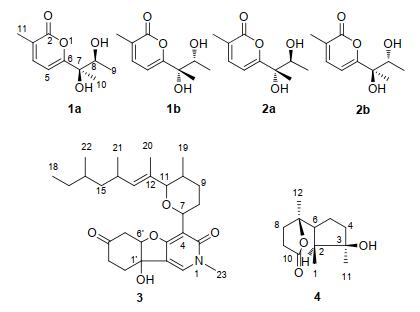
Figure 1.
Compounds 1~4 isolated from F. tricinctum
Four New 2-Pyrones from Fusarium tricinctum, an Endophytic Fungus of Ligusticum chuanxiong
Yumei Cao , Qixuan Kuang , Feng Ju , Yun Deng , Fang Deng , Yucheng Gu , Bo Ren , Dale Guo

2-Pyrone, a six-membered cyclic unsaturated ester, is common structural motif in many natural products[1] belonging to fungi of several genera including Alternaria, Aspergillus, Fusarium, Penicillium and Trichoderma. Compounds containing 2-pyrone sub-structures such as bufadienolides, fusapyrones, gibepyrones styrylpyrones and perpyrones also exhibit various activities including cytotoxicity, neurotoxicity as well as phytotoxicity.[2-6]
In the course of our ongoing search bioactive compounds of endophytes from Ligusticum chuanxiong, [7-8] Fusarium tricinctum was obtained from its root. And along with two known compounds, four new 2-pyrone derives sharing same planar structure of 6-(2, 3-dihydroxybutan- 2-yl)-3-methyl-2H-pyran-2-one were firstly isolated as two pairs of racemes by pre-HPLC and further as four epimers by subsequent chiral separation from the brown rice solid medium of F. tricinctum. Because the structures of these compounds contained a diol moiety on the branch (Figure 1) which was confirmed by spectroscopic analyses, induced circular dichroism (ICD) spectra based on a modified Snatzke's method were collected to elucidate their absolute configurations. The details of isolation, structure elucidation as well as bioactivity of these compounds are discussed below.
Compound 1 was obtained as yellow oil. The HRESIMS ion at m/z 199.0948 [M+H+] indicated its molecular formula should be C10H14O4 (calculated for C10H15O4 [M+H+], 199.0892). The IR bands indicated the presence of hydroxyl (3414 cm-1) and carbonyl (1701 cm-1) groups. The 1H NMR, 13C NMR and HSQC spectra displayed signals of three methyl [δH 2.04 (d, J=1.2 Hz, 3H, H-11), 1.46 (s, 3H, H-10) and 1.10 (d, J=6.5 Hz, 3H, H-9); δC 22.8 (C-10), 17.6 (C-9), 16.4 (C-11)]; an oxymethine [δH 3.91 (q, J=6.5 Hz, 1H, H-8); δC 72.5 (C-8)]; a carbonyl at δC 165.6 (C-2), two trisubstituted double bonds [δH 7.34 (dd, J=6.8, 1.2 Hz, 1H, H-4) and 6.44 (d, J=6.8 Hz, 1H, H-5); δC 167.8 (C-6), 142.1 (C-4), 123.6 (C-3), and 103.5 (C-5)] and an oxygen-bearing noprotoated carbon at δC 76.3 (C-7).
Detailed analysis of 1H-1H COSY data of 1 revealed the presence of two spin coupling systems (H-4/H-5 and H-8/H-9) as shown in Figure 2. The 1H detected heteronuclear multiple-bond coherence (HMBC) correlations of H-4/C-2, C-6 and H-5/C-3 confirmed the existence of a 2-pyrone moiety. Furthermore, the HMBC correlations of H-9/C-7, H-10/C-5, C-6, as well as H-11/C-2, C-3, C-4 confirmed the locations of above three methyl groups. The obvious downfield shifts at C-7 and C-8 combined with the molecular formula information indicated that the remaining two hydroxyl groups should be attached to C-7 and C-8 respectively. Thus, the gross structure of 1 was confirmed as shown in Figure 2.
Compound 2 was obtained as yellow oil. It was assigned the same molecular formula C10H14O4 as 1 on the basis of HRESIMS. The NMR spectra of 2 were similar to those of 1, which indicated that it was an epimer of 1 (Table 1).
HPLC analyses of compounds 1 and 2 with a chiralpak- IC column were carried out to explain their approximate- to-zero specific rotations and confirmed that there were two pairs of anticipated enantiomers. Subsequent HPLC preparation yielded the enantiomers 1a, 1b, 2a and 2b successfully.
Due to the presence of a vic-diol group in the branch of compounds 1 and 2, a modified Snatzke's method with dimolybdenum tetraacetate [Mo2(OAc)4] was applied to determine their absolute configurations. Specifically, the Cotton effects of Mo2-complex were determined by the absolute configuration of the diol group and the counterclockwise O—C—C—O torsional angle (negative sign of torsional angle) in the conformation of chiral Mo2-complex displayed negative signs of the bands Ⅱ and Ⅳ (approximately 300 nm) and vice versa.
As Mo2-erythro-vic-diols complex can generate two favored conformations which can lead to opposite optical rotations, Cotton effects produced by Mo2-threo-vic-diols complex should be stronger than that of Mo2-erythro-vic- diols complex when samples were measured at the same concentration.[9] And the diol moiety of compound 1 was confirmed as threo, and of compound 2 should be erythro by the means of above accordingly. Furthermore, the absolute configurations of these isomers were assigned by the optical rotation direction indicated by the C-9 and C7-O transpositional plane arrangement configuration. According to the favored conformation in the Mo2-complexes of 1 and 2 shown in Figure 3, the Mo2-complex of 1a in dimethyl sulfoxide (DMSO) showed a positive Cotton effects, and 1b showed a negative Cotton effects at approximately 302 nm (band Ⅳ), 2a showed a negative Cotton effects and 2b showed a positive Cotton effects at approximately 318 nm (band Ⅳ), inducing the fact that the absolute configuration of 1a should be 7S, 8S; of 1b should be 7R, 8R; of 2a should be 7S, 8R; and of 2b should be 7R, 8S.
Compounds 3 and 4 were identified as (-)-4, 6'-anhy- drooxysporidinone[10] and cyclonerodiol lactone[11] by spectroscopic analyses and comparing the spectral data with those reported.
A methyl thiazolyl tetrazolium (MTT) method with Taxol as a positive control was also applied to evaluate their cytotoxicity against HTC116, A549 and MV 4-11 cell lines. Unfortunately, none of them showed obvious growth inhibitions at the concentration of 20 μmol/L.
The rice medium of F. tricinctum gave all of four epimers sharing the same plane structure of 6-(2, 3-dihy- droxybutan-2-yl)-3-methyl-2H-pyran-2-one. Their absolute configurations were elucidated by comprehensive analyses of their spectroscopic spectra. A MTT method was also applied to evaluate their cytotoxicity against HTC116, A549 and MV 4-11 cell lines and none of them showed obvious growth inhibitions at the concentration of 20 μmol/L.
Optical rotations were determined on a Perkin-Elmer- 241 polarimeter (Perkin Elmer, Inc., Waltham, MA, USA) at room temperature. UV spectra were recorded on a Perkin-Elmer Lambda 35 UV-VIS spectrophotometer (Perkin Elmer, Inc., Waltham, MA, USA). IR spectra were measured by a Perkin-Elmer one FT-IR spectrometer (KBr) (Perkin Elmer, Inc., Waltham, MA, USA). The ICD spectrum was measured on a Chirascan circular dichroism spectrometer (Applied Photophysics Ltd., Leatherhead, UK). 1D and 2D NMR were carried out on Bruker- Ascend-400 and 800 MHz instruments (Bruker, Bremen, Germany) at 300 K, with TMS as internal standard. The HRESIMS data were obtained using a Q Exactive mass spectrometer (Thermo Fisher Scientific, MA, USA). Preparative HPLC was performed on a NP7000 serials pump (Hanbon Sci. & Tech., Jiangsu, China) equipped with a YMC ODS column (10 mm×250 mm, 5 μm, Akzo Nobel Pulp and Performance Chemicals AB, Bohus, Sweden) using a NU3000 serials UV detector (Hanbon Sci. & Tech., Jiangsu, China). A column Chiralpak IC column (4.6 mm×250 mm, 5 μm; Chiral Technologies, West Chester, PA, USA) was applied for chiral resolution. Column chromatographs (CC) were performed with silica gel (200~300 mesh, Qingdao Haiyang Chemical Co., Qingdao, China) and Sephadex LH-20 (GE-Healthcare Bio-Sciences AB, Uppsala, Sweden).
L. chuanxiong was collected in Aoping town, suburb of Pengzhou city, Sichuan Province, China, and taxonomically identified by prof. Fei Long of Chengdu University of TCM. A voucher specimen (access number: 2017042411) was deposited at the ministry of education key laboratory of standardization of Chinese herbal medicine, Chengdu university of TCM. F. tricinctum was isolated from the root of L. chuanxiong, and identified by morphological observation and sequence analyses of the ITS region of rDNA. The strain (GenBank accession No. MN216248) also was deposited at ministry of education key laboratory of standardization of Chinese herbal medicine, Chengdu university of TCM.
The fungal strain was inoculated into twenty-four 250 mL Erlenmeyer flasks, each containing 100 mLof liquid medium at 30 ℃ on an orbital shaker at 120 r/min for 3 d to produce the seed culture. Then large scale fermentation was carried out in 200 tissue culture flasks of 330 mL, each containing 2 g of peptone, 40 g of brown rice, 25 mL of water and 10 mL of seed culture for 40 d at room temperature.
Fermentation broth was ultrasonically extracted with 10 L of methanol for 3 times, and the extract was concentrated to give a crude extract. Then the extract was dispersed with water, and further extracted with EtOAc. The solvent was evaporated under vacuum and finally give an EtOAc extract (65.5 g).
The EtOAc extract was then fractionated by silica gel with petroleum ether and acetone mixture from 100:0 to 50:50 (V/V) to give twenty-seven fractions (Fr.1~Fr.27). The Fr.19 was further subjected to Sephadex LH-20 column chromatography (4 cm×180 cm; CHCl3/MeOH, V:V=1:1; about 1000 mL), to yield 5 subfractions (Fr.S1~Fr.S5). Fr.S2 (0.38 g) was purified by a preparative HPLC equipped with a YMC ODS-C18 column (10 cm×250 mm, s-5 μm; 210 nm) to afford 1 (7.9 mg), and the racemic mixture 1 was further purified by HPLC on a chiralpak-IC column (n-hexane/isopropanol, V:V=72:18, 0.8 mL/min) to give 1a (3.0 mg, tR=5.2 min) and 1b (3.9 mg, tR=5.6 min). Fr.S3 (0.27 g) was purified by a preparative HPLC equipped with a YMC ODS-C18 column (10 mm×250 mm, s-5 μm; 210 nm) to afford 2 (7.2mg), and the racemic mixture 2 was further purified by HPLC on a chiralpak-IC column (n-hexane/isopropanol, V:V=72:18, 0.8 mL/min) to give 2a (2.0 mg, tR=7.2 min) and 2b (2.8 mg, tR=8.6 min). The Fr.21 (1.21 g) was purified by a preparative HPLC equipped with a YMC ODS-C18 column (10 mm×250 mm, s-5 μm; 210 nm) to afford 3 (5.8 mg, tR=23 min), and the Fr.22 (1.40 g) was purified by a preparative HPLC equipped with a YMC ODS-C18 column (10 mm×250 mm, s-5 μm; 210 nm) to afford 4 (3.1 mg, tR=25 min).
6-((2S, 3S)-2, 3-Dihydroxybutan-2-yl)-3-methyl-2H- pyran-2-one (1a): 3.0 mg, light yellow oil. [α]20 D+89.6 (c 0.21, MeOH); UV (MeOH) λmax [log ε/(L•mol-1•cm-1)]: 298.7 (4.39), 218.8 (4.05) nm; 1H NMR (400 MHz, CD3OD) and 13C NMR (100 MHz, CD3OD) spectroscopic data are listed in Table 1; IR (KBr) ν: 3414.8, 1701.9, 1580.7, 1384.4, 1097.9 cm-1; HRESIMS calcd for C10H15O4 [M+H+] 199.0892, found 199.0948.
 下载:
导出CSV
下载:
导出CSV
| Position | 1a | 2b | |||
| δC | δH (J in Hz) | δC | δH (J in Hz) | ||
| 2 | 165.6 | 165.8 | |||
| 3 | 123.6 | 123.5 | |||
| 4 | 142.1 | 7.34, dd (6.8, 1.2) | 142.1 | 7.34, dd (6.8, 1.2) | |
| 5 | 103.5 | 6.44, d (6.8) | 103.5 | 6.46, d (6.8) | |
| 6 | 167.8 | 168.2 | |||
| 7 | 76.3 | 76.7 | |||
| 8 | 72.5 | 3.91, q (6.5) | 72.3 | 3.98, q (6.4) | |
| 9 | 17.6 | 1.10, d (6.5) | 17.0 | 1.18, d (6.4) | |
| 10 | 22.8 | 1.46, s | 22.8 | 1.37, s | |
| 11 | 16.4 | 2.04, d (1.2) | 16.4 | 2.04, d (1.2) | |
| a 400 MHz (1H NMR), 100 MHz (13C NMR), CD3OD; b 800 MHz (1H NMR), 200 MHz (13C NMR), CD3OD. | |||||
6-((2R, 3R)-2, 3-Dihydroxybutan-2-yl)-3-methyl-2H- pyran-2-one (1b): 3.9 mg, Light yellow oil. [α]20 D -89.6 (c 0.21, MeOH); UV, IR, NMR, MS and HRESIMS data are the same as those of 1a.
6-((2S, 3R)-2, 3-Dihydroxybutan-2-yl)-3-methyl-2H-pyran-2-one (2a): 2.0 mg, light yellow oil. [α]20 D -65.8 (c 0.28, MeOH); UV (MeOH) λmax [log ε/(L•mol-1•cm-1)]: 298.0 (4.14), 219.1 (3.89) nm; 1H NMR (800 MHz, CD3OD) and 13C NMR (200 MHz, CD3OD) spectroscopic data are listed in Table 1; IR (KBr) ν: 3354.9, 1704.9, 1580.5, 1384.1, 1095.3 cm-1; HRESIMS calcd for C10H15O4 [M+H+] 199.0892, found 199.0969.
6-((2R, 3S)-2, 3-Dihydroxybutan-2-yl)-3-methyl-2H- pyran-2-one (2b): 2.8 mg, light yellow oil. [α]20 D+65.8 (c 0.28, MeOH); UV, IR, NMR, MS and HRESIMS data are the same as those of 2a.
Generally, the electrostatic circular dichroism (ECD) spectra of these isomers were first recorded to establish background spectra, and then the ICD spectra of the Mo-complexes of these isomers with same concentration were offered according to the published procedure.[12]
HTC116, A549 and MV 4-11 cell lines were cultured in 96-well plates firstly, and then treated with above compounds at specified concentrations for 72 h, followed by stand MTT assays with Taxol as a positive control (which exhibited antiproliferative activity against the HTC116, A549 and MV4-11 cell lines with an IC50 value of (0.013±0.004), (0.014±0.005) and (0.039±0.016) μmol/L, respectively). The detailed process also were described in our previous work.[13]
Supporting Information Spectroscopic spectra including HRESIMS, IR, 1D and 2D NMR, and ICD of compounds 1 and 2. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
McGlacken, G. P.; Fairlamb, I. J. S. Nat. Prod. Rep. 2005, 22, 369. doi: 10.1039/b416651p
Bhat, Z. S.; Rather, M. A.; Maqbool, M.; UL Lah, H.; Yousuf, S. K.; Ahmad, Z. Biomed. Pharmacother. 2017, 91, 265. doi: 10.1016/j.biopha.2017.04.012
Lee, J.-S. Mar. Drugs 2015, 13, 1581. doi: 10.3390/md13031581
Furstner, A. Angew. Chem., Int. Ed. 2018, 57, 4215. doi: 10.1002/anie.201707260
Hou, X.-T.; Xu, Y.; Zhu, S.-M.; Zhang, Y.; Guo, L.-D.; Qiu, F.; Che, Y.-S. RSC Adv. 2020, 10, 15622. doi: 10.1039/D0RA02485F
Guo, D.-L.; Qiu, L.; Feng, D.; He, X.; Li, X.-H.; Cao, Z.-X.; Gu, Y.-C.; Mei, L.; Deng, F.; Deng, Y. Nat. Prod. Res. 2020, 34, 958. doi: 10.1080/14786419.2018.1544974
Li, X.-H.; Hu, H.-W.; Tan, L.; Wu, W.-L.; Cao, Z.-X.; Gu, Y.-C.; Deng, Y.; Guo, D.-L. Nat. Prod. Commun. 2018, 13, 1419.
Cao, Y.-M.; Guo, D.-L.; Jin, M.-Y.; Tan, L.; Yang, T.-L.; Deng, F.; Gu, Y.-C.; Li, X.-H.; Cao, Z.-X.; Deng, Y. Nat. Prod. Res. 2020, DOI: 10.1080/14786419.2020.1712385
李长伟, 崔承彬, 国际药学研究杂志, 2015, 42, 811. https://www.cnki.com.cn/Article/CJFDTOTAL-XDYC201502002.htmLi, C.-W.; Cui, C.-B. J. Int. Pharm. Res. 2015, 42, 811(in Chinese). https://www.cnki.com.cn/Article/CJFDTOTAL-XDYC201502002.htm
Zhan, J.-X.; Burns, A. M.; Liu, M.-P. X.; Faeth, S. H.; Gunatilaka, A. A. L. J. Nat. Prod. 2007, 70, 227. doi: 10.1021/np060394t
Hanson, J. R.; Hitchcock, P. B.; Nyfeler, R. J. Chem. Soc., Perkin Trans. 1 1975, 16, 1586.
Liu, Y.-F.; Yue, Y.-F.; Feng, L.-X.; Zhu, H.-J.; Cao, F. Mar. Drugs 2019, 17, 550. doi: 10.3390/md17100550
Li, S.; Chen, J.-F.; Qin, L.-L.; Li, X.-H.; Cao, Z.-X.; Gu, Y.-C.; Guo, D.-L.; Deng, Y. J. Asian Nat. Prod. Res. 2019, 22, 138.
Table 1. NMR spectroscopic data of compounds 1 and 2
| Position | 1a | 2b | |||
| δC | δH (J in Hz) | δC | δH (J in Hz) | ||
| 2 | 165.6 | 165.8 | |||
| 3 | 123.6 | 123.5 | |||
| 4 | 142.1 | 7.34, dd (6.8, 1.2) | 142.1 | 7.34, dd (6.8, 1.2) | |
| 5 | 103.5 | 6.44, d (6.8) | 103.5 | 6.46, d (6.8) | |
| 6 | 167.8 | 168.2 | |||
| 7 | 76.3 | 76.7 | |||
| 8 | 72.5 | 3.91, q (6.5) | 72.3 | 3.98, q (6.4) | |
| 9 | 17.6 | 1.10, d (6.5) | 17.0 | 1.18, d (6.4) | |
| 10 | 22.8 | 1.46, s | 22.8 | 1.37, s | |
| 11 | 16.4 | 2.04, d (1.2) | 16.4 | 2.04, d (1.2) | |
| a 400 MHz (1H NMR), 100 MHz (13C NMR), CD3OD; b 800 MHz (1H NMR), 200 MHz (13C NMR), CD3OD. | |||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们





 下载:
下载: