
图式 1.
叔丁基亚磺酰胺诱导手性胺的合成
Scheme 1.
Synthesis of chiral amines using tert-butanesulfinamide

1997年, Ellman课题组[1]报道了光学纯叔丁基亚磺酰胺的制备方法, 并利用叔丁基亚磺酰胺诱导合成了手性胺.之后以手性叔丁基亚磺酰胺为手性辅基不对称合成胺的方法受到化学家的广泛关注, 相关的研究蓬勃发展, 目前该策略已成为合成手性胺最广泛使用的方法之一[2].利用手性叔丁基亚磺酰胺诱导合成手性胺的通用步骤为:首先叔丁基亚磺酰胺和醛或酮缩合生成叔丁基亚磺酰亚胺, 新生成的叔丁基亚磺酰亚胺进一步与亲核试剂发生不对称1, 2-加成反应引入胺的α位立体中心; 或者叔丁基亚磺酰亚胺在碱性条件下去质子化, 生成的叔丁基亚磺酰基金属烯胺[3]进一步与亲电试剂发生加成反应, 可以引入胺的β位立体中心(Scheme 1).
利用叔丁基亚磺酰胺合成手性胺具有以下优势[2e,4]: (1)叔丁基亚磺酰胺在温和条件下可以与多种醛或酮直接缩合, 以高收率得到稳定的叔丁基亚磺酰亚胺; (2)由于叔丁基亚磺酰基的活化作用, 叔丁基亚磺酰亚胺具有较强的亲电性, 可与多种亲核试剂发生高效的加成, 从而转化为不同的含氮产物; 另外叔丁基亚磺酰基与金属配位可以提高加成反应的立体选择性; (3)叔丁基亚磺酰基也可以充当保护基, 与甲酰基保护基类似, 并且它在强碱和过渡金属催化的条件下都能稳定存在; (4)叔丁基亚磺酰基可以方便地在盐酸等条件下脱除, 得到胺类化合物, 反应通常效率很高.
总结了近些年利用手性叔丁基亚磺酰胺作为手性辅基诱导生成手性胺中间体在天然产物不对称全合成中的应用.根据引入立体中心的方式进行分类, 包括叔丁基亚磺酰亚胺的1, 2-加成反应和叔丁基亚磺酰基金属烯胺与亲电试剂的加成反应两部分.
叔丁基亚磺酰亚胺在多种亲核加成反应中都有应用, 在此分为三部分介绍:叔丁基亚磺酰亚胺与亲核试剂的1, 2-加成反应、叔丁基亚磺酰亚胺的还原和SmI2促进的叔丁基亚磺酰亚胺的还原偶联反应.
由于叔丁基亚磺酰基的吸电子效应, 叔丁基亚磺酰亚胺表现出较强的亲电性, 可与多种亲核试剂发生1, 2-加成反应.此外, 化学家还发展了金属催化的叔丁基亚磺酰亚胺的加成反应.根据叔丁基亚磺酰亚胺与不同的亲核试剂发生1, 2-加成反应, 在天然产物全合成中的应用进行介绍, 主要分为四部分:包括叔丁基亚磺酰亚胺与格氏试剂的1, 2-加成反应、叔丁基亚磺酰亚胺与有机锂试剂的, 2-加成反应、金属催化的1, 2-加成反应以及与其他碳负离子的1, 2-加成反应.
2006年, Rozwadowska课题组[5]报道了(S)-(-)- salsolidine的全合成.他们利用叔丁基亚磺酰亚胺与格氏试剂发生立体选择性的1, 2-加成反应, 成功引入分子内唯一的手性中心.
3, 4-二甲氧基苯甲醛(1)与(R)-叔丁基亚磺酰胺在Ti(OiPr)4作用下缩合生成叔丁基亚磺酰亚胺2, 随后与甲基溴化镁发生加成反应, 以高收率和立体选择性得到化合物3, dr值为98:2.化合物3在盐酸作用下脱除叔丁基亚磺酰基后, 与溴代乙醛缩二乙醇(4)发生氮烷基化反应得到化合物5.最后在酸性条件下发生分子内的傅克羟烷基化反应, 生成的环化产物在NaBH4和三氟醋酸(TFA)作用下脱氧得到(S)-(-)-salsolidine, ee值为98% (Scheme 2).

该课题组通过线性5步转化, 并以24%的总收率和98%的ee值合成了(S)-(-)-salsolidine.该策略丰富了合成异喹啉骨架的方法, 同时也可以用于合成一些1, 2, 3, 4-四氢咔啉(THBC)生物碱.
2015年, 林国强课题组[6]发展了一种高效不对称合成反式-5-羟基-6-烯基/炔基-2-哌啶酮的方法, 并用该方法实现了(+)-swainsonine的合成.采用的关键策略是手性叔丁基亚磺酰亚胺与炔基格氏试剂的1, 2-加成反应.
起始物6先后与乙炔溴化镁和Boc2O反应, 两步一锅法制备得到哌啶酮化合物7.在Lindlar催化剂催化下将7结构中的炔键氢化同时脱除Boc保护基, 生成的产物与烯丙基溴发生氮烷基化反应得到双烯化合物8.在第二代Grubbs催化剂的催化下化合物8发生关环复分解(RCM)反应, 构建五元环, 得到双环化合物9.化合物9发生立体选择性的双羟化反应生成二醇化合物10.最后化合物10经过LiAlH4还原和脱除叔丁基二甲基硅烷基(TBS)即可得到(+)-swainsonine.此外, 林国强课题组[7]应用该策略还合成了streptopyrrolidine (Scheme 3).
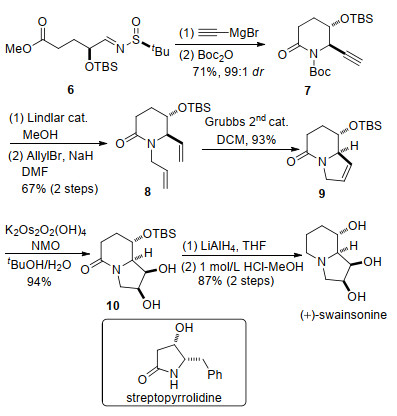
该课题组开发的合成反式-5-羟基-6-烯基/炔基-2-哌啶酮的方法具有较好的立体选择性, 并且多种类型的格氏试剂均适用于此反应.该方法具有应用于更多复杂化生物碱合成的潜力.
2016年, Love课题组[8]利用叔丁基亚磺酰亚胺的立体选择性加成制备了关键手性炔丙基胺中间体, 继而完成了K777的合成.
以苯丙醛11为起始物, 与(S)-叔丁基亚磺酰胺在KHSO4作用下缩合得到叔丁基亚磺酰亚胺12.亚胺12与炔基格氏试剂13发生不对称的加成反应生成14, 再经过4步保护基转化得到炔丙基胺15.在Wilkinson催化剂作用下, 苯硫酚与炔烃15加成得到乙烯基硫化物16, 16经过m-CPBA氧化为砜基化合物17.在TFA作用下化合物17脱除Boc保护基, 最后与手性酸18发生缩合得到K777 (Scheme 4).
作者使用叔丁基亚磺酰亚胺合成K777, 避免了使用昂贵的L-苯基丁氨酸作为起始原料, 提高了合成的经济性.
2017年, Amat课题组[9]发展了一种合成5, 7-顺式二取代十氢喹啉的方法, 并应用该方法合成了天然产物(-)-cermizine B.作者通过叔丁基亚磺酰亚胺与烯丙基格氏试剂的不对称1, 2-加成反应, 构建了一个氮杂手性中心.
在InCl3催化作用下, 烯酮19和烯丙基硅20发生Sakurai反应生成化合物21.化合物21和(R)-苯基甘氨醇在酸性条件下缩合生成单一的三环顺式稠合内酰胺22, 随后经过三氯化钌催化的氧化切断, 生成的醛基化合物与(R)-叔丁基亚磺酰胺缩合得到叔丁基亚磺酰亚胺23.烯丙基溴化镁与亚胺23发生高立体选择性的加成反应, 立体专一性地得到加成产物24.化合物24脱去叔丁基亚磺酰基后, 与丙烯酰氯25发生酰胺化反应生成双烯化合物26.化合物26经过RCM反应得到二氢吡啶酮衍生物27, 最后经过4步转化完成对氧化态的调整, 可以得到天然产物(-)-cermizine B.该课题组[10]应用同样的策略合成了(+)-serratezomine E和(+)-lucidu- line (Scheme 5).

Amat课题组应用叔丁基亚磺酰亚胺的高立体选择性1, 2-加成反应和RCM反应高效实现了手性哌啶环的构建.该策略丰富了包含手性哌啶环天然产物的合成方法.
2018年, Taghizadeh课题组[11]以(S)-叔丁基亚磺酰胺作为手性辅基引入(S)-ketamine结构中唯一的手性中心, 继而完成了(S)-ketamine的合成.
用乙二醇对1, 2-环己二酮28进行单保护, 与(S)-叔丁基亚磺酰胺缩合生成叔丁基亚磺酰亚胺30.接下来2-氯苯基溴化镁31和叔丁基亚磺酰亚胺30发生高立体选择性的1, 2-加成反应构建氮杂季碳手性中心, 得到化合物32, dr值为9:1.最后通过2步脱保护转化实现了(S)-ketamine的合成(Scheme 6).

Taghizadeh课题组报道的通过芳基格式试剂对叔丁基亚磺酰亚胺的加成, 构建手性苄胺的策略, 拓展了苄位手性季碳中心的构建方法.
应用叔丁基亚磺酰亚胺与格氏试剂的1, 2-加成反应作为关键合成策略完成的分子还有Pozo课题组[12a]合成的(-)-pinidinol、张洪彬课题组[12b]合成的(+)-nocar- dioazine B、Prasad课题组[12c]合成的lentiginosine和Methyldihydropalustramate以及Muñiz课题组[12d]合成的nicotine.
|
|
2011年, Reisman课题组[13]报道了手性苯醌单缩酮亚磺酰亚胺的制备方法, 并将其应用到天然产物(-)-3- demethoxyerythratidinone的全合成中.采用的关键策略是手性叔丁基亚磺酰亚胺与金属有机物发生高立体选择性1, 2-加成反应引入氮杂螺环立体中心.
苯醌单缩酮33与(R)-叔丁基亚磺酰胺缩合得到叔丁基亚磺酰亚胺34.亚胺34与芳基锂发生高立体选择性的1, 2-加成, 经过一锅法分子内氮烷基化引入螺环手性中心, 得到化合物36, dr值为98:2.化合物36与锡试剂37在零价钯促进下发生Stille偶联反应, 在酸作用下脱除叔丁基亚磺酰基, 氨基与烯醇醚在酸性条件下发生缩合形成氮杂五元环, 生成化合物38.最终还原38生成(-)-3-demethoxyerythratidinone (Scheme 7).

Reisman课题组发展了手性苯醌单缩酮亚磺酰亚胺与金属有机化合物的加成反应, 构建了4-氨基环己二烯酮的季碳手性中心, 利用该方法高效地实现了(-)-3-de- methoxyerythratidinone的螺环手性中心的构建, 并最终以6步26%的收率完成了(-)-3-demethoxyerythrati- dinone的合成.此方法为合成含有4-氨基环己二烯酮片段的天然产物提供了新思路.
2017年, Morris课题组[14]报道了萘基异喹啉类生物碱dioncophylline E的全合成.其核心策略是芳基三乙酸铅与萘酚通过Pinhey-Barton ortho-arylation反应连接.通过对叔丁基亚磺酰亚胺的高立体选择性加成反应引入C(3)位手性中心, 同时引入构建异喹啉环所需的氮原子.
作者从苯酚化合物39出发, 经过4步转化得到芳基三乙酸铅40, 再与萘酚41发生偶联反应得到联芳化合物42.接下来需要构建异喹啉环, 化合物42经Bn保护后, 其结构中的甲基在二异丙基氨基锂(LDA)条件下去质子化生成碳负离子, 与亚胺43发生高立体选择性的加成反应生成化合物44, dr值为97:3.化合物44用二异丁基氢化铝(DIBAL-H)还原后, 在盐酸作用下脱除叔丁基亚磺酰基, 生成的胺基与醛基缩合构建异喹啉45.在MeLi和CeCl3存在条件下[15], 亚胺45发生立体选择性的亲核加成反应构建C(1)位手性中心, dr值大于97:3, 最后在BCl3条件下脱除Bn保护基, TFA酸化合成dioncophylline E•TFA (Scheme 8).

Morris课题组报道的手性叔丁基亚磺酰亚胺与苄基负离子发生不对称加成反应, 高效地构建1, 3-二取代四氢异喹啉中的两个手性中心.
2019年, Prasad课题组[16]报道了手性叔丁基亚磺酰亚胺与二苯基烯丙基亚胺负离子发生高立体选择性的加成反应, 生成手性的1, 2-二胺基化合物.他们利用该方法经过6步转化完成了(-)-epiquinamide的合成.
二苯基烯丙基亚胺47在nBuLi作用下发生去质子化形成碳负离子, 再与5-溴戊醛衍生的叔丁基亚磺酰亚胺46发生高立体选择性的加成反应, 引入(-)-epiquina- mide结构中的两个立体中心.在NaH作用下发生分子内SN2反应得到哌啶化合物48, dr值为96:4.化合物48经过4步官能团转化生成双烯化合物49, 经过RCM反应构建二氢哌啶环, 最后还原双键实现了天然产物的(-)-epiquinamide的合成(Scheme 9).

作者报道的手性叔丁基亚磺酰亚胺与2-氮杂烯丙基负离子的不对称加成反应, 可以构建1, 2-二胺基中间体, 为合成包含1, 2-二胺基特征结构的生物碱提供了引入手性中心的方法.
2006年, 秦勇课题组[17]报道了保护的羟基乙酸酯与叔丁基亚磺酰亚胺的加成反应, 并应用该方法实现了β-内酰胺型紫杉醇侧链前体的合成.
在双(三甲基硅基)胺基锂(LiHMDS)作用下, Boc保护的羟基乙酸苄酯50首先生成烯醇负离子, 然后与叔丁基亚磺酰亚胺51发生高立体择性的Mannich反应, 引入两个连续立体中心, 得到化合物52, ee值高达99%.在Pearlman催化剂催化下氢解酯52可制得直链型紫杉醇侧链前体53, 再经过分子内缩合得到光学纯的β-内酰胺型紫杉醇侧链前体54.环丁酰胺54可以用于紫杉醇的半合成(Scheme 10).
作者发展的叔丁基亚磺酰亚胺的高立体选择性1, 2-加成反应可以高效地合成紫杉醇侧链前体, 可应用于紫杉醇衍生物的合成.
2015年, Njardarson课题组[18]报道了一种基于叔丁基亚磺酰亚胺的不对称[3+2]环化反应构建3-吡咯啉的新方法, 并应用该方法实现了(-)-supinidine, (-)-iso- retronecanol和(+)-elacomine的简洁合成.
他们以醛55作为原料, 与(S)-叔丁基亚磺酰胺在Ti(OiPr)4作用下缩合生成叔丁基亚磺酰亚胺56. 4-溴巴豆酸乙酯(57)在LDA作用下去质子化生成的碳负离子与亚胺56发生1, 2-加成反应后, 生成氮负离子中间体发生SN2反应, 经过5-exo-tet环化生成3-吡咯啉化合物58.化合物58在新制备的HCl作用下脱除叔丁基亚磺酰基后, 在Et3N作用下发生分子内SN2反应构建吡咯环, 以DIBAL-H作为还原剂, 还原酯基实现了(-)-supinidine的合成.在Pd/C和H2条件下, (-)-supinidine发生立体选择性还原, 得到(−)-isoretronecanol.另外, 叔丁基亚磺酰亚胺59与4-溴巴豆酸乙酯(57)在LDA作用下发生不对称的[3+2]环化反应生成3-吡咯啉化合物60, 再与芳香胺61发生胺解得到碘代芳基酰胺62.化合物62在零价钯促进下发生分子内Heck环化反应, 构建季碳手性中心, 生成螺环氧化吲哚63.化合物63在锌粉及冰醋酸条件下脱除叔丁基亚磺酰基, 同时还原生成的亚胺后, 再在BBr3条件下脱除甲基可以实现天然产物(+)-elacomine的合成(Scheme 11).

Njardarson课题组发展的不对称[3+2]环化反应构建3-吡咯啉的新方法, 丰富了叔丁基亚磺酰亚胺在构建杂环化合中的应用, 拓展了杂环上的手性取代基构建的方法.
2017年, 张洪彬课题组[19]发展了吲哚取代的叔丁基亚磺酰亚胺与二氧杂环己酮之间的选择性vinylogous Mannich反应, 并以该反应作为关键步骤完成了(-)- vindorosine的不对称全合成.他们通过vinylogous Mannich反应引入C(19)位和C(5)位两个手性中心, 其中C(19)位手性中心在串联Heathcock/aza-Prins环化反应中诱导C(12)位手性中心的生成.
他们以醛64与(S)-叔丁基亚磺酰胺为原料, 发生缩合生成叔丁基亚磺酰亚胺65.在LiHMDS作用下, dioxinone 66生成的烯醇负离子与亚胺65发生vinylogous Mannich反应, 引入C(19)位和C(5)位两个立体中心, 生成化合物67和68, dr值为7.6:1.在I2条件下化合物67脱除叔丁基亚磺酰基[20], 经过3步氮原子上的官能团转化得到化合物70.化合物70在NaI条件下发生卤素交换后, 在Ag(OTf)2作用下发生分子内串联Heathcock/aza-Prins环化反应构建C和E两个环, 同时引入C(12)位和C(2)位的手性中心, 生成四环化合物71.化合物71经过氧化切断端烯和吲哚氮上引入保护基得到72, 在1, 5-二氮杂二环[5.4.0]十一烯-5 (DBU)条件下发生分子内aldol反应构建D环, 引入C(6)位手性中心得到五环骨架化合物73.化合物73经过3步转化得到74, 经过Luche还原C(4)位得到预期外的单一异构体醇75.利用Mitsunobu反应将化合物75的C(4)位手性异构[21]后, 经过m-CPBA氧化烯胺引入C(3)位羟基[22], 还原胺化引入吲哚氮原子上的甲基取代基, 得到化合物76.最后经过2步还原及酯化反应完成了(-)-vindorosine的合成(Scheme 12).
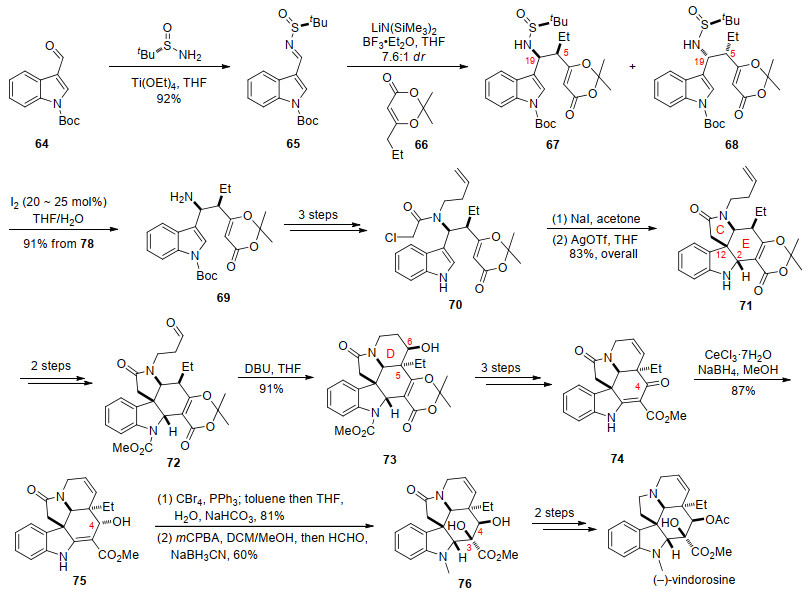
张洪彬课题组开发的基于吲哚取代的叔丁基亚磺酰亚胺的克级vinylogous Mannich反应, 在吲哚3位引入手性边链进行修饰后, 通过串联Heathcock/aza- Prins环化反应, 高效构建了C和E两个环得到化合物71. 71作为关键中间体可用于合成其他吲哚生物碱.
2017年, Brown课题组[23]以叔丁基亚磺酰亚胺和戊二酸衍生物的双imino-aldol反应为关键步骤, 经过线性5步完成了(+)-β-isosparteine不对称合成.
戊二酸酯77在LDA作用下形成双烯醇负离子, 与2 equiv.的叔丁基亚磺酰亚胺78发生双imino-aldol反应[24]一步生成4个立体中心, 得到预期的化合物79b以及副产物79a.化合物79b在I2作用下脱去叔丁基亚磺酰基后, 在Na2CO3作用下发生酰胺化反应, 生成化合物80.在KOH条件下化合物80发生分子内双SN2反应构建2个哌啶环, 最后还原酰胺合成(+)-β-isosparteine.此外, Brown课题组[25]应用imino-aldol反应还合成了(-)-tashiromine, (-)-epilupinine和(+)-allomatrine (Scheme 13).

Brown课题组利用叔丁基亚磺酰亚胺的双imino- aldol反应可以高效地在链状底物中构建多个手性中心, 增加了手性链状化合物的合成方法.
应用叔丁基亚磺酰亚胺与有机锂试剂的1, 2-加成反应作为关键合成策略完成的分子还有Senanayake课题组[26a]合成的(R)-sibutramine、Baran课题组[26b]合成的(+)-araiosamine C、魏邦国课题组[26c]合成的(+)- halofuginone以及Sarpong课题组[26d]合成的(-)-daphlo- ngamine H和(-)-isodaphlongamine H.
|
|
2010年, Ellman课题组[27]报道了(-)-aurantio- clavine的不对称全合成.合成中以Rh(Ⅰ)催化的叔丁基亚磺酰亚胺的高立体选择性1, 2-加成反应为关键步引入(-)-aurantioclavine结构中唯一的立体中心.
作者从4-溴色醇(82)出发, 吲哚C(4)位发生甲酰化反应得到醛83, 83与(R)-叔丁基亚磺酰胺缩合得到叔丁基亚磺酰亚胺84, 再经过吲哚和一级羟基的保护得到化合物85.亚胺85和N-甲基亚氨基二乙酸硼酸酯86在Rh(Ⅰ)催化下, 发生不对称的烯基化反应生成化合物87, 反应取得非常好的立体选择性, dr值为97:3.化合物87在NaH条件下发生分子内SN2反应构建氮杂环庚烷, 再经过2步脱保护得到ee值大于99%的(―)- aurantioclavine (Scheme 14).

作者发展的Rh(Ⅰ)催化的叔丁基亚磺酰亚胺的不对称烯基化反应, 丰富了叔丁基亚磺酰亚胺的加成反应类型.
2017年, 孙兴文课题组[28]发展了钯催化的手性叔丁基亚磺酰亚胺的高立体选择性1, 2-加成反应, 可以合成手性纯的β-芳基高烯丙基胺, 并利用该方法引入C(4a)位和C(10a)位两个立体中心, 成功实现了(+)-lycori- cidine和(+)-7-deoxypancratistatin的全合成.
他们以碘代核糖衍生物88为原料, 与nBuLi作用发生消除后, 与(S)-叔丁基亚磺酰胺缩合得到叔丁基亚磺酰胺亚胺89.接下来在Pd(PPh3)4和ZnEt2条件下, 亚胺89和醋酸肉桂醇酯衍生物90发生串联的不对称1, 2-加成反应及分子内的胺解反应, 构建了B环, 引入了C(4a)位和C(10a)位两个立体中心, 得到单一异构体91.化合物91通过RCM反应构建C环得到化合物92后, 再经过双羟化和脱叔丁基亚磺酰基生成二醇93. SOCl2与二醇93反应生成环状亚砜化合物, 接着在RuCl3和NaIO4条件下氧化得到环硫酸酯94. 94在nBu4NI的作用下, 在位阻小的C(1)位发生开环生成碘代物, 再在酸性条件下发生反式消除, 同时水解邻二羟基缩酮保护, 完成了(+)-lycoricidine的合成.另外, 环硫酸酯94在PhCOONa条件下同样在C(1)位发生开环反应, 酸性条件下水解缩酮, 碱性条件下水解酯基后完成了(+)-7- deoxy-pancratistatin的合成(Scheme 15).

孙兴文课题组发展了钯催化的醋酸肉桂醇酯与手性叔丁基亚磺酰亚胺的不对称1, 2-加成反应, 避免使用不稳定的肉桂基卤化物, 扩展了制备光学纯的β-芳基高烯丙基胺的底物范围.
另外, 该课题组应用类似的策略完成了天然产物(-)-α-lycorane和(-)-zephyranthine的全合成, 以及(+)-clivonine的形式合成[29].
|
|
2006年, Chemla课题组[30]通过手性叔丁基亚磺酰亚胺的1, 2-加成反应一步构建了2个立体中心, 从而实现了(-)-1-hydroxyquinolizidinone的不对称合成(Scheme 16).

5-氯戊醛95为起始物与(S)-叔丁基亚磺酰胺缩合可得到叔丁基亚磺酰亚胺96, 其与联烯锌试剂97发生不对称的1, 2-加成反应, 引入胺的α位和β位2个立体中心, 得到炔烃化合物98, dr值为24:1.在NaH条件下, 化合物98发生分子内SN2反应构建哌啶环, 并脱除TMS保护基, 得到单一异构体99.接着化合物99经过4步转化实现了(-)-1-hydroxyquinolizidinone的合成.借鉴黄培强课题组[31]报道的策略可以完成(-)-epiquin- amide和(-)-homopumiliotoxin 223G的形式合成.
Chemla课题组从(S)-叔丁基亚磺酰胺出发, 经过7步转化, 以25.2%的总产率高效合成(-)-1-hydroxy- quinolizidinone.利用叔丁基亚磺酰亚胺可以引入天然产物所需的氮原子, 并高选择性地完成立体中心的构建.
Chemla课题组通过该策略还完成了(-)-α-conhy- drine、(+)-6-epi-castanospermine和(-)-balanol的合成以及(+)-CP-99, 994、(+)-CP-99, 994和(+)-CP-122, 721的形式合成[32].
|
|
2010年, 赵刚课题组[33]报道了(-)-fasicularin和(-)-lepadiformine A的不对称全合成, 作者利用手性叔丁基亚磺酰亚胺与烯丙基锌发生不对称1, 2-加成反应引入C(10)位立体中心.
以易得的手性酮100为起始物, 与(R)-叔丁基亚磺酰胺缩合得到101.烯丙基溴和锌原位生成的烯丙基锌与亚胺101通过螯合作用, 经过椅式过渡态, 发生不对称烯丙基化反应构建C(10)位氮杂季碳手性中心, 得到化合物102, dr值为12:1.化合物102经过3步官能团转化生成化合物103, 经过Ley氧化生成醛后自发环化得到化合物104.接着经过4步转化生成化合物105a和105b.在NaH作用下, 化合物105a发生SN2反应构建氮杂五元环化合物106, 经过5步转化生成化合物107.在钠萘条件下化合物107脱除Ts保护基, 经过分子内环化生成不稳定的烯胺108, 通过Luche还原生成单一异构体的化合物109.用四丁基氟化铵(TBAF)脱除硅基保护基, 最后在Mitsunobu反应条件下引入硫氰基, 完成了(-)-fasicularin的合成.另外, 用L-selectride还原化合物107, 可以得到预期构型的化合物110, dr值为6:1.化合物110在钠萘条件下脱除Ts保护基, 然后在4-二甲氨基吡啶(DMAP)催化作用下发生分子内类Mitsunobu环化反应构建氮杂六元环, 最后脱除保护基合成(-)-lepadiformine A (Scheme 17).

赵刚课题组使用手性辅基叔丁基亚磺酰胺, 利用亚磺酰基与锌配位, 以较好的非对映选择性引入氮杂季碳手性中心, 这对该家族的其他氮杂螺三环生物碱不对称全合成有一定借鉴意义.
2011年, Andrade课题组[34]报道了(-)-leuconicine A和(-)-leuconicine B的不对称全合成.他们利用手性叔丁基亚磺酰亚胺的烯丙基化反应构建C(21)位立体中心, 后续通过该立体中心诱导C(7)位立体中心的生成; 连续一锅螺环化/分子内aza-Baylis-Hillman反应构建ABCE骨架; 串联acylation/Knoevenagel反应构筑F环; 分子内Heck环化反应构建D环, 继而实现(-)-leuconicine A和(-)-leuconicine B的全合成.
起始物111和(R)-叔丁基亚磺酰胺在Ti(OEt)4作用下缩合生成叔丁基亚磺酰亚胺, 与原位产生的烯丙基铟试剂反应[35], 以高收率和较好的立体选择性得到高烯丙基胺112, dr值为10:1.化合物112经过脱除亚磺酰基保护并修饰胺基以及引入边链等5步转化得到化合物113.在AgOTf和2, 6-二叔丁基-4-甲基吡啶(DTBMP)的作用下, 化合物113发生分子内螺环化反应引入C(7)位螺环手性中心, 随后在DBU作用下发生aza-Baylis- Hillman反应构建E环, 同时引入C(2)位手性中心, 得到具有ABCE四环结构的化合物115.化合物115经过4步转化调整分子的氧化态, 得到的化合物116与酰氯117在Et3N作用下发生串联acylation/Knoevenagel反应构建F环, 生成化合物118.在钯催化下, 化合物118发生分子内Heck反应构筑D环, Raney Ni还原环外双键可以得到(-)-leuconicine B. (-)-leuconicine B经过氨解可以转化为(-)-leuconicine A (Scheme 18).

作者使用连续一锅螺环化/分子内aza-Baylis-Hill- man反应构建ABCE骨架, 串联acylation/Knoevenagel反应构筑F环, 分子内Heck环化反应构建D环, 高效地构建了(-)-leuconicine A和(-)-leuconicine B的骨架.
2011年, González-Gómez课题组[36]报道了(-)- aphanorphine的全合成.他们利用手性叔丁基亚磺酰亚胺的烯丙基化反应实现仲胺立体中心的构建, 继而通过后续的转化合成(-)-aphanorphine.
起始物4-甲氧基苯乙醛(120)与(R)-叔丁基亚磺酰胺在Ti(OEt)4作用下缩合生成叔丁基亚磺酰亚胺, 再与原位产生的烯丙基铟试剂反应, 以高收率和立体选择性得到高烯丙基胺121, dr值为93:7.化合物121与m-CPBA作用同时发生双键的环氧化和亚砜的氧化得到砜基化合物122, 在KI和K2CO3条件下发生氮原子进攻环氧开环构筑氮杂五元环, 得到化合物123.在AlCl3作用下123发生Friedel-Crafts烷基化得到三环化合物125, 最后经过3步反应实现氮和氧原子上官能团的修饰得到(-)-aphanorphine (Scheme 19).

该课题组合成(-)-aphanorphine使用的策略为不对称合成桥环化合物提供了新思路.
此外, González-Gómez课题组[37]利用类似的策略还合成了tetraponerines T3, T4和(-)-tylophorine.
|
|
2013年, Evans课题组[38]报道了(-)-nakadomarin A的全合成. (-)-Nakadomarin A可以由内酰胺134和双环[6.3.0]内酰胺130两个片段连接、转化得到.片段130中的手性中心控制了后续所有手性中心的产生, 片段130中的手性中心则是通过叔丁基亚磺酰亚胺发生锌促进的烯丙基化反应构建.
作者首先合成双环[6.3.0]内酰胺130.丙烯醛126和(R)-叔丁基亚磺酰胺在Ti(OEt)4作用下缩合生成叔丁基亚磺酰亚胺127.亚胺127与原位生成的有机锌试剂发生aza-Barbier类型的反应[39]生成化合物129, 酸解脱除叔丁基亚磺酰基, 在NaOH条件下发生内酰胺化后, 再与1-碘-5-己烯发生氮烷基化, 最后在第二代Grubbs催化剂的催化下发生烯烃的关环复分解(RCM)反应构建八元环, 生成双环[6.3.0]内酰胺130.
另外, 3-糠醛131经过2步转化得到化合物132, 与再烯丙醇发生分子间Heck偶联反应生成醛基化合物, 经过Wittig反应延长碳链, 生成以Z-烯烃为主的产物, 然后经过脱保护、Still-Gennari改进的Horner-Wadsworth- Emmons反应[40], 生成Z式α, β-不饱和酯133.化合物133经过3步转化生成内酰胺134.内酰胺134和双环[6.3.0]内酰胺130在叔丁基二甲硅基三氟甲磺酸酯(TBSOTf)促进下发生双Michael加成反应, 生成化合物135和预期构型136, 构建了(-)-nakadomarin A的骨架.化合物136在Tf2O和2, 6-二叔丁基-4-甲基吡啶条件下活化酰胺, 与呋喃环发生分子内成环, 随后在Red-Al作为还原剂的条件下得到(-)-nakadomarin A (Scheme 20).

Evans课题组报道的合成策略巧妙地利用叔丁基亚磺酰亚胺作为起始物, 通过加成反应引入手性中心, 继而控制后续手性中心的产生, 最终完成了(-)-nakado- marin A的合成.该策略体现了叔丁基亚磺酰亚胺在全合成中引入重要手性中心的高效性.
2013年, Andrade课题组[41]报道了(-)-melotenine A的首次不对称全合成.合成中通过手性叔丁基亚磺酰亚胺的烯丙基化反应构建C(21)位立体中心, 且该立体中心诱导C(7)位立体中心的生成, 通过后续的转化实现了(-)-melotenine A的全合成.
起始物111和(R)-叔丁基亚磺酰胺在Ti(OEt)4作用下缩合生成叔丁基亚磺酰亚胺, 再与原位产生的烯丙基铟试剂反应引入C(21)位立体中心, 并以高收率和较好的立体选择性得到高烯丙基胺112, dr值为10:1.化合物112经过4步转化引入边链以及脱除亚磺酰基保护得到化合物137.在Mitsunobu反应条件下, 化合物137发生螺环化实现C(7)位螺环手性中心的构建, 在DBU加热的条件下构建E环, 引入C(2)位手性中心, 生成具有ABCE四环结构的化合物138.该反应可能是经历了aza-Baylis-Hillman或是vinylogous Mannich/olefin isomerization的过程.在LDA条件下, 化合物138结构中的α, β-不饱和酯的γ位去质子化形成碳负离子, 之后与乙醛发生aldol反应, 催化氢化双键, 2, 3-二氯-5, 6-二氰-1, 4-苯醌(DDQ)氧化生成烯胺139.化合物139经过3步转化后得到化合物140, 最后在nBuLi条件下发生分子内Piers环化构建D环, 在PPh3和I2的条件下[14]实现区域选择性脱水, 完成了(-)-melotenine A的合成(Scheme 21).

作者开发了新的环化顺序构建(-)-melotenine A的ABCE四环骨架, 之后Piers螺环化反应构建D环最终构建天然产物的环系骨架.其中叔丁基亚磺酰亚胺作为手性源, 参与了关键手性中心的构建, 该策略为不对称合成氮杂并环骨架提供了很好的借鉴.
2014年, Davies课题组[42]报道了(−)-nakinadine A的全合成.他们以苯乙酸甲酯和手性叔丁基亚磺酰亚胺发生的Mannich-type反应作为关键步骤, 引入C(2)和C(3)位的两个立体中心, 首次实现了不对称合成(-)-nakina- dine A, 并确定了其绝对构型.
以11-溴-1-十一醇141为起始原料, 经过5步转化延长碳链并引入吡啶基团得到化合物142, 再与(R)-叔丁基亚磺酰胺缩合得到叔丁基亚磺酰亚胺143.甲基溴化镁作为碱与苯乙酸甲酯作用生成烯醇负离子, 然后与亚胺143发生立体选择性的1, 2-加成反应得到化合物144和预期构型产物145, 二者比例为20:80.酸性条件下, 145脱除叔丁基亚磺酰基, 再与醛基化合物146发生还原胺化反应生成化合物147, 最后经过酯基的水解以及纯化完成了天然产物(-)-(2S, 3R, Z)-nakinadine A的合成.该课题组[43]应用该方法还合成了nakinadines D, E, F (Scheme 22).

Davies课题组采用叔丁基亚磺酰亚胺的不对称Mannich-type反应为关键步骤, 经过11步线性转化, 以9%的收率得到了天然产物(-)-nakinadine A.
2015年, 黄培强课题组[44]以叔丁基亚磺酰亚胺的不对称vinylogous Mannich反应为关键步骤, 完成了pandamarilactonines A~C的全合成.
他们以(R)-叔丁基亚磺酰胺与4-氯丁醛(148)为原料, 在CuSO4促进下缩合生成叔丁基亚磺酰亚胺149.呋喃150与149发生不对称vinylogous Mannich反应, 得到以化合物syn-151为主的产物, dr值为95:5.化合物syn-151在盐酸作用下脱去叔丁基亚磺酰基, 再在K2CO3作用下发生分子内SN2反应, 可以构建吡咯环生成化合物152.在这个过程中C(15)位手性中心发生了一定程度的消旋化.化合物152和片段153经过氮烷基化反应得到pandamarilactonines A~C (Scheme 23).黄培强课题组利用叔丁基亚磺酰亚胺与呋喃发生vinylo- gous Mannich反应, 引入相邻的2个手性中心, 高效构建了5-吡咯烷基-呋喃-2-酮片段, 继而合成pandamari- lactonines A~C.

黄培强课题组使用叔丁基亚磺酰亚胺的不对称vinylogous Mannich反应还完成了(+)-absouline和(-)- deoxoprosophylline的全合成以及L-deoxyallonojirimycin和L-3-epi-fagomine的形式合成[45].
2016年, 孙兴文课题组[46]利用aza-Barbier烯丙基化反应一步引入两个相邻的立体中心, 以生成的产物作为通用中间体完成了amathaspiramides B, D, F的不对称全合成.
酮154和(S)-叔丁基亚磺酰胺缩合生成叔丁基亚磺酰亚胺155, 再与原位产生的烯丙基溴化锌发生烯丙基化反应引入C(5)位和C(9)位2个相邻的立体中心, 生成157a和157b, dr值为10:1.从期望构型化合物157a出发, 在盐酸作用下脱除亚磺酰基, 在Et3N条件下发生内酰胺化得到化合物158, 随后经过2步官能团转化生成159.之后化合物159发生甲基化, 氧化切断端烯生成醛, 与胺基发生环化合成amathaspiramide B.另外化合物159直接经过臭氧切断、胺基缩合生成amathaspira- mide D和160.选择性还原酰胺160可以得到天然产物amathaspiramide F (Scheme 24).

作者通过芳基烯丙基溴化物与叔丁基亚磺酰亚胺发生aza-Barbier烯丙基化反应高效地合成具有两个相邻立体中心的中间体, 之后经过4步克级转化并以63%的收率合成amathaspiramide D; 另外分别经过5步、4步转化可以分别合成amathaspiramide B和amatha- spiramide F.
|
|
2017年, 贾彦兴课题组[47]报道了8个ergot生物碱的全合成.他们利用手性叔丁基亚磺酰亚胺的炔丙基化反应引入C(5)位立体中心; 使用钯催化的Larock吲哚环化反应[48]或串联钯催化的分子内Larock吲哚环化/ Tsuji-Trost烯丙基化反应, 可以高效完成环系骨架的构建.
他们从溴化物161出发, 经过5步转化得到叔丁基亚磺酰亚胺162, 与原位生成的联烯锌试剂[49]发生炔丙基化反应引入C(5)位立体中心, 生成163a和163b.化合物163a和163b可以分别转化成不同的ergot生物碱.首先在Pd(OAc)2和Me-Phos的催化体系中, 化合物163a发生串联分子内Larock吲哚环化/Tsuji-Trost烯丙基化反应一步构建BCD三个环, 生成具有四环核心骨架的化合物164, 再经过3步转化对氮原子和吲哚2位取代基进行修饰, 得到化合物165, 再在Raney Ni和H2条件下还原双键, 以5:3的比例得到天然产物festu- clavine和pyroclavine. Festuclavine在N-溴代丁二酰亚胺(NBS)作用下发生C(2)位溴代生成pibocin A. Festuclavine在t-BuOCl条件下进行氯代后, 再与异戊二烯基-9-BBN[50]反应生成9-deacetoxyfumigaclavine C.另外pyroclavine经过相同的过程完成了fumigaclavine G的合成.化合物163b在Pd(OAc)2和Me-Phos的催化体系中只能发生分子内Larock吲哚环化反应构建B和C两个环, 生成三环化合物166.化合物166经过4步转化关D环并完成对取代基的修饰得到167, 在Crabtree催化剂及H2条件下还原双键, 最终以1:1.2的比例得到costa- clavine和epi-costaclavine (Scheme 25).

另外, 四环化合物164发生Mukaiyama反应[51]生成醇168a和168b, 其中168a经过3步转化完成官能团修饰得到天然产物dihydrosetoclavine, 而168b经过类似的3步转化得到iso-dihydrosetoclavine (Scheme 26).
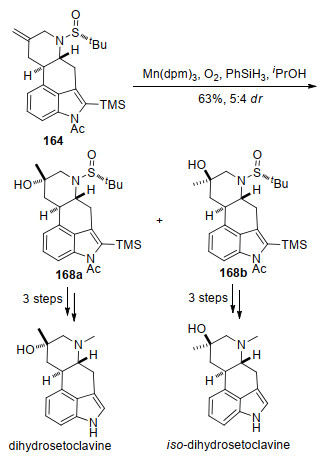
作者通过叔丁基亚磺酰胺诱导的不对称炔丙基化反应构建了关键的C(5)氮杂手性中心, 并通过串联钯催化的分子内Larock吲哚环化/Tsuji-Trost烯丙基化反应一步高效地构建了ergot生物碱的环系骨架, 从而多样性地合成了ergot生物碱.
除此之外, 应用叔丁基亚磺酰亚胺与碳负离子的1, 2-加成反应, Zafarian课题组[52a-52b]合成了neodysidenin和sintokamides A, B, D, E; Ganesan课题组[52c]合成了azumamides A, E; Huang课题组[52d]合成(+)-poran- theridine; Wolfe课题组[52e]合成了merobatzelladine B; 韩世清课题组[52f]合成了(+)-negamycin.
2019年, 齐湘兵课题组[53]报道了(-)-alstofolinine A的全合成.作者通过对叔丁基亚磺酰亚胺的还原引入关键的氮杂手性中心, 利用呋喃氧化重排和吲哚亲核加成反应为核心策略构建出了天然产物骨架.
作者从化合物169出发, 与(S)-叔丁基亚磺酰胺缩合得到叔丁基亚磺酰亚胺, 经过(+)-二异松蒎烯基硼烷还原引入首个立体中心, 得到仲胺170, dr值为20:1. m-CPBA与化合物170作用后, 发生氧化重排及分子内亲核加成得到化合物171.随后171经过氮上引入甲基、还原双键、发生Claisen缩合及将羰基转化为烯醇OTf得到化合物172.在零价钯促进下, 172与三丁基锡甲醇发生Stille偶联后, 再发生分子内的酯化得到化合物173.最后173经过还原双键和氮原子上引入甲基得到(-)-alstofolinine A (Scheme 27).

作者通过手性的叔丁基亚磺酰亚胺的还原得到手性的磺酰胺, 该手性控制了aza-Achmatowicz重排和吲哚亲核环化反应的立体化学, 生成单一的氮杂双环[3.3.1]壬烷骨架化合物, 为不对称合成氮杂双环[3.3.1]壬烷骨架的天然产物提供了一种高效的方法.
应用叔丁基亚磺酰亚胺的还原引入手性中心合成的天然产物的例子还包括李闯创课题组[54a]合成的(+)-(aS, 7R)-colchicine、Ryan课题组[54b]合成的mexile- tine和周伟澄课题组[54c]合成的maravrioc.
|
|
2005年, 林国强课题组[55]发展了SmI2促进的叔丁基亚磺酰亚胺与醛的还原偶联反应.他们将该方法应用于D-erythro-sphinganine的全合成.
作者以手性的叔丁基亚磺酰亚胺175与正十六醛(174)为原料, 在SmI2作用下高立体选择性地实现了还原偶联得到β-氨基醇176.接着在锂/萘和盐酸条件下分别脱除苄基和亚磺酰基得到D-erythro-sphinganine.这是已知的该化合物最简洁的合成方法.同时他们也合成了(3R, 4S)-statine (Scheme 28).

作者开发了一种高效且实用的合成手性β-氨基醇的方法, 这种方法对于手性β-氨基醇化合物的制备是非常高效的.该方法在天然产物的不对称合成中应该会取得更广泛的应用.
2009年, 林国强课题组[56]报道了(+)-febrifugine的不对称合成.关键步骤为SmI2促进的手性叔丁基亚磺酰亚胺与醛的还原偶联构建β-氨基醇.
作者从化合物177出发, 经过Swern氧化, 生成的醛与叔丁基亚磺酰胺缩合得到化合物178.醛基化合物179与叔丁基亚磺酰亚胺178在二碘化钐促进下发生还原偶联反应得到β-氨基醇180, dr值为93.5:4.0:2.0:0.5.化合物180经过两步氮和氧原子上的保护基修饰得到化合物181, TBAF脱除伯醇硅保护基后将其活化为甲磺酸酯, 再在NaH作用下发生分子内氮烷基化反应生成化合物182.化合物182经过3步转化将邻二醇结构转化为环氧得到化合物183, 随后与4-喹唑酮钾盐反应发生环氧开环生成化合物184.化合物184经过二级醇的氧化及氮上保护基的脱除可以转化为天然产物(+)-febrifugine (Scheme 29).
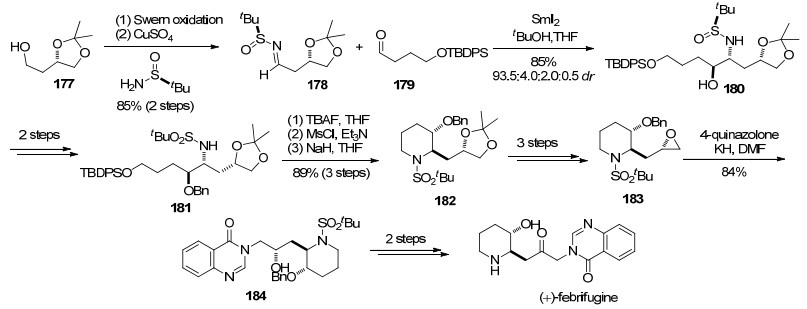
作者发展的SmI2促进的叔丁基亚磺酰亚胺与γ-羟基醛衍生物的还原偶联, 为3-羟基取代的哌啶骨架的不对称合成提供了一种高效的方法.
2016年, 魏邦国课题组[57]报道了对epohelmin A的不对称全合成.作者利用手性叔丁基亚磺酰亚胺在SmI2条件下与醛的还原偶联, 构建β-氨基醇.
他们以醛185和叔丁基亚磺酰亚胺186为原料, 经过还原偶联得到β-氨基醇187.化合物187经过三步保护基的转化得到化合物188, 再经过3步转化延长碳链得到化合物189.接下来化合物189与甲基膦酸酯加成得到化合物190.化合物190脱除PMB保护基以后, 与己醛发生Horner-Wadsworth-Emmons反应生成化合物191.用MsCl活化羟基, 脱除Boc基团后发生分子内双氮烷基化反应, 最后脱除硅基可以得到epohelmin A.同时该小组也合成了epohelmin B (Scheme 30).
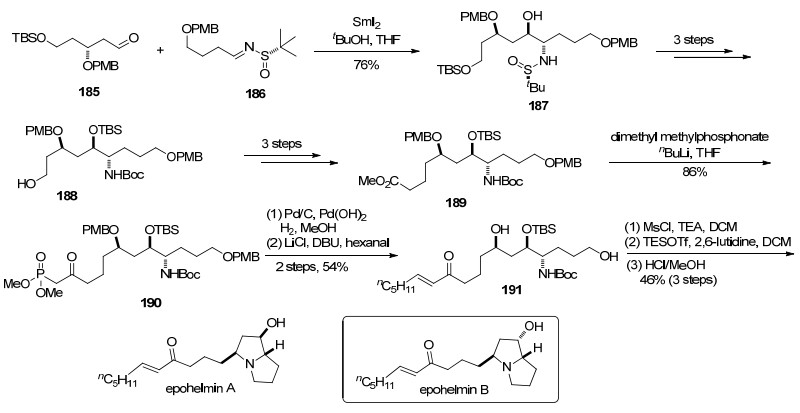
另外, 利用此还原偶联方法, 林国强课题组还合成了(-)-deoxoprosophylline[58a]; 魏邦国课题组合成了D-ribo-phytosphingosine和dolastatin 10[58b, 58c].
叔丁基亚磺酰亚胺转化为叔丁基亚磺酰基金属烯胺以后, 可以与亲电试剂反应, 立体选择性地生成新的化学键.相比于叔丁基亚磺酰亚胺的1, 2-加成反应诱导胺的α位立体中心的生成, 叔基亚磺酰基金属烯胺与亲电试剂的加成反应可以诱导胺的β位立体中心的生成.反应过程中新产生的叔丁基亚磺酰亚胺可以继续发生1, 2-加成反应构建胺的α位立体中心, 进而得到含有α和β两个手性中心手性胺类化合物.
2004年, Ellman课题组[59]报道了手性叔丁基亚磺酰亚胺的高立体选择性α-烷基化反应.作者利用该方法引入C(6)位立体中心, 最终实现了天然产物(6R, 7S)-7- amino-7, 8-dihydro-α-bisabolene的首次不对称合成.
以原酸酯192和(R)-叔丁基亚磺酰胺为原料, 在MgSO4作用下进行缩合后, 再与NaI发生卤素交换生成碘代物, 在异丙烯基溴化镁及CuI条件下发生偶联反应, 得到化合物193.化合物193和吗啡啉在催化量的NaCN作用下反应得到194, 在六甲基二硅氨基钾(KHMDS)条件下发生去质子化生成金属烯胺, 再与烯丙基溴发生烷基化引入C(6)位手性中心, 生成单一的非对映异构体195.化合物195与甲基铈试剂发生加成反应, 再经过RCM反应生成环己烯196.化合物196和锂试剂197发生亲核加成反应生成单一的非对映异构体产物, 实现C(7)位氮杂季碳手性中心的构建.最后脱除叔丁基亚磺酰基合成(6R, 7S)-7-amino-7, 8-dihydro-α-bisabolene (Scheme 31).

Ellman课题组发展了一种在手性叔丁基亚磺酰亚胺的α位引入手性烷基链的反应, 并且利用新生成的叔丁基亚磺酰亚胺参与后续的1, 2-加成反应, 该方法可以应用于包含α和β手性中心的胺的合成, 为连续手性中心的构建提供了一种高效的方法.
2006年, Ellman课题组[60]利用叔丁基亚磺酰胺参与的不对称加成反应完成了tubulysin D的首次全合成. Tubulysin D的结构可以分为4个氨基酸片段: (2R)-N-甲基-2-哌啶甲酸(Mep)、异亮氨酸(Ile)、tubuvaline (Tuv)和tubuphenylalanine (Tup).
Tup片段的合成以苯乙醛(198)为原料, 与(R)-叔丁基亚磺酰胺缩合生成叔丁基亚磺酰亚胺199, 再在SmI2促进下与2-甲基丙烯酸甲酯200发生还原偶联反应, 引入2个立体中心得到化合物201, 酸解脱除亚磺酰基生成胺盐酸盐Tup (202).对于tubulysin D的合成, 在LDA条件下, (S)-叔丁基亚磺酰酮亚胺203去质子化生成叔丁基亚磺酰基金属烯胺, 然后与噻唑啉醛204发生不对称加成反应引入亚胺的β位立体中心, 生成化合物205, dr值为92:8.用NaBH4立体选择性还原亚胺205, dr值为91:1, 在酸性条件下生成1, 3-氨基酸盐酸盐Tuv (206). Tuv (206)和α-叠氮酰氯207发生酰胺化反应, 用三乙基硅基(TES)保护仲醇, 氮烷基化引入侧链, 生成叠氮化合物208.在Pd/C及H2条件下还原化合物208中的叠氮后, 与五氟苯酚活化的Mep酯发生胺解, 接着脱除TES保护基, Me3SnOH水解甲酯[61]得到羧酸209.氟苯酚活化羧酸209后, 与Tup (202)发生胺解, 最后乙酰化合成tubulysin D (Scheme 32).

作者采用手性叔丁基亚磺酰胺的高立体选择性反应构建了tubulysin D结构中的四个手性中心并完成了tubulysin D的首次不对称全合成, 同时报道了首例亚胺和不饱和羰基化合物的不对称偶联反应.
2013年, Andrade课题组[62]报道了aspidosperma生物碱(-)-aspidospermidine, (-)-tabersonine和(-)-vin- cadifformine的不对称全合成.他们利用叔丁基亚磺酰亚胺与不饱和酯发生串联Michael/Mannich/氮烷基化反应一步构建两个立体中心, 并形成E环; 通过RCM反应构建D环, Bosch-Rubiralta螺环化反应构建C环.
以醛210为原料, 在Ti(OEt)4作用下与(S)-叔丁基亚磺酰胺缩合生成叔丁基亚磺酰亚胺211.化合物211在LiHMDS作用下发生去质子化, 生成叔丁基亚磺酰基金属烯胺中间体, 再与2-乙基丙烯酸酯212发生Michael加成反应形成烯醇化物, 然后发生分子内Mannich反应构建E环, 最后加入烯丙基溴淬灭反应, 发生氮烷基化生成四氢咔唑化合物213, dr值为11:1.化合物213经过3步转化将C(5)酯基转化为乙烯基, 得到化合物214, 然后在第二代Grubbs催化剂的催化下发生RCM反应得到四环化合物215, 再经过2步转化生成化合物216.在t-BuOK条件下, 化合物216发生Bosch-Rubiralta螺环化反应构建C环得到五环骨架化合物.在LDA作用下, 217生成金属烯胺, 再经Mander试剂淬灭即可得到(−)-tabersonine.在Adams催化剂的催化下进行氢化可以将(-)-tabersonine转化为(-)-vincadifformine.另外, 化合物217经过氢化可以合成天然产物(-)-aspidosper- midine (Scheme 33).

Andrade课题组发展了手性叔丁基亚磺酰亚胺到烯胺的转化, 并实现了串联Michael/Mannich/氮烷基化反应构建连续的手性中心的新方法.
2015年, 秦勇课题组[63]报道了(-)-2-hydroxyisos- chizogamine及isoschizogamine的全合成, 他们通过叔丁基亚磺酰胺衍生物对不饱和酯基化合物的加成构建了第一个关键的手性中心.
叔丁基亚磺酰亚胺218在LiHMDS作用下发生去质子化, 生成叔丁基亚磺酰基金属烯胺中间体, 再与二酯化合物219发生Michael加成反应, 引入C(7)位手性中心, 生成化合物220, dr值为5:1.使用硼烷还原亚胺同时实现脱氧可以将220转化为221, 经过后续6步转化构建B环, 得到化合物222.化合物222在三氟甲磺酸的作用下脱去氨基上的保护基, 同时与酮羰基缩合形成烯胺结构, 完成C环的构建, 得到化合物223.在NaH作用下化合物223与不饱和炔酮发生Michael加成反应引入C(20)位手性, 之后进行酰胺化实现D环的构建, 得到化合物224.此过程中C(7)位的立体中心很好地诱导了C(20)位季碳手性中心的生成. 224经过七步官能团转化得到化合物225, 在Meerwin试剂作用下将酰胺异构为亚胺, 自发进行环化反应然后进行还原, 得到天然产物的骨架化合物226, 最后进行4步官能团转化可以得到(-)-2-hydroxyisoschizogamine及(-)-isoschizo- gamine (Scheme 34).

秦勇课题组通过叔丁基亚磺酰亚胺生成的金属烯胺与不饱和二酯化合物的不对称Michael加成反应, 引入C(7)位立体中心.该立体中心很好地诱导了C(20)位季碳手性的构建, 为不对称合成(-)-2-hydroxyisoschi- zogamine和(−)-2-hydroxyisoschizogamine奠定了基础.
2019年, Andrade课题组[64]应用组内发展的叔丁基亚磺酰亚胺与不饱和酯发生串联Michael/Mannich环化反应[62], 引入了三个连续的手性中心, 该立体中心诱导了后续螺环化反应生成新的立体中心, 进而完成了(+)-epi-condyfoline的首次全合成.
他们从简单易得的叔丁基亚磺酰亚胺227出发, 在LiHMDS作用下拔氢形成叔丁基亚磺酰基金属烯胺, 再与内酯228发生串联Michael/Mannich环化反应实现三个连续立体中心的构建, 生成四环内酯229, dr值为6.5:1.化合物229经过2步转化得到化合物230, 再在LiHMDS条件下发生分子内SN2反应构建哌啶环, 得到化合物231.化合物231经过6步官能团转化得到232, 接着在DMTSF (233)作用下发生螺环化反应[65]构建吡咯环, 生成五环骨架化合物234, 最后经过2步氧化态的调整完成了(+)-epi-condyfoline的全合成(Scheme 35).

作者报道的手性叔丁基亚磺酰基金属烯胺发生串联Michael/Mannich环化反应, 为合成包含四氢-β-咔啉骨架的天然产物提供了新的思路.
叔丁基亚磺酰基金属烯胺与亲电试剂的加成反应合成天然产物的例子还包括Suna课题组[66a]合成的(-)-14-epi-pseudotabersonine和(+)-pseudotabersonine以及Floreancig课题组[66b]合成的(−)-andrachcinidine.
|
|
综上所述, 以叔丁基亚磺酰胺为手性辅基诱导生成立体中心, 在天然产物全合成中具有广泛的应用.该策略在全合成中的应用大都可以以较好的立体选择性得到期望的手性中心.该反应常应用于构建天然产物中的叔碳手性中心, 而构建季碳手性中心的例子相对较少.所以其在氮杂季碳手性中心的构建及相应的天然产物合成研究方面还需进一步拓展.目前发现的包含氮杂季碳手性中心的天然产物极为丰富, 如erythrina, hasubanan, acutumine和ecteinascidins等生物碱, 期望将来有更多叔丁基亚磺酰胺在含有氮杂季碳手性中心的天然产物全合成中的应用报道.
Liu, G.; Cogan, D. A.; Ellman, J. A. J. Am. Chem. Soc. 1997, 119, 9913.
(a) Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984.
(b) Ellman, J. A. Pure Appl. Chem. 2003, 75, 39.
(c) Lin, G. Q.; Xu, M. H.; Zhong, Y. W.; Sun, X. W. Acc. Chem. Res. 2008, 41, 831.
(d) Ferreira, F.; Botuha, C.; Chemla, F.; Pérez-Luna, A. Chem. Soc. Rev. 2009, 38, 1162.
(e) Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 11, 3600.
(f) Liu, Q.; Hou, W. F.; Jing, Y. R.; Zhou, W.; Zhao, X. J. Prog. Pharm. Sci. 2013, 37, 493(in Chinese).
(刘强, 侯文峰, 景云荣, 周微, 赵小军, 药学进展, 2013, 37, 493.)
(g) Zhu, Y. J.; Huan, Y. L.; Leng, W. D.; Wu, C. X.; Wang, B. Chin. J. Pharm. 2017, 48, 621(in Chinese).
(朱怡君, 宦玉亮, 冷卫东, 伍成祥, 王兵, 中国医药工业杂志, 2017, 48, 621.)
(a) Kochi, T.; Tang, T. P.; Ellman, J. A. J. Am. Chem. Soc. 2002, 124, 6518.
(b) Kochi, T.; Tang, T. P.; Ellman, J. A. J. Am. Chem. Soc. 2003, 125, 11276.
Xu, H. C.; Chowdhury, S.; Ellman, J. A. Nat. Protoc. 2013, 8, 2271.
Kościołowicz, A.; Rozwadowska, M. D. Tetrahedron:Asymmetry 2006, 17, 1444.
Si, C.-M.; Mao, Z.-Y.; Dong, H.-Q.; Du, Z.-T.; Wei, B.-G.; Lin, G.-Q. J. Org. Chem. 2015, 80, 5824.
Si, C.-M.; Mao, Z.-Y.; Liu, Y.-W.; Du, Z.-T.; Wei, B.-G.; Lin, G.-Q. Org. Chem. Front. 2015, 2, 1485.
Kiemele, E. R.; Wathier, M.; Bichler, P.; Love, J. A. Org. Lett. 2016, 18, 492.
Pinto, A.; Griera, R.; Molins, E.; Fernández, I.; Bosch, J.; Amat, M. Org. Lett. 2017, 19, 1714.
Pinto, A.; Piccichè, M.; Griera, R.; Molins, E.; Bosch, J.; Amat, M. J. Org. Chem. 2018, 83, 8364.
Taghizadeh, M. J.; Gohari, S. J. A.; Javidan, A.; Moghimi, A.; Iman, M. J. Iran. Chem. Soc. 2018, 15, 2175.
(a) Fustero, S.; Monteagudo, S.; Sánchez-Roselló, M.; Flores, S.; Barrio, P.; del Pozo, C. Chem.-Eur. J. 2010, 16, 9835.
(b) Shen, X., Zhao; J., Xi, Y.; Chen, W.; Zhou, Y.; Yang, X.; Zhang, H. J. Org. Chem. 2018, 83, 14507.
(c) Prasad, K. R.; Rangari, V. A. Tetrahedron 2019, 75, 130496.
(d) Del Castillo, E.; Muñiz, K. Org. Lett. 2019, 21, 705.
Chuang, K. V.; Navarro, R.; Reisman, S. E. Chem. Sci. 2011, 2, 1086.
Toop, H. D.; Brusnahan, J. S.; Morris, J. C. Angew. Chem., Int. Ed. 2017, 56, 8536.
Liu, H. J.; Shia, K. S.; Shang, X.; Zhu, B. Y. Tetrahedron 1999, 55, 3803.
Uphade, M. B.; Reddy, A. A.; Khandare, S. P.; Prasad, K. R. Org. Lett. 2019, 21, 9109.
Wang, Y.; He, Q. F.; Wang, H. W.; Zhou, X.; Huang, Z. Y.; Qin, Y. J. Org. Chem. 2006, 71, 1588.
Chogii, I.; Njardarson, J. T. Angew. Chem., Int. Ed. 2015, 54, 13706.
Chen, W.; Yang, X.; Tan, W.; Zhang, X.; Liao, X.; Zhang, H. Angew. Chem., Int. Ed. 2017, 56, 12327.
Chen, W.; Ren, J.; Wang, M.; Dang, L.; Shen, X.; Yang, X.; Zhang, H. Chem. Commun. 2014, 50, 6259.
(a) Zhang, S. X.; Shen, X. L.; Li, Z. Q.; Zou, L. W.; Wang, F. Q.; Zhang, H. B.; Shao, Z. H. J. Org. Chem. 2013, 78, 11444.
(b) Han-ya, Y.; Inui, T.; Yokoshima, S.; Tokuyama, H.; Fukuyama, T. Chem. Pharm. Bull. 2016, 64, 800.
Kobayashi, S.; Ueda, T.; Fukuyama, T. Synlett 2000, 883.
Al-Saffar, F. M.; Brown, R. C. D. Org. Lett. 2017, 19, 3502.
Tang, T. P.; Ellman, J. A. J. Org. Chem. 2002, 67, 7819.
(a) Cutter, A. C.; Miller, I. R.; Keily, J. F.; Bellingham, R. K.; Light, M. E.; Brown, R. C. Org. Lett. 2011, 13, 3988.
(b) Watkin, S. V.; Camp, N. P.; Brown, R. C. Org. Lett. 2013, 15, 4596.
(a) Han, Z.; Krishnamurthy, D.; Senanayake, C. H. Org. Process Res. Dev. 2006, 10, 327.
(b) Tian, M.; Yan, M.; Baran, P. S. J. Am. Chem. Soc. 2016, 138, 14234.
(c) Wang, C.; Liu, Y. W.; Zhou, Z., Si; C. M., Sun, X.; Wei, B. G. Tetrahedron 2018, 74, 2158.
(d) Hugelshofer, C. L.; Palani, V.; Sarpong, R. J. Am. Chem. Soc. 2019, 141, 8431.
Brak, K.; Ellman, J. A. Org. Lett. 2010, 12, 2004.
Cai, S. L.; Yuan, B. H.; Jiang, Y. X.; Lin, G. Q.; Sun, X. W. Chem. Commun. 2017, 53, 3520.
Chen, Y.-J.; Cai, S.-L.; Wang, C.-C.; Cheng, J.-D.; Kramer, S.; Sun, X.-W. Chem.-Asian J. 2017, 12, 1309.
Voituriez, A.; Ferreira, F.; Perezluna, A.; Chemla, F. Org. Lett. 2007, 9, 470.
Huang, P. Q.; Guo, Z. Q.; Ruan, Y. P. Org. Lett. 2006, 8, 1435.
(a) Voituriez, A.; Ferreira, F.; Chemla, F. J. Org. Chem. 2007, 72, 5358.
(b) Louvel, J.; Botuha, C.; Chemla, F.; Demont, E.; Ferreira, F.; Pérez-Luna, A. Eur. J. Org. Chem. 2010, 2921.
(c) Louvel, J.; Chemla, F.; Demont, E.; Ferreira, F.; Pérez-una, A.; Voituriez, A. Adv. Synth. Catal. 2011, 353, 2137.
(d) Hélal, B.; Ferreira, F.; Botuha, C.; Chemla, F.; Perez-Luna, A. Synlett 2009, 3115.
Mei, S.; Zhao, G. Eur. J. Org. Chem. 2010, 1660.
Sirasani, G.; Andrade, R. B. Org. Lett. 2011, 13, 4736.
González-Gómez, J. C.; Medjahdi, M.; Foubelo, F.; Yus, M. J. Org. Chem. 2010, 75, 6308.
Medjahdi, M.; González-Gómez, J. C.; Foubelo, F.; Yus, M. Eur. J. Org. Chem. 2011, 2230.
(a) Bosque, I.; González-Gómez, J. C.; Guijarro, A.; Foubelo, F.; Yus, M. J. Org. Chem. 2012, 77, 10340.
(b) Anton-Torrecillas, C.; González-Gómez, J. C. Org. Biomol. Chem. 2014, 12, 7018.
Bonazzi, S.; Cheng, B.; Wzorek, J. S.; Evans, D. A. J. Am. Chem. Soc. 2013, 135, 9338.
Shen, A.; Liu, M.; Jia, Z. S.; Xu, M. H.; Lin, G. Q. Org. Lett. 2010, 12, 5154.
Still, W. C.; Gennari, C. Tetrahedron Lett. 1983, 24, 4405.
Zhao, S.; Sirasani, G.; Vaddypally, S.; Zdilla, M. J.; Andrade, R. B. Angew. Chem., Int. Ed. 2013, 52, 8309.
Davies, S. G.; Fletcher, A. M.; Roberts, P. M.; Shah, R. S.; Thompson, A. L.; Thomson, J. E. Org. Lett. 2014, 16, 1354.
Davies, S. G.; Fletcher, A. M.; Shah, R. S.; Roberts, P. M.; Thomson, J. E. J. Org. Chem. 2015, 80, 4017.
Ye, J.; Zhang, Y.; Liu, Y.; Zhang, J.; Ruan, Y.; Huang, P. Org. Chem. Front. 2015, 2, 697.
(a) Guo, L. D.; Liang, P.; Zheng, J. F.; Huang, P. Q. Eur. J. Org. Chem. 2013, 2230.
(b) Ye, J. L.; Chen, H.; Zhang, Y. F.; Huang, P. Q. Org. Chem. Front. 2016, 3, 683.
Cai, S. L.; Song, R.; Dong, H. Q.; Lin, G. Q.; Sun, X. W. Org. Lett. 2016, 18, 1996.
Liu, H.; Zhang, X.; Shan, D.; Pitchakuntla, M.; Ma, Y.; Jia, Y. Org. Lett. 2017, 19, 3323.
Larock, R. C.; Yum, E. K.; Refvik, M. D. J. Org. Chem. 1998, 63, 7652.
(a) Botuha, C.; Chemla, F.; Ferreira, F.; Perez Luna, A.; Roy, B. New J. Chem. 2007, 31, 1552.
(b) Chemla, F.; Ferreira, F.; Gaucher, X.; Palais, L. Synthesis 2007, 1235.
(c) Chemla, F.; Ferreira, F. Synlett 2006, 2613.
Schkeryantz, J. M.; Woo, J. C. G.; Siliphaivanh, P.; Depew, K. M.; Danishefsky, S. J. J. Am. Chem. Soc. 1999, 121, 11964.
Bonazzi, S.; Cheng, B.; Wzorek, J. S.; Evans, D. A. J. Am. Chem. Soc. 2013, 135, 9338.
(a) Beaumont, S.; Ilardi, E. A.; Monroe, L. R.; Zakarian, A. J. Am. Chem. Soc. 2010, 132, 1482.
(b) Gu, Z.; Zakarian, A. Angew. Chem., Int. Ed. 2010, 49, 9702.
(c) Wen, S.; Carey, K. L.; Nakao, Y.; Fusetani, N.; Packham, G.; Ganesan, A. Org. Lett. 2007, 9, 1105.
(d) Wang, J.; Li, W.; Liu, L.; Wang, B.; Zhou, Y.; Huang, S.; Wang, X. Org. Chem. Front. 2019, 6, 1599.
(e) Babij, N. R.; Wolfe, J. P. Angew. Chem., Int. Ed. 2012, 51, 4128.
(f) Zhang, S.-J.; Li, X.-T.; Wang, Y.; Zheng, Y.-C.; Han, S.-Q.; Yu, H.-L.; Huang, S.-H. Chin. J. Org. Chem. 2020, 40, 521(in Chinese).
(张世举, 李晓彤, 王燕, 郑宇璁, 韩世清, 郁惠蕾, 黄莎华, 有机化学, 2020, 40, 521.)
Zhang, L.; Zhang, Y.; Li, W.; Qi, X. Angew. Chem., Int. Ed. 2019, 58, 4988.
(a) Chen, B.; Liu, X.; Hu, Y.-J.; Zhang, D.-M.; Deng, L.; Lu, J.; Min, L.; Ye, W.-C.; Li, C.-C. Chem. Sci. 2017, 8, 4961.
(b) Ryan, D. A.; Okolotowicz, K. J.; Mercola, M.; Cashman, J. R. Tetrahedron Lett. 2015, 56, 4195.
(c) Zhu, Y.; Li, H.; Lin, K.; Wang, B.; Zhou, W. Synth. Commun. 2019, 49, 1721.
Zhong, Y.-W.; Dong, Y.-Z.; Fang, K.; Izumi, K.; Xu, M.-H.; Lin, G.-Q. J. Am. Chem. Soc. 2005, 127, 11956.
Wang, R.; Fang, K.; Sun, B.-F.; Xu, M.-H.; Lin, G.-Q. Synlett 2009, 2301.
Si, C.-M.; Liu, Y.-W.; Mao, Z.-Y.; Han, P.; Du, Z.-T.; Wei, B.-G. Tetrahedron 2016, 72, 8091.
(a) Liu, R. C.; Wei, J. H.; Wei, B. G.; Lin, G. Q. Tetrahedron: Asymmetry 2008, 19, 2731.
(b) Xarnod, C.; Huang, W.; Ren, R. G.; Liu, R. C.; Wei, B. G. Tetrahedron 2012, 68, 6688.
(c) Zhou, W.; Nie, X.-D.; Zhang, Y.; Si, C.-M.; Zhou, Z.; Sun, X.; Wei, B.-G. Org. Biomol. Chem. 2017, 15, 6119.
Kochi, T.; Ellman, J. A. J. Am. Chem. Soc. 2004, 126, 15652.
Peltier, H. M.; Mcmahon, J. P.; Patterson; A, W.; Ellman, J. A. J. Am. Chem. Soc. 2006, 128, 16018.
Nicolaou, K. C.; Estrada, A. A.; Zak, M.; Lee, S. H.; Safina, B. S. Angew. Chem., Int. Ed. 2005, 44, 1378.
Zhao, S.; Andrade, R. B. J. Am. Chem. Soc. 2013, 135, 13334.
Wang, X.; Xia, D.; Tan, L.; Chen, H.; Huang, H.; Song, H.; Qin, Y. Chem.-Eur. J. 2015, 21, 14602.
Kokkonda, P.; Andrade, R. B. Org. Lett. 2019, 21, 9594.
Amat, M.; Alvarez, M.; Bonjoch, J.; Casamitjana, N.; Gràcia, J.; Lavilla, R.; Garcías, X.; Bosch, J. Tetrahedron Lett. 1990, 31, 3453.
(a) Kazak, M.; Priede, M.; Shubin, K.; Bartrum, H. E., Poisson, J. F.; Suna, E. Org. Lett. 2017, 19, 5356.
(b) Jung, H. H.; Floreancig, P. E. J. Org. Chem. 2007, 72, 7359.

图式 2 Rozwadowska课题组关于(S)-(-)-salsolidine的合成
Scheme 2 Synthesis of (S)-(-)-salsolidine by Rozwadowska group

图式 3 林国强课题组关于(+)-swainsonine的全合成
Scheme 3 Total synthesis of (+)-swainsonine by Lin group

图式 5 Amat课题组关于(-)-cermizine B的全合成
Scheme 5 Total synthesis of (-)-cermizine B by Amat group

图式 6 Taghizadeh课题组关于(S)-ketamine的全合成
Scheme 6 Total synthesis of (S)-ketamine by Taghizadeh group

图式 7 Reisman课题组关于(−)-3-demethoxyerythratidinone的全合成
Scheme 7 Total synthesis of (-)-3-demethoxyerythratidinone by Reisman group

图式 8 Morris课题组关于dioncophylline E•TFA的全合成
Scheme 8 Total synthesis of dioncophylline E•TFA by Morris group

图式 9 Prasad课题组关于(-)-epiquinamide的全合成
Scheme 9 Total synthesis of (-)-epiquinamide by Prasad group

图式 11 Njardarson课题组关于(-)-supinidine, (-)-isoretro- necanol和(+)-elacomine的全合成
Scheme 11 Total synthesis of (-)-supinidine, (-)-isoretrone- canol and (+)-elacomine by Njardarson group

图式 12 张洪彬课题组关于(-)-vindorosine的全合成
Scheme 12 Total synthesis of (-)-vindorosine by Zhang and co-workers

图式 13 Brown课题组关于(+)-β-isosparteine的全合成
Scheme 13 Total synthesis of (+)-β-isosparteine by Brown group

图式 14 Ellman课题组关于(-)-aurantioclavine的全合成
Scheme 14 Total synthesis of (-)-aurantioclavine by Ellman group

图式 15 孙兴文课题组关于(+)-lycoricidine和(+)-7-deoxypancratistatin的全合成
Scheme 15 Total synthesis of (+)-lycoricidine and (+)-7-deoxypancratistatin by Sun group

图式 16 Chemla课题组关于(-)-1-hydroxyquinolizidinone的全合成
Scheme 16 Total synthesis of (-)-1-hydroxyquinolizidinone by Chemla group

图式 17 赵刚课题组关于(-)-fasicularin和(-)-lepadiformine A的全合成
Scheme 17 Total synthesis of (-)-fasicularin and (-)-lepadiformine A by Zhao group

图式 18 Andrade课题组关于(-)-leuconicine A和(-)-leuconicine B的全合成
Scheme 18 Total synthesis of (-)-leuconicine A and (-)-leuconicine B by Andrade group

图式 19 González-Gómez课题组关于(-)-aphanorphine的全合成
Scheme 19 Total synthesis of (-)-aphanorphine by González-Gómez and co-workers

图式 20 Evans课题组关于(-)-nakadomarin A的全合成
Scheme 20 Total synthesis of (-)-nakadomarin A by Evans group

图式 21 Andrade课题组关于(-)-melotenine A的全合成
Scheme 21 Total synthesis of (-)-melotenine A by Andrade and co-workers

图式 22 Davies课题组关于(-)-nakinadine A的全合成
Scheme 22 Total synthesis of (-)-nakinadine A by Davies group

图式 23 黄培强课题组关于pandamarilactonines A~C的全合成
Scheme 23 Total synthesis of pandamarilactonines A~C by Huang group

图式 24 孙兴文课题组关于amathaspiramides B, D, F的全合成
Scheme 24 Total synthesis of amathaspiramides B, D, F by Sun and co-workers

图式 25 贾彦兴课题组关于festuclavine, pyroclavine, costaclavine, epi-costaclavine, pibocin A, 9-deacetoxyfumigaclavine C和fumigaclavine G的全合成
Scheme 25 Total synthesis of festuclavine, pyroclavine, costaclavine, epi-costaclavine, pibocin A, 9-deacetoxyfumigaclavine C and fumigaclavine G by Jia group

图式 26 贾彦兴课题组关于dihydrosetoclavine的全合成
Scheme 26 Total synthesis of dihydrosetoclavine by Jia group

图式 27 齐湘兵课题组关于(-)-alstofolinine A的全合成
Scheme 27 Total synthesis of (-)-alstofolinine A by Qi group

图式 28 林国强课题组关于D-erythro-sphinganine的全合成
Scheme 28 Total synthesis of D-erythro-sphinganine by Lin group

图式 29 林国强课题组关于(+)-febrifugine的不对称合成
Scheme 29 Asymmetric synthesis of (+)-febrifugine by Lin group

图式 30 魏邦国课题组关于epohelmin A的不对称合成
Scheme 30 Asymmetric synthesis of epohelmin A by Wei group

图式 31 Ellman课题组关于(6R, 7S)-7-amino-7, 8-dihydro-α-bisabolene的合成
Scheme 31 Total synthesis of (6R, 7S)-7-amino-7, 8-dihydro-α-bisabolene by Ellman group

图式 32 Ellman课题组关于tubulysin D的全合成
Scheme 32 Total synthesis of tubulysin D by Ellman group

图式 33 Andrade课题组关于(-)-aspidospermidine, (-)-tabersonine和(-)-vincadifformine的全合成
Scheme 33 Total synthesis of (-)-aspidospermidine, (-)-tabersonine and (-)-vincadifformine by Andrade group

图式 34 秦勇课题组关于(-)-2-hydroxyisoschizogamine和isoschizogamine的全合成
Scheme 34 Total synthesis of (-)-2-hydroxyisoschizogamine and isoschizogamine by Qin and co-workers

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:











 下载:
下载:



 下载:
下载: