯生在细菌胞外的提前释放可能是偶联物保持抗菌活性的根本原因.
Miller课题组[71, 88]受沙霉素独特的作用机制启发, 设计了一系列铁载体-抗生素偶联物, 它们分别是通过特异性模拟沙霉素三异羟肟酸铁载体与不同作用机制的抗生素氯碳头孢68(以细菌细胞壁为靶标的抗生素)、环丙沙星54(以细菌DNA螺旋酶和拓扑异构酶为靶标的抗生素)和三氯生67(以烯脂酰载体蛋白还原酶为靶标的抗生素)结合形成的偶联物.他们发现, 在几乎所有测试的菌株中, 偶联物54和68与它们的母体抗生素氯碳头孢和环丙沙星相比, 抗菌活性降低, 这可能是由于它们缺乏相应的抗生素释放机制所致.进一步分析发现, 与酰胺作为连接部分的偶联物54和68相比, 以酚酯作为连接部分的偶联物67具有更高的水解不稳定性, 极有可能在胞外环境中提前释放游离三氯生, 发挥其抗菌作用.综上, 这说明连接部分对偶联物转运进入细菌胞内发挥抗菌活性起重要作用, 连接基团需具有某些特定条件下分解的特性, 以保证偶联物可以进入细菌细胞, 并在细胞内使药物释放.
2005年Miller课题组[91]报道了以具有抑制真菌生长的化合物desketoneoenactin (DE)作为药效基团, 合成了三个连接不同铁载体的偶联物69, 70和71(图 19). Desketoneoenactin具体作用机制并不明确, 推测可能是因为分子结构中含有疏水基团和亲水基团, 化合物具有两性, 可以插入到细胞膜中, 通过破坏细胞膜的完整性来杀死真菌[92].偶联物69中铁载体部分为高铁色素铁载体类似物(ferrichrome, FCH), 偶联物70中铁载体部分为鸟氨酸铁载体类似物(ornithine-based trihydroxamate, ORNI), 偶联物71中的铁载体部分为异氰脲酸酯类铁载体类似物(isocyanurate-based hydroxamate, ISO).初步活性探究发现, 缺铁条件下偶联物69, 70和71对白色念珠菌的MIC值依次为3.1, 12.5和25 µmol/L.遗憾的是, 该系列偶联物的抗菌活性均低于desketoneoenactin (MIC值为0.2 µmol/L).进一步机制探究中发现白色念珠菌更易识别高铁色素铁载体, 结构中含有高铁色素铁载体类似物的偶联物69更易于被真菌摄取, 抗菌活性也优于其它两种偶联物.不同真菌菌株对铁载体可能存在特异选择性, 铁载体直接影响到偶联物的抗菌活性, 所以根据不同真菌菌株, 选取不同种类铁载体是真菌类抗生素偶联物设计中必需考虑的关键因素.
综上, 连接部分的稳定性对偶联物发挥抗菌活性起重要作用.如果连接部分在胞外水解, 与母体抗生素相比, 偶联物的抗菌活性会下降甚至消失.所以, 连接部分的选择是“特洛伊木马”策略抗生素成功的关键因素.由于菌株对不同类型铁载体的摄取具有特异性, 因此, 科研工作者在设计偶联物时也应该评价目标菌株对不同种类铁载体的摄取能力, 筛选出易被目标菌株利用的铁载体作为偶联物中的药物递送部分.
“特洛伊木马”分子策略作为新作用模式的抗生素在实验和临床方面都取得了一定的进展, 并具有巨大的研发潜能与应用前景. “特洛伊木马”分子偶联物是由三个部分组成:铁载体(结构式中红色部分)、连接基团(结构式中黑色部分)和抗生素(结构式中蓝色部分)[89, 93-94].在分子设计中应综合考虑这三个组成部分中的每一组分对抗菌效果的影响, 进行系统选择和优化, 以便在膜渗透和抗生素释放方面产生最佳抗菌活性.大量研究显示, 这些部分的选择和优化具有一定的困难性, 这也是迄今为止只有极少数成功的“特洛伊木马”抗生素出现的原因.铁载体-抗生素偶联物发展的第一个难点是寻找适用于“特洛伊木马”策略的抗生素, 大多数作用于细菌细胞质内靶标的抗生素偶联的“特洛伊木马”偶联物(41, 47)通常显示出低于母体抗生素的抗菌活性.这种限制主要存在两种可能,第一种是由于抗生素偶联的铁载体和细菌自身分泌的铁载体与细菌膜转运蛋白的转运选择性差异而导致携带抗生素的铁载体很难到达细胞质中的作用靶点[22, 29, 59-63, 95]; 另一种可能是因为外周胞质中细菌天然铁载体释放铁离子阻挡偶联物进入细菌发挥抗菌活性.所以, 对于革兰氏阴性菌, 大多数铁载体-抗生素偶联物只能结合到位于外周胞质的青霉素结合蛋白, 这也解释了为什么迄今为止大多数成功的“特洛伊木马”策略都选择作用在细菌细胞壁和细胞膜等细菌外周胞质靶标的抗生素, β-内酰胺抗生素(47, 33, 49)就是最好的例子.铁载体-抗生素偶联物发展的另一个难点是寻找适用于“特洛伊木马”策略的连接基团.理想的连接基团不仅能在细胞外部条件下稳定, 也能在铁吸收系统的主动转运过程中稳定, 并在细菌内使铁载体-抗生素偶联物裂解释放抗生素.但迄今为止发现的连接基团还远未达到理想状态.大部分可水解的连接基团, 由于其不稳定性容易导致抗生素过早释放.与不可水解的连接基团相比, 可水解的连接基团可使偶联物在体外和转运过程中更加稳定, 然后在细菌内使偶联物释放抗生素, 提高偶联物的抗菌活性.但过于稳定的连接基团又增加了抗生素释放的难度, 还需要进一步的优化改造(55~57)[60-65, 71-76]. Miller课题组关于铁载体-噁唑烷酮类偶联物的最新研究进展向我们提供了一个成功的例子, β-内酰胺类化合物(47, 49)也可以作为一个理想的连接基团, 将其应用于其它以胞质为靶点的铁载体-抗生素偶联物中.在“特洛伊木马”策略中还要考虑的铁载体-抗生素偶联物的第三部分是铁载体.迄今为止, 已经描述了大量的铁载体-抗生素偶联物, 但与天然铁载体相比, 人工合成的儿茶酚或异羟肟酸酯类铁载体是最有效的铁载体类似物(20, 22~24)[38-42, 88, 93, 96].混合型铁载体相较于单一型铁载体展现出更优的药物递送能力, 更有助于偶联物发挥抗菌作用.所以, 优化“特洛伊木马”分子偶联物中抗生素、连接基团和铁载体三个组成部分, 可能是未来铁载体类抗生素研究的主要方向.
铁载体-抗生素药物设计策略为新型作用机制类抗生素药物研发提供了新的发展方向, 与母体抗生素相比, “特洛伊木马”策略偶联物的选择性和抗菌活性的提高还可以降低抗生素耐药性的风险.迄今为止, 最成功的铁载体-抗生素是作用于细菌周质靶标的抗生素β-内酰胺类的头孢地尔, 该铁载体偶联抗生素的发现会继续刺激对铁载体-抗生素偶联物的进一步研究.作者认为, β-内酰胺-铁载体偶联物虽然一直是科学家和制药企业的药物研发热门, 但非β-内酰胺抗生素-铁载体偶联物似乎仍然是学术上的追求.铁载体-抗生素偶联物的潜在化学药物研发空间巨大, 相信不久的将来, 其将会是未来新型作用模式抗生素的重要来源.
Zaman, S. B.; Hussain, M. A.; Nye, R. Mehta, V.; Mamun, K. T.; Hossain, N. Cureus 2017, 9, 1403.
Brown, E. D.; Wright, G. D. Nature 2016, 529, 336. doi: 10.1038/nature17042
Blair, J. M.; Webber, M. A.; Baylay, A. J.; Ogbolu, D. O.; Piddock, L. J. Nat. Rev. Microbiol. 2015, 13, 42. doi: 10.1038/nrmicro3380
Rossiter, S. E.; Fletcher, M. H.; Wuest, W. M. Chem. Rev. 2017, 117, 2415.
Lewis, K. Nat. Rev. Drug Discovery 2013, 12, 371. doi: 10.1038/nrd3975
Abouelhassan, Y.; Garrison, A, T.; Yang, H.; Chavez-Riveros, A.; Burch, G. M.; Huigens, R. W. I. J. Med. Chem. 2019, 6, 7618.
Soares, M. P.; Weiss, G. EMBO Rep. 2015, 16, 1482. doi: 10.15252/embr.201540558
Hider, R. C.; Kong, X. Nat. Prod. Rep. 2010, 27, 637. doi: 10.1039/b906679a
Gorska, A.; Sloderbach, A.; Marszall, M. P. Trends Pharmacol. Sci. 2014, 35, 442. doi: 10.1016/j.tips.2014.06.007
Page, M. G. Ann. N. Y. Acad. Sci. 2013, 1277, 115. doi: 10.1111/nyas.12024
Ferguson, A. D.; Braun, V.; Fiedler, H. P.; Coulton, J. W.; Diederichs, K.; Welte, W. Protein Sci. 2000, 9, 956. doi: 10.1110/ps.9.5.956
Clarke, T. E.; Braun, V.; Winkelmann, G.; TariL, W.; Vogel, H. J. J. Biol. Chem. 2002, 277, 13966. doi: 10.1074/jbc.M109385200
Braun, V.; Pramanik, A.; Gwinner, T.; Koberle, M.; Bohn, E. Biometals 2009, 22, 3. doi: 10.1007/s10534-008-9199-7
Juarez-Hernandez, R. E.; Miller, P. A.; Miller, M. J. ACS Med. Chem. Lett. 2012, 3, 799. doi: 10.1021/ml300150y
Vertesy, L.; Aretz, W.; Fehlhaber, H. W.; Kogler H. Helv. Chim. Aata 1995, 78, 46. doi: 10.1002/hlca.19950780105
Roosenberg, J. M.; Miller, M. J. J. Org. Chem. 2000, 65, 4833 doi: 10.1021/jo000050m
Braun, V. K.; Günthner, H.; Zimmermann, L. J. Bacteriol. 1983, 156, 308. doi: 10.1128/JB.156.1.308-315.1983
Saha, M.; Sarkar, S.; Sarkar, B.; Sharma, B. K.; Bhattacharjee, S.; Tribedi, P. Environ. Sci. Pollut. Res. Int. 2016, 23, 3984. doi: 10.1007/s11356-015-4294-0
Bilitewski, U.; Blodgett, J. Duhme-Klair, A.K.; Dallavalle, S. Laschat, S.; Routledge, A.; Schobert, R. Angew. Chem., Int. Ed. 2017, 56, 14360. doi: 10.1002/anie.201701586
Abouelhassan, Y.; Garrison, A. T.; Yang, H.; Chavez-Riveros, A.; Burch, G. M.; Huigens, R. R. J. Med. Chem. 2019, 62, 7618. doi: 10.1021/acs.jmedchem.9b00370
Madsen, J. L.; Johnstone, T. C.; Nolan, E. M. J. Am. Chem. Soc. 2015, 137, 9117. doi: 10.1021/jacs.5b04557
Ji, C.; Miller, P. A.; Miller, M. J. J. Am. Chem. Soc. 2012, 134, 9898. doi: 10.1021/ja303446w
Mislin, G. L.; Schalk, I. J. Metallomics 2014, 6, 408. doi: 10.1039/C3MT00359K
Muller, G.; Barclay, S. J.; Raymond, K. N. J. Biol. Chem. 1985, 260, 13916.
Sayer, J. M.; Emery, T. F. Biochemistry 1968, 7, 184. doi: 10.1021/bi00841a023
Krewulak, K. D.; Vogel, H. J. Biochim. Biophys. Acta 2008, 1778, 1781. doi: 10.1016/j.bbamem.2007.07.026
Miethke, M.; Marahiel, M. A. Microbiol. Mol. Biol. Rev. 2007, 71, 413. doi: 10.1128/MMBR.00012-07
Winkelmann, G. Biometals 2007, 20, 379. doi: 10.1007/s10534-006-9076-1
Mollmann, U.; Heinisch, L.; Bauernfeind, A.; Kohler, T.; Ankel-Fuchs, D. Biometals 2009, 22, 615. doi: 10.1007/s10534-009-9219-2
Ballouche, M.; Cornelis, P.; Baysse, C. Recent Pat. Anti-Infect. Drug Discovery 2009, 4, 190.
Murphy-Benenato, K. E.; Bhagunde, P. R.; Chen, A; Davis, H. E.; Durand-Reville, T. F.; Ehmann, D. E. J. Med. Chem. 2015, 58, 2159. doi: 10.1021/jm5012484
Dolence, E. K.; Minnick, A. A.; Miller, M. J. J. Med. Chem. 1990, 33, 461. doi: 10.1021/jm00164a001
Mckee, J. A.; Sharma, S. K.; Miller, M. J. Bioconjugate Chem. 1991, 2, 281. doi: 10.1021/bc00010a013
Ramurthy, S.; Miller, M. J. J. Org. Chem. 1996, 61, 4120. doi: 10.1021/jo9600621
Minnick, A. A.; Mckee, J. A.; Dolence, E. K.; Miller, M. J. Antimicrob. Agents Chem. 1992, 36, 840. doi: 10.1128/AAC.36.4.840
Ghosh, A.; Ghosh, M.; Niu, C.; Malouin, F.; Moellmann, U.; Miller, M. J. Chem. Biol. 1996, 3, 1011. doi: 10.1016/S1074-5521(96)90167-2
Ghosh, M.; Miller, M. J. Bioorg. Med. Chem. 1996, 4, 43. doi: 10.1016/0968-0896(95)00161-1
Kinzel, O.; Tappe, R.; Gerus, I.; Budzikiewicz, H. J. Antibiot. 1998, 51, 499. doi: 10.7164/antibiotics.51.499
Heinisch, L.; Wittmann, S.; Stoiber, T.; Berg, A.; Ankel-Fuchs, D.; Mollmann, U. J. Med. Chem. 2002, 45, 3032. doi: 10.1021/jm010546b
Heinisch, L.; Wittmann, S.; Stoiber, T.; Scherlitz-Hofmann, I.; Ankel-Fuchs, D.; Mollmann, U. Arzneim. Forsch. 2003, 53, 188.
Ji, C.; Miller, P. A.; Miller, M. J. J. Am. Chem. Soc. 2012, 134, 9898. doi: 10.1021/ja303446w
Ji, C.; Miller, M. J. Bioorg. Med. Chem. 2012, 20, 3828. doi: 10.1016/j.bmc.2012.04.034
Miller, M. J.; Zhu, H.; Xu, Y.; Wu, C.; Walz, A. J.; Vergne, A.; Roosenberg, J. M.; Moraski, G.; Minnick, A. A.; Mckee-Dolence, J.; Hu, J.; Fennell, K.; Kurtdolence, E.; Dong, L.; Franzblau, S.; Malouin, F.; Mollmann, U. Biometals 2009, 22, 61. doi: 10.1007/s10534-008-9185-0
Cimarusti, C. M.; Sykes, R. B. Med. Res. Rev. 1984, 4, 1. doi: 10.1002/med.2610040103
Bush, K.; Freudenberger, J. S.; Sykes, R.B. Antimicrob. Agents Chem. 1982, 22, 414. doi: 10.1128/AAC.22.3.414
Cusnir, R.; Imberti, C.; Hider, R.C.; Blower, P. J.; Ma, M. T. Int. J. Mol. Sci. 2017, 18, 1161. doi: 10.3390/ijms18061161
Barbachyn, M. R.; Tuominen, T. C. J. Antibiot. 1990, 43, 1199. doi: 10.7164/antibiotics.43.1199
Han, S.; Zaniewski, R. P.; Marr, E. S.; Lacey, B. M.; Tomaras, A. P.; Evdokimov, A.; Miller, J. R.; Shanmugasundaram, V. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 22002. doi: 10.1073/pnas.1013092107
Flanagan, M. E.; Brickner, S. J.; Lall, M.; Casavant, J.; Deschenes, L.; Finegan, S. M.; George, D. M.; Granskog, K.; Hardink, J. R.; Huband, M. D.; Thuy, H.; Lamb, L.; Marra, A.; Mitton-Fry, M.; Mueller, J. P.; Mullins, L. M.; Noe, M. C.; O'Donnell, J. P.; Pattavina, D.; Penzien, J. B.; Schuff, B. P.; Sun. J.; Whipple, D. A.; Young, J.; Gootz, T. D. ACS Med. Chem. Lett. 2011, 2, 385. doi: 10.1021/ml200012f
Mcpherson, C. J.; Aschenbrenner, L. M.; Lacey, B. M.; Fahnoe, K. C.; Lemmon, M. M.; Finegan, S. M.; Tadakamalla, B.; O'Donnell, J. P.; Mueller, J. P.; Tomaras, A. P. Antimicrob. Agents Chemother. 2012, 56, 6334. doi: 10.1128/AAC.01345-12
Tomaras, A. P.; Crandon, J. L.; Mcpherson, C. J.; Nicolau, D. P. Antimicrob. Agents Chemother. 2015, 59, 2439. doi: 10.1128/AAC.04172-14
Sato, T.; Yamawaki, K. Clin. Infect. Dis 2019, 69, 529. doi: 10.1093/cid/ciz825
Page, M. G.; Dantier, C.; Desarbre, E. Antimicrob. Agents Chemother. 2010, 54, 2291. doi: 10.1128/AAC.01525-09
Sato, T.; Yamawaki, K. Clin. Infect. Dis. 2019, 69, S538. doi: 10.1093/cid/ciz826
Yamano, Y.; Nishikawa, T.; Komatsu, Y. Appl. Microbiol. Biotechnol. 1994, 40, 892. doi: 10.1007/BF00173995
Aoki, T. Yoshizawa, H.; Yamawaki, K.; Yokoo, K.; Sato, J.; Hisakawa, S.; Hasegawa, Y.; Kusano, H.; Sano, M.; Sugimoto, H.; Nishitani, Y.; Sato, T.; Tsuji, M.; Nakamura, R.; Nishikawa, T.; Yamano, Y. Eur. J. Med. Chem. 2018, 155, 847. doi: 10.1016/j.ejmech.2018.06.014
Bird, T. G.; Arnould, J. C.; Bertrandie, A.; Jung, F. H. J. Med. Chem. 1992, 35, 2643. doi: 10.1021/jm00092a015
Ghosh, M.; Miller, M. J. Bioorg. Med. Chem. 1995, 3, 1519. doi: 10.1016/0968-0896(95)00134-3
Poras, H.; Kunesch, G.; Barriere, J. C.; Berthaud, N.; Andremont, A. J. Antibiot. 1998, 51, 786. doi: 10.7164/antibiotics.51.786
Jones, R. N.; Johnson, D. M.; Erwin, M. E. Antimicrob. Agents Chemother. 1996, 40, 720. doi: 10.1128/AAC.40.3.720
Bassetti, M.; Baguneid, M.; Bouza, E.; Dryden, M.; Nathwani, D.; Wilcox, M. Clin. Microbiol. Infect. 2014, 204, 3.
Mendes, R. E.; Hogan, P. A.; Streit, J. M.; Jones, R. N.; Flamm, R. K. Antimicrob. Agents Chemother. 2015, 59, 2454. doi: 10.1128/AAC.04784-14
Paulen, A.; Gasser, V.; Hoegy, F.; Perraud, Q.; Pesset, B.; Schalk, I. J.; Mislin, G. L. A. Org. Biomol. Chem. 2015, 13, 11567. doi: 10.1039/C5OB01859E
Paulen, A.; Hoegy, F.; Roche, B.; Schalk, I. J.; Mislin, G. L. A. Bioorg. Med. Chem. Lett. 2017, 27, 4867. doi: 10.1016/j.bmcl.2017.09.039
Noel, S.; Gasser, V.; Pesset, B.; Hoegy, F.; Rognan, D.; Schalk, I. J.; Mislin, G. L. A. Org. Biomol. Chem. 2011, 9, 8288. doi: 10.1039/c1ob06250f
Liu, R.; Miller, P. A.; Vakulenko, S. B.; Stewart, N. K.; Boggess, W. C.; Miller, M. J. J. Med. Chem. 2018, 61, 3845. doi: 10.1021/acs.jmedchem.8b00218
Schalk, I. J. J. Med. Chem. 2018, 61, 3842. doi: 10.1021/acs.jmedchem.8b00522
Rivault, F.; Liebert, C.; Burger, A.; Hoegy, F.; Abdallah, M. A.; Schalk, I. J.; Mislin, G. L. A. Bioorg. Med. Chem. Lett. 2007, 17, 640. doi: 10.1016/j.bmcl.2006.11.005
Hennard, C.; Truong, Q. C.; Desnottes, J. F.; Paris, J. M.; Moreau, N. J.; Abdallah, M. A. J. Med. Chem. 2001, 44, 2139. doi: 10.1021/jm990508g
Barrett, J. F.; Bernstein, J. I.; Krause, H. M.; Hilliard, J. J.; Ohemeng, K. A. Anal. Biochem. 1993, 214, 313. doi: 10.1006/abio.1993.1493
Wencewicz, T. A.; Long, T. E.; Moellmann, U.; Miller, M. J. Bioconjugate Chem. 2013, 24, 473. doi: 10.1021/bc300610f
Fardeau, S.; Dassonville-Klimpt, A.; Audic, N.; Sasaki, A.; Pillon, M.; Baudrin, E.; Mullie, C.; Sonnet, P. Bioorg. Med. Chem. 2014, 22, 4049. doi: 10.1016/j.bmc.2014.05.067
Milstien, S.; Cohen, L. A. J. Am. Chem. Soc. 1972, 94, 9158. doi: 10.1021/ja00781a029
Ji, C.; Miller, M. J. Bioorg. Med. Chem. 2012, 20, 3828. doi: 10.1016/j.bmc.2012.04.034
Wilhelm, S.; Tommassen, J.; Jaeger, K. E. J. Bacteriol. 1999, 181, 6977. doi: 10.1128/JB.181.22.6977-6986.1999
Zheng, T.; Nolan, E. M. Bioorg. Med. Chem. Lett. 2015, 25, 4987. doi: 10.1016/j.bmcl.2015.02.034
Gupta, D.; Gupta, S. V.; Lee, K.; Amidon, G. L. Mol. Pharmaceutics 2009, 6, 1604. doi: 10.1021/mp900084v
Neumann, W.; Sassone-Corsi, M.; Raffatellu, M.; Nolan, E. M. J. Am. Chem. Soc. 2018, 140, 5193. doi: 10.1021/jacs.8b01042
Zheng, T.; Bullock, J. L.; Nolan, E. M. J. Am. Chem. Soc. 2012, 134, 18388. doi: 10.1021/ja3077268
Neumann, W.; Nolan, E. M. J. Biol. Inorg. Chem. 2018, 23, 1025. doi: 10.1007/s00775-018-1588-y
Taylor, S. D.; Palmer, M. Bioorg. Med. Chem. 2016, 24, 6253. doi: 10.1016/j.bmc.2016.05.052
Ghosh, M.; Miller, P. A.; Mollmann, U.; Claypool, W. D.; Schroeder, V. A.; Wolter, W. R.; Suckow, M.; Yu, H.; Li, S.; Huang, W.; Zajicek, J.; Miller, M. J. J. Med. Chem. 2017.; 60, 4577. doi: 10.1021/acs.jmedchem.7b00102
Ghosh, M.; Lin, Y. M.; Miller, P. A.; Mollmann, U.; Boggess, W. C.; Miller, M. J. ACS Infect. Dis. 2018, 4, 1529. doi: 10.1021/acsinfecdis.8b00150
Randall, C. P.; Mariner, K. R.; Chopra, I.; O'Neill, A. J. Antimicrob. Agents Chemother. 2013, 57, 637. doi: 10.1128/AAC.02005-12
Kraaij, C.; Vos, W. M.; Siezen, R. J.; Kuipers, O. P. Nat. Prod. Rep. 1999, 16, 575. doi: 10.1039/a804531c
Willey, J. M.; Donk, W.A. Annu. Rev. Microbiol. 2007, 61, 477. doi: 10.1146/annurev.micro.61.080706.093501
Yoganathan, S.; Sit, C. S.; Vederas, J. C. Org. Biomol. Chem. 2011, 9, 2133. doi: 10.1039/c0ob00846j
Wencewicz, T. A.; Mollmann, U.; Long, T. E.; Miller, M. J. Biometals 2009, 22, 633. doi: 10.1007/s10534-009-9218-3
Maiden, M. M.; Hunt, A.; Zachos, M. P.; Gibson, J. A.; Hurwitz, M. E.; Mulks, M. H.; Waters, C. M. Antimicrob. Agents Chemother. 2018, 62, 96.
Heath, R. J.; Rubin, J. R.; Holland, D. R.; Zhang, E.; Snow, M. E.; Rock, C. O. J. Biol. Chem. 1999, 274, 11110. doi: 10.1074/jbc.274.16.11110
Bernier, G.; Girijavallabhan, V.; Murray, A.; Niyaz, N.; Ding, P.; Miller, M. J.; Malouin, F. Antimicrob. Agents Chemother. 2005, 49, 241. doi: 10.1128/AAC.49.1.241-248.2005
Yamamoto, K.; Shiinoki, Y.; Nishio, M.; Matsuda, Y.; Inouye, Y.; Nakamura, S. J. Antibiot. 1990, 43, 1012. doi: 10.7164/antibiotics.43.1012
Mollmann, U.; Ghosh, A.; Dolence, E. K.; Dolence, J. A.; Ghosh, M.; Miller, M. J.; Reissbrodt, R. Biometals 1998, 11, 1. doi: 10.1023/A:1009266705308
Rivault, F.; Liebert, C.; Burger, A.; Hoegy, F.; Abdallah, M. A.; Schalk, I. J.; Mislin, G. L. Bioorg. Med. Chem. Lett. 2007, 17, 640. doi: 10.1016/j.bmcl.2006.11.005
Souto, A.; Montaos, M. A.; Balado, M.; Osorio, C. R.; Rodriguez, J.; Lemos, M. L.; Jimenez, C. Bioorg. Med. Chem. 2013, 21, 295. doi: 10.1016/j.bmc.2012.10.028
Milner, S. J.; Seve, A.; Snelling, A. M.; Thomas, G. H.; Kerr, K. G.; Routledge, A.; Duhme-Klair, A. K. Org. Biomol. Chem. 2013, 11, 3461. doi: 10.1039/c3ob40162f

图 1 铁载体介导的铁转运系统及“特洛伊木马”策略[8]
Figure 1 Siderophore-mediated iron acquisition and its exploitation by "Trojan horses"

图 9 合成类铁载体结合β-内酰胺和头孢菌素的结构
Figure 9 Structures of synthetic β-lactams and cephalosporins antibiotic-siderophore conjugates

图 10 头孢地尔(33)、头孢他啶(35)、头孢吡肟(36)和37的结构式
Figure 10 Structures of cefiderocol (33), ceftazidime (35), cefepime (36) and 37

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:













 下载:
下载:
















 下载:
下载: