
图式 1.
光催化烷基硼化合物的活化模式
Scheme 1.
Activated models of alkyl boron compounds under photoredox condition

有机硼化合物作为重要的有机合成中间体, 被广泛地应用于医药、农药、配体以及有机光电材料的合成等领域中.一直以来, 其合成与转化受到广泛的关注, 其中部分反应因为巨大的应用价值已经成为人名反应, 如Suzuki-Miyaura交叉偶联反应[1]、Brown硼氢化反应[2]、Chan-Lam反应[3]以及Zweifel烯基化反应[4]等.
在各类有机硼化合物中, 烷基硼化合物因其合成方法丰富及结构多样而一直受到化学家们的高度关注与重视.过渡金属催化的Suzuki-Miyaura偶联反应主要实现了芳基硼类化合物的转化[5].对于烷基硼而言, 因碳硼键极性较小, 在金属催化的偶联反应中难以进行转金属化过程并且存在潜在的β-氢消除副反应.因此, 尽管目前有少量的报道, 金属催化烷基硼的偶联反应仍具有很大的挑战, 基于四配位硼1, 2-迁移反应已经成为烷基硼转化的重要方法[6].近些年来, 自由基化学得到迅速的发展, 并成为了一类前景良好并且具有潜在普适性的有机合成方法.可见光作为一种清洁可持续能源, 其参与高效的自由基引发体系不仅具有优异的经济性, 也符合绿色化学的发展潮流[7].目前从反应机理上看, 光催化烷基硼化合物脱硼生成烷基自由基模式主要为两类: (1)烷基四配位硼化合物被激发态的光催化剂单电子氧化释放出烷基自由基以及三配位硼(Scheme 1, a); (2)三配位硼化合物在外加自由基作用下, 发生自由基交换, 释放出烷基自由基(Scheme 1, b).生成的烷基自由基随后被不同的自由基受体捕获, 进一步转化形成不同的化学键. 2015年, Blanchard和Bisseret等[8]综述了光催化下有机硼化合物的合成以及转化的相关研究. 2018年, Li, Liu和Yan等[9]共同综述了烷基硼化合物的自由基转化, 介绍了氧化剂介导下烷基硼化合物的转化以及可见光催化下烷基三氟硼酸钾在C—C成键上的应用.尽管上述文献中从不同角度包含了可见光催化烷基硼化合物的部分转化反应, 但是我们注意到目前光催化烷基硼化合物的转化尚无系统的总结.基于此, 本文从成键类型包括碳碳成键、碳氧成键、碳硫成键以及碳氢成键出发, 综述烷基硼化合物在可见光促进下脱硼衍生化的研究进展.
C—C成键反应是目前有机合成研究最为广泛的反应.通过可见光催化介导烷基硼化合物脱硼来实现碳碳键偶联, 往往具有条件温和及官能团兼容性广的优点.
2014年, 陈以昀课题组[10]报道了首例光催化烷基三氟硼酸钾的脱硼炔基化反应, 实现了烷基硼化合物脱硼构建C(sp3)—C(sp)键(Scheme 2).该反应具有优异的普适性, 不仅可以兼容一级、二级以及三级硼化合物, 同时烷基硼酸也反应良好.此外, 该反应还可以兼容敏感的官能团如卤素(Br, I)、炔基、醛酮羰基以及羟基等.值得一提的是, 该反应在中性水缓冲液中也可以进行, 并且向体系中添加氨基酸、核苷、低聚糖、核酸、蛋白质和细胞裂解物均不会影响反应.通过一系列控制实验证明, 三价的有机碘试剂BI-OH释放出BI自由基, 该自由基氧化激发态Ru(Ⅱ)生成Ru(Ⅲ)引发了整个反应.生成的Ru(Ⅲ)与烷基硼化合物(烷基三氟硼酸钾或者烷基硼酸)发生单电子氧化生成烷基自由基以及初始Ru(Ⅱ)光催剂.随后烷基自由基与炔BI发生自由基加成并随后消除得到目标产物, 同时释放出氧化激发态Ru(Ⅱ)的BI自由基实现了反应的循环.

2016年, 许华建课题组[11]以Ru(bpy)3(PF6)2作为催化剂, 对甲基苯磺酰腈(TsCN)作为氰基来源, BI-OAc和三氟醋酸(TFA)作为添加剂, 实现了烷基三氟硼酸钾的脱硼氰基化反应(Scheme 3, a).该反应在空气中即可进行, 条件温和.结合烯烃硼氢化反应, 作者通过三步以61%的收率得到烯烃反马氏氰基化产物.值得一提的是, 烷基频哪醇硼酸酯作为底物时, 反应也能以30%的收率得到目标产物.同年, Molander课题组[12]以Mes- Acr+作为光催化剂, N, N-二甲基甲酰胺(DMF)作为溶剂, 使用同样的氰源实现了一级以及二级烷基三氟硼酸钾的脱硼氰基化(Scheme 3, b).相比较而言, 该反应更为简单且不需要任何添加剂, 同时避免了过渡金属的使用.

近期, 刘超课题组[13]报道了光催化的烷基频哪醇硼酸酯脱硼炔基化反应.他们以廉价易得的NaOMe或者NaOH作为烷基硼酸酯的活化试剂, 使其成为可以被激发态光催化剂单电子氧化的四配位硼前体, 并通过核磁11B谱实验证明了四配位硼中间体的形成.进一步通过自由基捕获实验、电子顺磁共振(EPR)实验以及荧光淬灭实验证明了该四配位硼前体为自由基源.该反应不需要过渡金属, 底物兼容性好.此外, 对于二硼化合物, 反应能选择性对位阻大的一侧碳硼键进行炔基化(Scheme 4).
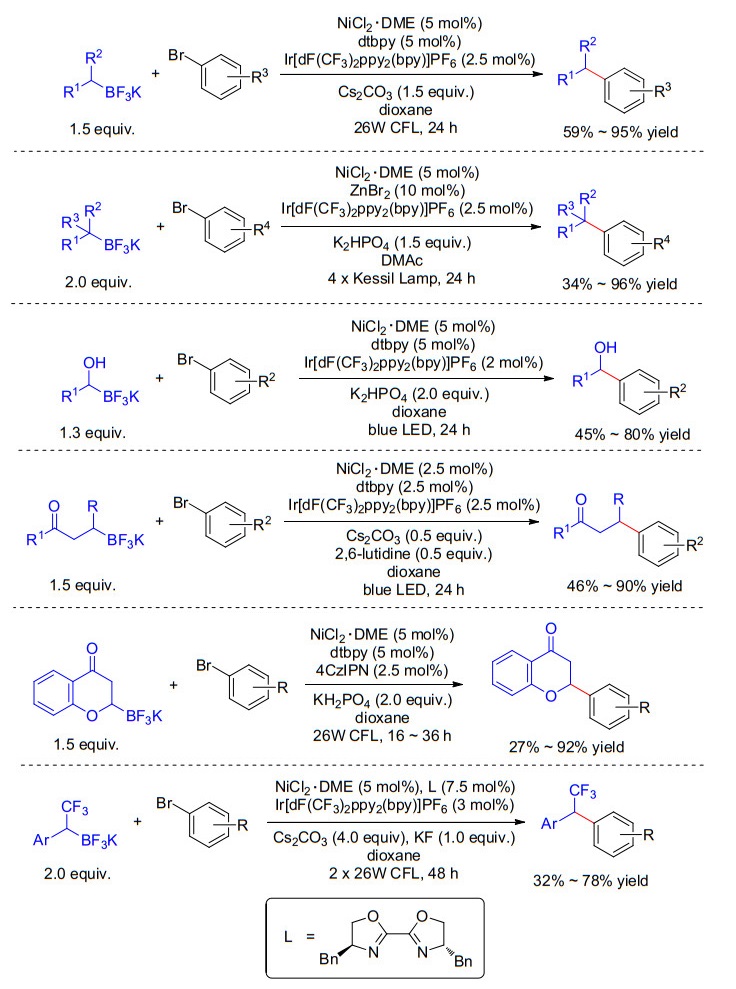
2014年, Molander课题组[14]报道了首例光/镍协同催化苄基三氟硼酸钾与芳基溴化物的偶联反应(Scheme 5, a).相比于传统的过渡金属催化模式来说, 该反应具有反应条件温和及底物兼容性好的特点.该反应可能的机理为:苄基三氟硼酸钾被激发态Ir光催化剂单电子氧化为苄基自由基; 同时, Ir光催化剂被还原生成还原态Ir催化剂, 还原态的Ir光催化剂将体系中Ni(Ⅰ)催化剂单电子还原为Ni(0)物种; 进一步, 生成的Ni(0)物种与芳基卤化物氧化加成得到芳基镍物种A, 芳基镍物种A捕捉体系中苄基自由基生成Ni(Ⅲ)中间体B, 随后Ni(Ⅲ)中间体B发生还原消除得到目标产物并生成初始Ni(Ⅰ)物种.此外, 当使用手性Box类配体时, 能以50%的ee值得到目标产物, 表明该转化可实现立体选择性控制(Scheme 5, b).随后, Molander小组与Kozlowski小组[15]合作通过密度泛函理论(DFT)计算来进一步验证了该反应的机理.在此工作基础上, Molander课题组进一步发展了二级烷基三氟硼酸钾[16]、三级烷基三氟硼酸钾[17]的可见光镍协同催化的脱硼芳基化反应, 为烷基三氟硼酸钾提供了一种条件温和且反应高效的芳基化策略.该策略还可被应用到α-羟基三氟硼酸钾[18]、α-三氟甲基三氟硼酸钾[19]、β-酰基三氟硼酸钾[20]以及α-烷氧基三氟硼酸钾[21]的脱硼芳基化反应.这些反应均表现出良好的底物普适性(Scheme 6).

2016年, Ley课题组[22]首次报道了流动光催化苄基硼酸酯脱硼芳基化的方法(Scheme 7).反应机理研究表明, 吡啶类的Lewis碱如4-二甲氨基吡啶(DMAP)等可以与苄基硼酸酯形成四配位硼化合物, 该四配位硼化合物可被激发态的Ir催化剂单电子氧化生成苄基自由基.进一步通过镍催化和芳基卤代烃偶联.值得一提的是, 含氰基的氮杂芳烃既作为活化试剂又作为原料, 同苄基自由基发生脱氰基偶联反应并且不需要镍作为共催化剂.相较于的烷基三氟硼酸钾, 烷基硼酸酯来源更加广泛, 更具有经济性.

Minisci反应为碳自由基亲核加成到缺电子杂环上, 随后发生C—H键取代反应[23].光催化作为产生亲核自由基的一种强有力的手段已经被应用于该反应[24].作为一种化学性质稳定并且来源广泛的自由基源, 烷基硼化合物的Minisci反应用于含氮杂环化合物衍生化的反应成为研究的重点. 2016年, 陈弓课题组[25]报道了以Ru(bpy)3Cl2作为光催化剂, 一级以及二级烷基硼酸作为烷基源的光催化含氮杂环芳烃碳氢键烷基化反应(Scheme 8).该反应具有优异的底物兼容性和选择性, 可以实现如奎宁、咖啡因、喜树碱、泛昔洛韦、法舒地尔等药物分子或者天然产物定点烷基化, 为药物后续开发提供了可能.实验以及DFT计算表明该反应的关键步骤为BI-OAc被激发态的Ru(Ⅱ)还原成邻碘苯甲酰氧自由基, 该自由基具有亲硼的性质, 可以和烷基硼酸发生反应生成烷基自由基.随后烷基自由基对含氮杂环发生亲核加成, 进一步芳构化得到目标产物.

2017年, 许华建课题组[26]以Ru配合物作为光催化剂, 三氟乙酸作为添加剂, BI-OAc作为氧化剂实现了光催化吡啶氮氧化物C(2)位碳氢键的烷基化反应(Scheme 9, a).该反应底物普适性好, 可兼容酯、酰胺、醚、氰基、酮、炔烃和卤化物等.通过该策略, 他们以三步59%的收率合成药物分子环吡酮. 2017年, Molander课题组[27]报道了无金属光催化的烷基三氟硼酸钾作为自由基源的Minisci反应(Scheme 9, b).他们用此方法实现了温和条件下菲啰啉配体以及联吡啶配体高区域选择性高收率烷基化反应. 2019年, 徐海超课题组[28]采用光电共催化的策略也同样实现了烷基三氟硼酸钾的Minisci反应(Scheme 9, c).相比较而言, 该反应不需要外加氧化剂并且可以兼容各类一级、二级以及三级烷基三氟硼酸钾.此外, 他们通过该方法对伏立康唑、喜树碱、法舒地尔和奎宁进行了选择性烷基化.

2016年, Molander课题组[29]报道了可见光镍协同催化α烷氧基三氟硼酸钾的脱硼酰基化反应.该反应机理为:酰氯和金属镍形成酰基镍物种, 随后与烷氧基三氟硼酸钾脱硼生成的烷氧基自由基发生氧化加成, 最后还原消除生成目标产物.该策略发展了一种新的酰基成键模式.随后他们进一步拓展了该活化模式的普适性, 分别实现了在可见光金属镍共催化条件下, 烷基三氟硼酸钾与酰氯[30]、酰胺[31]及羧酸[32]的偶联反应, 这些反应均具有良好的底物兼容性并且反应条件温和, 实现了以不同酰基源化合物为原料构建酮类化合物(Scheme 10).

2017年, Doyle小组和Rovis课题组[33]合作报道了光镍协同催化内消旋酸酐和苄基三氟硼酸盐发生对映选择性去对称化反应, 制备了带有两个相邻手性中心的酮酸化合物(Scheme 11).该反应对苄基三氟硼酸钾兼容性好, 但是对于酸酐兼容性较差, 当酸酐的β位有取代基时, 产率和选择性出现明显的降低.最后, 作者通过差向异构化实验发现催化剂与苄基三氟硼酸钾的相对量对反应立体选择性有着明显的影响.

2018年, 李朝军课题组[34]发展了一种新颖的方法实现了烷基以及芳基三氟硼酸钾的甲酰化反应(Scheme 12).在该体系下, 2, 3-丁二酮不仅作为底物(烷基或者芳基三氟硼酸钾)的活化试剂, 同时还作为酰基源参与反应.该反应条件温和, 不需要光催化剂, 同时使用水作为溶剂, 具有绿色环境友好的特点.作者推测反应机理可能为:烷基(芳基)三氟硼酸钾在溶液中部分以二氟烷基(芳基)硼烷的形成存在, 其可以和光活化的2, 3-丁二酮氧自由基发生SH2(双分子均裂取代机理)过程释放出烷基自由基.

2015年, 陈以昀课题组[35]在可见光催化下, 以Ru络合物为催化剂, 以BI-OAc为氧化剂, 实现了烷基三氟硼酸钾和α, β-不饱和羧酸的偶联反应(Scheme 13, a).该反应底物适用范围广, 官能团兼容性好, 且高选择性地得到反式产物.通过对反应机理的研究他们发现该反应的关键中间体为α, β-不饱和羧酸RCH=CHCOOH与BI-OAc生成的产物BI-OOCCH=CHR, 该中间体捕获体系中烷基硼化合物生成的烷基自由基, 随后消除得到产物并释放出二氧化碳气体以及BI自由基. 2016年, Molander课题组[12]发展了非过渡金属光催化剂Na2- Eosin Y催化的烷基三氟硼酸钾的烯基化反应(Scheme 13, b).在该体系下, 不需要添加氧化剂, 以芳香烯基砜作为自由基受体.该反应同样具有较好的底物兼容性以及高的立体选择性.

2013年, Akita课题组[36]以Ir作为光催化剂, 实现了α烷氧基三氟硼酸钾脱硼构建C(sp3)—C(sp3)键反应(Scheme 14, a).该反应经历α-烷氧基三氟硼酸钾的单电子氧化生成烷氧基自由基, 并被贫电子烯烃捕获生成新的自由基, 随后被还原为碳负离子并质子解生成对应的目标产物.值得一提的是, 该反应在太阳光下也能很好地进行.随后, 基于此工作, 他们进一步发展了光催化Boc保护α-氨基三氟硼酸钾[37]的烷基化反应, 并合成了具有生物活性的γ-氨基丁酸(GABA)衍生物(Scheme 14, b).在此工作基础上, α-硫醚三氟硼酸钾[38]也被成功地应用于该策略(Scheme 14, c). 2015年, 他们小组[39]对该反应进一步改进, 以廉价易得的常见染料MesAcr+为光催化剂, 实现了普通烷基三氟硼酸钾的烷基化反应(Scheme 14, d).此外, Yoshimi课题组[40]报道了菲作为光催化剂, 对苯二腈作为共催化剂实现了芳基硼酸类化合物与丙烯腈反应构建烷基腈化合物.他们发现环己基硼酸也可在该条件下转化为对应的烷基腈化合物.

2017年, Molander课题组[41]以α-三氟甲基芳基乙烯类化合物为自由基受体, 以烷基三氟硼酸钾或者烷基硅试剂作为自由基源, 实现了1, 1-二氟烯烃类化合物的构建.该方法具有优异的底物普适性, 可兼容酯基, 一级、二级以及三级胺, 醇羟基, 醛基, 炔基等多种官能团.该策略为合成二氟乙烯类化合物提供了一种强有力的方法(Scheme 15).
Meggers课题组[42]报道了在光照条件下, 以手性八面体Rh络合物和MesAcr+作为催化剂, 烷基三氟硼酸钾为自由基前体, 含吡唑或者咪唑导向基团的α, β-不饱和酮作为自由基受体, 通过自由基加成随后质子解得到目标产物, 实现了含导向基团的贫电子烯烃的手性烷基化加成反应(Scheme 16).该反应具有良好的底物兼容性, 可兼容一级、二级以及三级烷基硼化合物, 值得一提的是, 该反应具有优异的立体选择性.随后他们和Wiest课题组[43]合作通过DFT计算发现反应手性控制的关键在于α, β-不饱和2-酰基咪唑和手性Rh催化剂迅速形成配合物C, C作为体系中的烷基自由基捕获剂.该反应机理为: α, β-不饱和2-酰基咪唑和手性Rh催化剂迅速形成配合物C, C捕获体系中光生烷基自由基得到含Rh配合物自由基D, D进一步被体系中还原态光催化剂淬灭得到烯醇Rh化合物E, E与水反应得到目标化合物中间体F, 随后F与α, β-不饱和2-酰基咪唑发生配体交换得到目标产物以及参与反应的前体C.

2018年, Hanna课题组[44]实现了温和条件下光催化以烷基三氟硼酸钾为烷基化试剂的N-苄叉苯胺烷基化.该反应主要为烷基自由基对苄亚胺的加成(Scheme 17, a). 2019年, Molander课题组[45]报道了温和条件下光催化烷基三氟硼酸钾的Petasis反应(Scheme 17, b).该反应主要通过烷基自由基与原位生成的苄亚胺反应来实现转化的.此外, 该反应具有良好的官能团兼容性, 可以实现药物分子消炎疼、非诺贝特以及磺胺二甲氧嘧啶的衍生化.不仅如此, 具有潜在胰高血糖素受体调节作用药物的关键中间体也能用该方法合成.

2018年, 肖文精课题组[46]实现了光镍协同催化的苄基氮杂环丙烷的开环合成β-取代胺化合物(Scheme 18).在该体系中, 以苄基三氟硼酸钾作为自由基源, 选择性地加成在苄基氮杂环丙烷的苄位.然而, 该反应底物存在一定的限制性, 脂肪烷基三氟硼酸钾无法发生该转化.

2018年, 陈以昀课题组[47]报道了光催化合成α-季碳中心环酮以及呋喃酮类化合物.该反应以烷基硼酸作为自由基源.反应的关键在于烯丙醇与环碘试剂(BI-OAc)形成配合物活化烯丙醇(Scheme 19).

2019年, 陈以昀课题组[48]报道了无需光催化剂同时无需添加剂的光催化烷基硼化合物(硼酸或四氟硼酸钾)对α-酮酸的自由基加成反应(Scheme 20).该反应可以在有机溶剂与水混合溶剂中进行, 并成功避免了α-酮酸自身发生自由基脱羧, 也避免产物发生进一步的碳-碳键断裂.通过对反应机理的研究, 他们发现α-酮酸与烷基硼酸可以形成全新的分子复合物.该复合物具有可见光吸收并且在可见光照射下, 可发生脱硼生成烷基自由基.此外, 该复合物还可促进烷基自由基对酮酸的加成并抑制产物碳-碳键断裂.另外, 使用流动光反应装置可以合成克级规模的乳酸类药物分子关键中间体.据此, 他们实现了以硼试剂如硼酸三甲酯作为Lewis酸催化烷基二氢吡啶衍生物与α-酮酸的自由基加成反应.

2019年, 韩满意与王敏课题组[49]以烷基硼酸为原料, 采用BI-OAc作为活化试剂, 以丙烯酰胺为自由基捕获剂, 构建了3, 3-二取代吲哚酮结构(Scheme 21).该反应具有条件温和及不需要金属催化剂的特点.此外, 在该体系下烯基硼酸也能转化为相应的产物, 但是芳基硼无法实现该转化.
目前光催化的自由基反应多数实现了二组分反应, 实现三组分参与的反应更具有挑战性. 2019年, Molander课题组[50]报道了有机染料光催化的脱氢丙氨酸烷基化氟化反应, 合成多种α-氟取代氨基酸衍生物(Scheme 22).该反应可兼容炔基、酮羰基、酰胺基团、酯基以及磺酰胺基团等多种官能团.该反应的机理为光催化烷基三氟硼酸钾生成烷基自由基, 随后被脱氢丙氨酸捕捉并被Selectfluor淬灭得到目标产物.

2019年, Molander课题组[51]报道了光镍协同催化的芳基溴、烷基三氟硼酸钾以及乙烯基频哪醇硼酸酯的三组分反应合成烷基硼化合物(Scheme 23).该反应具有条件温和及底物普适性好的特点.该反应的机理为:光催化烷基三氟硼酸钾生成烷基自由基, 随后烷基自由基被烯基硼捕获生成α-硼酸酯自由基, 该自由基被Ni(0)捕获生成Ni(Ⅰ)中间体并与芳基溴氧化加成生成Ni(Ⅲ)化合物, Ni(Ⅲ)化合物进一步发生还原消除得到目标产物.基于该策略, 他们以71%的收率制备革兰氏阳性细菌胸苷酸激酶抑制剂(TK-666)的重要中间体.

2017年, Ley课题组[52]基于烷基硼酸酯以及烷基硼酸的Lewis酸性, 采用催化量的Lewis碱作为活化试剂, 在光照条件下使用Ir催化剂催化其生成烷基自由基.以贫电子烯烃这一常见自由基受体为捕获剂, 实现了C(sp3)—C(sp3)键的构建(Scheme 24, a).作者发现常见的Lewis碱如DMAP、奎宁环、3-羟基奎宁环以及三苯基膦均可实现烷基硼酸酯的活化.在该催化体系下, 烷基硼酸底物兼容性较烷基硼酸酯好. 2018年, 他们小组[53]对反应进行了优化, 采用Mes-Acr-4作为光催化剂, 通过流动反应装置, 提高了反应效率.基于此, 他们通过两步合成四种中枢神经系统GABA家族药物.随后2019年, 他们[54]继续以该策略实现了苄基频哪醇硼酸酯与芳香醛酮的加成反应, 高效地合成了二级以及三级醇类化合物(Scheme 24, b).

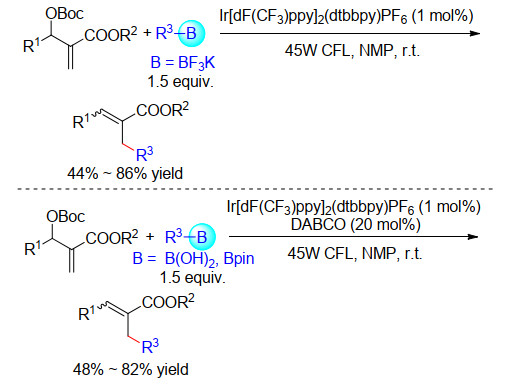
2018年, 许孝良课题组[55]实现了光催化烷基三氟硼酸酯与Baylis-Hillman反应产物衍生物的偶联反应, 合成了一系列α, β-不饱和羧酸酯衍生物.该反应具有良好的底物兼容性.遗憾的是, 该反应产物的立体(E/Z)选择性稍差.随后2019年, 他们小组[56]以烷基硼酸酯或者烷基硼酸作为自由基源实现了类似的转化.在该体系下, 催化量的碱1, 4-二氮杂二环[2.2.2]辛烷(DABCO)作为三配位硼的活化试剂(Scheme 25).
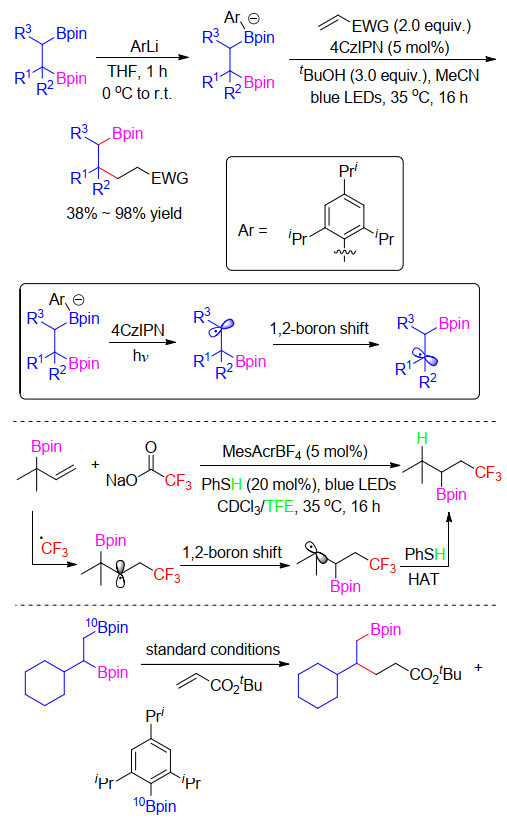
2019年, Aggarwal课题组[57]报道了烷基频哪醇硼酸酯脱硼环丁烷化反应(Scheme 26).作者发现相比于烷基三氟硼酸钾, 苯基锂与烷基硼酸酯形成的四配位硼具有更低的氧化电势, 可以被廉价易得的有机染料4CzIPN单电子氧化生成烷基自由基, 同时伴随着苯基频哪醇硼酸酯副产物的生成.基于此, 当以γ-碘代烯酸酯作为自由基捕获剂时, 通过自由基加成以及脱碘环化反应得到目标环丁烷类化合物.此外, 当加入不同碳数的末端卤代烯酸酯, 三环、五环、六环以及七环均可被构建.与Ley课题组报道方法相比, 该方法具有更加良好的底物兼容性, 可以兼容多种一级、二级以及三级烷基频哪醇硼酸酯, 并且多种药物以及天然产物硼化衍生物也可以以良好的收率得到目标产物.

在此工作基础上, Aggarwal课题组[58]报道了1, 2-二硼酸酯的选择性烷基化反应(Scheme 27).他们发现在光催化1, 2-双硼酸酯的脱硼化反应中, 等化学计量的活化试剂芳基锂优先同位阻小的硼形成四配位硼, 随后发生光催化脱硼过程生成β-硼酸酯烷基自由基, 该自由基迅速经历1, 2-硼转移过程形成热力学稳定的多取代基大位阻的烷基自由基, 随后烷基自由基被贫电子烯烃捕捉, 从而允许空间受阻的硼酸酯发生选择性转化.该反应不需要金属催化剂并且底物能兼容大多数不同取代基.此外, 作者通过一系列实验来验证该反应的机理, 当以1, 2, 3-含伯硼、仲硼以及叔硼化合物作为底物时, 热力学控制有利于最稳定的叔碳自由基中心作为反应位点.当用三氟甲基亚磺酸钠生成三氟甲基自由基处理烯丙基硼酸酯, 目标产物发生1, 2-硼迁移. DFT计算也支持了1, 2-迁移过程.此外, 通过硼同位素实验证明了该迁移过程并且证实形成四配位硼位点位于位阻小的硼酸酯上.
目前, 实现烷基硼化合物脱硼构建C—O键常用的方法是过量氧化剂氧化, 如H2O2/NaOH、NaBO4等. 2012年, Akita课题组[59]报道了第一例光催化烷基硼酸盐的脱硼C—O成键反应(Scheme 28).该反应机理为:激发态的Ir光催化剂单电子氧化烷基硼酸盐化合物生成烷基自由基, 随后烷基自由基被四甲基哌啶氧化物(TEMPO)捕获生成目标产物.尽管该反应体系的底物具有一定的局限性, 如不能将简单的一级以及二级烷基三氟硼酸盐类化合物转化为相应的目标产物, 但是该反应的发现为烷基硼化合物的活化提供了一种新的策略, 同时也表明四配位硼络合物可在光催化条件下作为一种良好的自由基前体.

早在2012年, 肖文精课题组[60]就实现了空气氛围下, 光催化氧化芳基硼酸生成酚的反应.然而, 烷基硼化合物的相应转化却缺乏报道. 2018年, 张博课题组[61]以有机染料玫瑰红为光催化剂, 乙醇为溶剂, 在空气氛围温和条件下将烷基以及芳基硼化合物氧化为对应的醇或者酚(Scheme 29).在该催化体系下, 空气中的氧气作为氧化剂.当以手性硼化合物为底物时, 反应得到对应的手性醇, 表明该体系下, 烷基硼化合物并没有形成碳中心自由基.机理研究表明, 该体系可能是光催化原位生成双氧水参与了有机硼的氧化过程.

目前, 通过光催化实现烷基硼化合物脱硼构建碳-硫键的报道较少. 2018年, 吴劼课题组[62]首次报道了通过光催化烷基三氟硼酸钾、DABCO•(SO2)2和贫电子取代烯烃的三组分反应合成烷基磺酰基化合物的方法(Scheme 30, a).该反应可以兼容不同的烷基三氟硼酸钾, 同时含有多种敏感官能团如羟基、酮羰基以及硝基的贫电子烯烃在该反应条件下也可被兼容.作者认为该反应的机理为激发态光催化剂将烷基三氟硼酸钾氧化为烷基自由基, 随后烷基自由基捕获体系中二氧化硫生成砜自由基, 进一步砜自由基与烯烃加成并氧化还原态光催化剂生成对应的碳负离子, 最后质子化得到目标产物.基于此工作, 他们[63]又报道了类似条件下生成的砜自由基与烯丙基溴反应得到烯丙基砜(Scheme 30, b).此外, 该小组[64]还报道了砜自由基与芳香末端炔烃反应得到的烯基砜产物, 不同的是, 该反应需要Cu(OTf)2作为共催化剂.铜催化剂捕获砜自由基生成砜铜物种, 该物种与芳香末端炔烃加成生成烯基铜物种, 随后质子化得到目标产物(Scheme 30, c). 2020年, 该小组[65]进一步以烷基三氟硼酸钾作为自由基源, 并通过Na2S2O5捕获生成对应的砜自由基, 最后与炔溴反应得到炔砜类化合物(Scheme 30, d).

烷基碳硼键脱硼质子化具有一定的研究价值. 2019年, Studer课题组[66]报道了光催化的烷基硼酸酯脱硼氢化反应(Scheme 31).他们使用苯基锂作为亲核试剂将烷基硼化合物预先制备为四配位硼络合物, 随后以苯硫酚作为氢源, Ir配合物作为光催化剂实现了烷基频哪醇硼酸酯的氢化反应.当以烯烃为原料时, 结合硼氢化反应以及Matteson同系化反应, 实现了烯烃反马氏甲基化反应.此外, 他们进一步拓展了该反应的应用价值, 以Boc保护四氢吡咯为原料, 通过简单的四步转化合成了吲哚里西啶209B中间体和可卡因关键中间体.
综上所述, 近些年来通过光催化的手段活化烷基硼化合物极大地丰富了其转化类型, 并且反应具有条件温和及官能团兼容性广的优点.然而, 该领域依旧存在一系列挑战需要解决.首先, 目前烷基硼化合物大多原料为烷基三氟硼酸钾, 其合成需要通过其他硼化合物的进一步转化制备.因此, 以易制备易分离的烷基硼酸酯直接作为自由基源具有重要的研究意义以及更好的经济性.仅有的几例烷基硼酸酯的活化存在着底物普适性较差、活化方式单一的问题.故而, 发展更为廉价、催化量、普适性好烷基硼酸酯活化试剂显得十分有必要.其次, 受制于自由基受体的种类, 光催化烷基硼化合物脱硼成键类型依旧十分有限, 进一步拓展其成键类型具有十分重要的意义.最后, 目前关于该类反应的不对称催化体系十分少见, 如何有效控制反应的立体选择性仍然是一个巨大的挑战.
Miyaura, N.; Yamada, K.; Suzuki, A. Tetrahedron Lett. 1979, 20, 3437. doi: 10.1016/S0040-4039(01)95429-2
Brown, H. C.; Tierney, P. A. J. Am. Chem. Soc. 1958, 80, 1552. doi: 10.1021/ja01540a011
(a) Chan, D. M. T.; Monaco, K. L.; Wang, R.-P.; Winters, M. P. Tetrahedron Lett. 1998, 39, 2933.
(b) Evans, D. A.; Katz, J. L.; West, T. R. Tetrahedron Lett. 1998, 39, 2937.
(c) Lam, P. Y. S.; Clark, C. G.; Saubern, S.; Adams, J.; Winters, M. P.; Chan, D. M. T.; Combs, A. Tetrahedron Lett. 1998, 39, 2941.
Zweifel, G.; Arzoumanian, H.; Whitney, C. C. J. Am. Chem. Soc. 1967, 89, 3652. doi: 10.1021/ja00990a061
Lennox, A. J.; Lloyd-Jones, G. C. Chem. Soc. Rev. 2014, 43, 412. doi: 10.1039/C3CS60197H
(a) Leonori, D.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2015, 54, 1082.
(b) Sandford, C.; Aggarwal, V. K. Chem. Commun. 2017, 53, 5481.
(a) Chen, Y.; Lu, L.-Q.; Yu, D.-G.; Zhu, C.-J.; Xiao, W.-J. Sci. China: Chem. 2018, 62, 24.
(b) Chen, D.; Liu, J.; Zhang, X.; Jiang, H.; Li, J. Chin. J. Org. Chem. 2019, 39, 3353(in Chinese).
(陈丹, 刘剑沉, 张馨元, 蒋合众, 李加洪, 有机化学, 2019, 39, 3353.)
(c) Dai, X.; Xu, X.; Li, X. Chin. J. Org. Chem. 2013, 33, 2046(in Chinese).
(戴小军, 许孝良, 李小年, 有机化学, 2013, 33, 2046.)
Duret, G.; Quinlan, R.; Bisseret, P.; Blanchard, N. Chem. Sci. 2015, 6, 5366. doi: 10.1039/C5SC02207J
Duan, K.; Yan, X.; Liu, Y.; Li, Z. Adv. Synth. Catal. 2018, 360, 2781. doi: 10.1002/adsc.201701626
Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 2280. doi: 10.1021/ja413208y
Dai, J. J.; Zhang, W. M.; Shu, Y. J.; Sun, Y. Y.; Xu, J.; Feng, Y. S.; Xu, H. J. Chem. Commun. 2016, 52, 6793. doi: 10.1039/C6CC01530A
Heitz, D. R.; Rizwan, K.; Molander, G. A. J. Org. Chem. 2016, 81, 7308. doi: 10.1021/acs.joc.6b01207
Shi, D.; Xia, C.; Liu, C. CCS Chem. 2020, 2, 1718.
Tellis, J. C.; Primer, D. N.; Molander, G. A. Science 2014, 345, 433. http://www.onacademic.com/detail/journal_1000036301985210_f589.html
Gutierrez, O.; Tellis, J. C.; Primer, D. N.; Molander, G. A.; Kozlowski, M. C. J. Am. Chem. Soc. 2015, 137, 4896. doi: 10.1021/ja513079r
Primer, D. N.; Karakaya, I.; Tellis, J. C.; Molander, G. A. J. Am. Chem. Soc. 2015, 137, 2195. doi: 10.1021/ja512946e
Primer, D. N.; Molander, G. A. J. Am. Chem. Soc. 2017, 139, 9847. doi: 10.1021/jacs.7b06288
Alam, R.; Molander, G. A. J. Org. Chem. 2017, 82, 13728. doi: 10.1021/acs.joc.7b02589
Ryu, D.; Primer, D. N.; Tellis, J. C.; Molander, G. A. Chem.-Eur. J. 2016, 22, 120. doi: 10.1002/chem.201504079
Tellis, J. C.; Amani, J.; Molander, G. A. Org. Lett. 2016, 18, 2994. doi: 10.1021/acs.orglett.6b01357
Matsui, J. K.; Molander, G. A. Org. Lett. 2017, 19, 436. doi: 10.1021/acs.orglett.6b03448
Lima, F.; Kabeshov, M. A.; Tran, D. N.; Battilocchio, C.; Sedelmeier, J.; Sedelmeier, G.; Schenkel, B.; Ley, S. V. Angew. Chem., Int. Ed. 2016, 55, 14085. doi: 10.1002/anie.201605548
Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinummo, M. Tetrahedron 1971, 27, 3575. doi: 10.1016/S0040-4020(01)97768-3
Proctor, R. S. J.; Phipps, R. J. Angew. Chem., Int.Ed. 2019, 58, 13666. doi: 10.1002/anie.201900977
Li, G. X.; Morales-Rivera, C. A.; Wang, Y. X.; Gao, F.; He, G.; Liu, P.; Chen, G. Chem. Sci. 2016, 7, 6407. doi: 10.1039/C6SC02653B
Zhang, W. M.; Dai, J. J.; Xu, J.; Xu, H. J. J. Org. Chem. 2017, 82, 2059. doi: 10.1021/acs.joc.6b02891
Matsui, J. K.; Primer, D. N.; Molander, G. A. Chem. Sci. 2017, 8, 3512. doi: 10.1039/C7SC00283A
Yan, H.; Hou, Z. W.; Xu, H. C. Angew. Chem., Int. Ed. 2019, 58, 4592. doi: 10.1002/anie.201814488
Amani, J.; Sodagar, E.; Molander, G. A. Org. Lett. 2016, 18, 732. doi: 10.1021/acs.orglett.5b03705
Amani, J.; Molander, G. A. J. Org. Chem. 2017, 82, 1856. doi: 10.1021/acs.joc.6b02897
Amani, J.; Alam, R.; Badir, S.; Molander, G. A. Org. Lett. 2017, 19, 2426. doi: 10.1021/acs.orglett.7b00989
Amani, J.; Molander, G. A. Org. Lett. 2017, 19, 3612. doi: 10.1021/acs.orglett.7b01588
Stache, E. E.; Rovis, T.; Doyle, A. G. Angew. Chem., Int. Ed. 2017, 56, 3679. doi: 10.1002/anie.201700097
Liu, W.; Liu, P.; Lv, L.; Li, C. J. Angew. Chem., Int. Ed. 2018, 57, 13499. doi: 10.1002/anie.201807181
Huang, H.; Jia, K.; Chen, Y. Angew. Chem., Int. Ed. 2015, 54, 1881. doi: 10.1002/anie.201410176
Miyazawa, K.; Yasu, Y.; Koike, T.; Akita, M. Chem. Commun. 2013, 49, 7249. doi: 10.1039/c3cc42695e
Miyazawa, K.; Koike, T.; Akita, M. Adv. Synth. Catal. 2014, 356, 2749. doi: 10.1002/adsc.201400556
Li, Y.; Miyazawa, K.; Koike, T.; Akita, M. Org. Chem. Front. 2015, 2, 319. doi: 10.1039/C4QO00352G
Chinzei, T.; Miyazawa, K.; Yasu, Y.; Koike, T.; Akita, M. RSC Adv. 2015, 5, 21297. doi: 10.1039/C5RA01826A
Iwata, Y.; Tanaka, Y.; Kubosaki, S.; Morita, T.; Yoshimi, Y. Chem. Commun. 2018, 54, 1257. doi: 10.1039/C7CC09140K
Lang, S. B.; Wiles, R. J.; Kelly, C. B.; Molander, G. A. Angew. Chem., Int. Ed. 2017, 56, 15073. doi: 10.1002/anie.201709487
Huo, H.; Harms, K.; Meggers, E. J. Am. Chem. Soc. 2016, 138, 6936. doi: 10.1021/jacs.6b03399
Tutkowski, B.; Meggers, E.; Wiest, O. J. Am. Chem. Soc. 2017, 139, 8062. doi: 10.1021/jacs.7b01786
Plasko, D. P.; Jordan, C. J.; Ciesa, B. E.; Merrill, M. A.; Hanna, J. M. Photochem. Photobiol. Sci. 2018, 17, 534. doi: 10.1039/C8PP00061A
Yi, J.; Badir, S. O.; Alam, R.; Molander, G. A. Org. Lett. 2019, 21, 4853. doi: 10.1021/acs.orglett.9b01747
Yu, X. Y.; Zhou, Q. Q.; Wang, P. Z.; Liao, C. M.; Chen, J. R.; Xiao, W. J. Org. Lett. 2018, 20, 421. doi: 10.1021/acs.orglett.7b03747
Liu, M.; Huang, H.; Chen, Y. Chin. J. Chem. 2018, 36, 1209. doi: 10.1002/cjoc.201800461
Xie, S.; Li, D.; Huang, H.; Zhang, F.; Chen, Y. J. Am. Chem. Soc. 2019, 141, 16237. doi: 10.1021/jacs.9b09099
Li, X.; Han, M.-Y.; Wang, B.; Wang, L.; Wang, M. Org. Biomol. Chem. 2019, 17, 6612. doi: 10.1039/C9OB01023H
Sim, J.; Campbell, M. W.; Molander, G. A. ACS Catal. 2019, 9, 1558. doi: 10.1021/acscatal.8b04284
Campbell, M. W.; Compton, J. S.; Kelly, C. B.; Molander, G. A. J. Am. Chem. Soc. 2019, 141, 20069. doi: 10.1021/jacs.9b08282
Lima, F.; Sharma, U. K.; Grunenberg, L.; Saha, D.; Johannsen, S.; Sedelmeier, J.; Van der Eycken, E. V.; Ley, S. V. Angew. Chem., Int. Ed. 2017, 56, 15136. doi: 10.1002/anie.201709690
Lima, F.; Grunenberg, L.; Rahman, H. B. A.; Labes, R.; Sedelmeier, J.; Ley, S. V. Chem. Commun. 2018, 54, 5606. doi: 10.1039/C8CC02169D
Chen, Y.; May, O.; Blakemore, D. C.; Ley, S. V. Org. Lett. 2019, 21, 6140. doi: 10.1021/acs.orglett.9b02307
Ye, H.; Ye, Q.; Cheng, D.; Li, X.; Xu, X. Tetrahedron Lett. 2018, 59, 2046. doi: 10.1016/j.tetlet.2018.04.035
Ye, H.; Zhao, H.; Ren, S.; Ye, H.; Cheng, D.; Li, X.; Xu, X. Tetrahedron Lett. 2019, 60, 1302. doi: 10.1016/j.tetlet.2019.04.015
Shu, C.; Noble, A.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2019, 58, 3870. doi: 10.1002/anie.201813917
Kaiser, D.; Noble, A.; Fasano, V.; Aggarwal, V. K. J. Am. Chem. Soc. 2019, 141, 14104. doi: 10.1021/jacs.9b07564
Yasu, Y.; Koike, T.; Akita, M. Adv. Synth. Catal. 2012, 354, 3414. doi: 10.1002/adsc.201200588
Zou, Y. Q.; Chen, J. R.; Liu, X. P.; Lu, L. Q.; Davis, R. L.; Jorgensen, K. A.; Xiao, W. J. Angew. Chem., Int. Ed. 2012, 51, 784. doi: 10.1002/anie.201107028
Weng, W. Z.; Liang, H.; Zhang, B. Org. Lett. 2018, 20, 4979. doi: 10.1021/acs.orglett.8b02095
Liu, T.; Li, Y.; Lai, L.; Cheng, J.; Sun, J.; Wu, J. Org. Lett. 2018, 20, 3605. doi: 10.1021/acs.orglett.8b01385
Ye, S.; Li, X.; Xie, W.; Wu, J. Asian J. Org. Chem. 2019, 8, 893. doi: 10.1002/ajoc.201900172
Liu, T.; Ding, Y.; Fan, X.; Wu, J. Org. Chem. Front. 2018, 5, 3153. doi: 10.1039/C8QO00965A
Gong, X.; Yang, M.; Liu, J.-B.; He, F.-S.; Wu, J. Org. Chem. Front. 2020, 7, 938.
Clausen, F.; Kischkewitz, M.; Bergander, K.; Studer, A. Chem. Sci. 2019, 10, 6210. doi: 10.1039/C9SC02067E

图式 1 光催化烷基硼化合物的活化模式
Scheme 1 Activated models of alkyl boron compounds under photoredox condition

图式 2 光催化烷基三氟硼酸钾的炔基化反应
Scheme 2 Photoredox catalyzed alkynylation of alkyltrifluo-roborates

图式 3 光催化烷基三氟硼酸钾的氰基化反应
Scheme 3 Photoredox catalyzed cyanylation of alkyltrifluoroborates

图式 5 光镍协同催化苄基三氟硼酸钾的芳基化反应
Scheme 5 Photoredox/nickel dual catalyzed arylation of benzylic trifluoroborates

图式 6 光镍协同催化烷基三氟硼酸钾的芳基化反应
Scheme 6 Dual photoredox/nickel catalyzed arylation of alkyltrifluoroborates

图式 7 光催化苄基频哪醇硼酸酯的芳基化反应
Scheme 7 Photoredox catalyzed arylation of benzylic pinacol boronates

图式 8 光催化烷基硼酸的Minisci反应
Scheme 8 Photoredox catalyzed Minisci reaction of alkyl boric acid

图式 9 光催化烷基三氟硼酸钾的Minisci反应
Scheme 9 Photoredox catalyzed Minisci reaction of alkyltrifluoroborates

图式 10 光镍协同催化的烷基三氟硼酸钾的酰基化反应
Scheme 10 Dual photoredox/nickel catalyzed acylation of alkyltrifluoroborates

图式 11 光镍协同催化内消旋酸酐的对映选择性去对称化反应
Scheme 11 Dual nickel-photoredox-catalyzed enantioselective desymmetrization of cyclic meso-anhydrides

图式 12 2, 3-丁二酮光共活化烷基三氟硼酸钾
Scheme 12 Dual diacetyl/photoredox catalyzed activation of organotrifluoroborate

图式 13 光催化烷基三氟硼酸钾烯基化反应
Scheme 13 Photoredox catalyzed alkenylation of alkyltrifluoroborates

图式 14 贫电子烯烃作为受体的烷基硼化合物光催化转化
Scheme 14 Photoredox-catalyzed transformation of alkyl boron compounds using electron-deficient alkenes as accepter

图式 16 光手性铑共催化烷基三氟硼酸钾对烯烃的手性加成
Scheme 16 Dual chiral rhodium complex/photoredox catalyzed enantioselective addition of alkyltrifluoroborates to alkenes

图式 17 光催化烷基三氟硼酸钾对亚胺的加成反应
Scheme 17 Photoredox-catalyzed alkyltrifluoroborates addition to imines

图式 18 光镍协同催化β取代胺的合成
Scheme 18 Dual photoredox/nickel-catalyzed synthesis of β‑substitued amines

图式 19 光催化烷基硼酸与烯丙醇的加成重排反应
Scheme 19 Photoredox the addition/rearrangement reaction of alkyl boric acid with allyl alcohols

图式 20 光催化烷基硼化合物对α-酮酸的加成反应
Scheme 20 Photoredox-catalyzed alkyl boron compounds addition to ketoacids

图式 22 光催化脱氢丙氨酸烷基化氟化反应
Scheme 22 Photoredox-catalyzed carbofluorination of dehydroalanine

图式 23 光催化烷基硼频哪醇硼酸酯的合成
Scheme 23 Photoredox catalyzed synthesis of alkyl pinacol boronates

图式 24 Lewis碱活化烷基硼化合物的光催化转化
Scheme 24 Photeredox conversion of alkyl boron compounds activated by Lewis base

图式 25 光催化α, β-不饱和羧酸酯的合成
Scheme 25 Photoredox catalyzed synthesis of α, β-unsaturated carboxylic esters

图式 26 光催化烷基频哪醇硼酸酯的环化反应
Scheme 26 Photoredox catalyzed cyclization of alkyl pinacol boronates

图式 27 光催化1, 2-二烷基硼酸酯的选择性转化
Scheme 27 Photoredox catalyzed selectively alkylarylation of 1, 2-bis-alkyl pinacol boronates

图式 28 光催化烷基硼酸盐的碳氧成键反应
Scheme 28 Construction of C—O bonds by Photoredox catalyzed alkylborates deboronation

图式 29 光催化空气氧化烷基硼化合物
Scheme 29 Photoredox catalyzed air oxidation of alkyl boron compounds

图式 30 光催化烷基三氟硼酸钾构建C—S键
Scheme 30 Construction of C—S bonds by photoredox catalyzed alkyltrifluoroborates

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




 下载:
下载:




 下载:
下载: