
图式 1.
C(sp3)—H的不对称氧化反应及面临的挑战
Scheme 1.
Challenges associated with asymmetric C(sp3)—H oxidation

C—H键直接氧化反应可在烷烃底物中引入氧原子, 将其直接变为醇、酮或醛等重要含氧化合物, 是非常重要的有机反应, 用途广泛[1].其中, 烷烃C—H键的不对称氧化反应是极具挑战性的反应之一, 由于同一分子中存在多种不同性质的C—H键, 实现C—H位点选择性或对映选择性的氧化尤为困难[2].同时, 不对称C—H氧化所形成的手性醇类底物, 极易在相同氧化体系下发生过度氧化而生成酮, 导致手性中心消除(Scheme 1).尽管C—H不对称氧化反应的研究已经持续了数十年, 但进展仍然缓慢, 有待深入研究和发展出高效催化剂.实现C—H键不对称氧化, 需要深入理解影响C—H氧化反应的因素, 从不同的参数和方向调控不对称C—H氧化反应.自然界生物酶能活化氧气, 形成高价金属-氧(M—oxo)中间体, 可在温和条件下实现C—H、C=C的(不对称)氧化反应, 是发展高效催化氧化的理想模型[3].本文将综述三十多年来模拟生物酶活性中心结构发展的小分子金属配合物及其催化的烷烃C—H键不对称氧化反应进展.
实现C—H氧化反应的第一步是C—H键的断裂.因此, 不同C—H键的解离能将直接影响C—H氧化反应的活性大小[4].比如, 苯环C—H键解离能(BDE)为472.6 kJ/mol, 苄位C—H键的解离能为357.5 kJ/mol, 三级C—H键的解离能为400.6 kJ/mol, 二级C—H键的解离能为410.6 kJ/mol, 一级C—H键的解离能为422.4 kJ/mol.值得注意的是, 一些特殊官能团的α位C—H键解离能的变化:比如, 环丙烷α位的C—H键的解离能为407.7 kJ/mol, 醚类底物氧原子α位的C—H键的解离能为389.3 kJ/mol, 哌啶中N原子α位的C—H键的解离能为379.7 kJ/mol.因此, 从C—H键的BDE大小考虑, 不同种类底物C—H氧化反应发生的活性顺序如Scheme 2所示.

分子中不同位置的C—H键具有不同的电性, 且由于氧化剂或者氧化活性中心的亲电性, 导致烃类化合物在发生C—H氧化反应时具有不同的活性位点.当分子中存在一些拉电子基团, 比如酯基、氰基、酰亚胺等基团时, C—H氧化反应往往发生在距离这些拉电子基团较远的位点.比如, 1996年, Asensio等[5]报道了使用甲基(三氟甲基)-1, 3-二环氧丙烷(2, TFDO)为氧化剂氧化含脂肪链的酯类化合物的C—H氧化反应(Scheme 3).该研究发现, 处于1中酯基远端的亚甲基C—H被氧化为酮, 而其他亚甲基几乎没有转化. 2007年, White等[6]也报道了分子结构中既含有三级C—H也含有酯基的脂肪链的C—H氧化反应.结果显示, 处于酯基远端的三级C—H更加容易被氧化, 而靠近酯基的三级C—H则反应活性更低.

胺和含氮杂环化合物不仅是重要的有机合成中间体, 而且在农药、天然产物以及活性药物分子中广泛存在.因此, 含氮化合物的C—H氧化反应也是C—H官能化反应中一个重要的反应类型.通常情况下, 氮原子α位的C—H键的解离能更小, 会优先发生反应, 比如生成亚胺等.但是如果先将有机胺质子化后形成相应的铵盐, 翻转含氮基团的电性, 远离氮原子的C—H键发生氧化将成为可能. 1993年, Asensio等[7]就利用四氟硼酸和正己基胺(11)制备了相应的铵盐12.该铵盐在氧化性条件下转化为相应的2-甲基四氢吡啶(14).这一反应中, 在铵盐的诱导下二级C—H键的氧化发生在远离铵盐的位点, 生成了相应的酮13, 而氨基的α位C—H键没有发生反应.随后, 13发生分子内的脱水反应从而生成2-甲基四氢吡啶(14)(Scheme 4, A). 2015年, Sanford等[8]使用相同的策略, 在1.1 equiv.硫酸的作用下, 实现了N-乙基吡咯烷(15)的C—H氧化反应(Scheme 4, B).这些实例也表明了诱导效应在C—H氧化反应的位点选择性中所起到的重要作用.
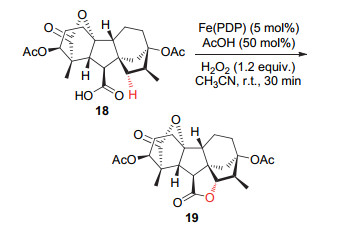
如果分子中存在一种官能团, 其可以改变由电性和诱导效应等因素控制的C—H键氧化位点选择性, 则可将该官能团视为此C—H氧化反应的导向基团[9].理想的状况是:这种导向基团在氧化性条件下较为稳定, 而在完成选择性C—H氧化后, 还可以在随后的转化反应中利用起来, 或可以方便地脱除. 2007年, White等[6]研究了羧酸作为导向基团控制复杂化合物C—H氧化位置选择性的反应(Scheme 5).他们选取了四氢赤霉酸18作为氧化反应的底物, 发现C—H氧化发生在与羧基邻近的二级C—H键, 生成了相应的内酯19, 而不是更加富电子的三级C—H键.同时, 如果将羧酸基团甲酯化以后, 四氢赤霉酸的C—H氧化反应的转化率将变低下且产物分布杂乱.这一例子清楚地证明了C—H氧化的反应位点可以由导向基团决定, 而不仅仅是由C—H键的BDE和电性来决定.
有机分子中与C—H键相邻或者邻近的位置有大位阻的基团时, 该C—H键发生氧化的活性和速率就会大大降低.如果两个C—H键的位阻大小差异相对较小, 也可以通过调整催化剂的空间结构来控制含多个类别的C—H的氧化反应.早在1992年, Waegell等[10]就发现了位阻因素对C—H氧化反应的影响结果(Scheme 6).柏木烷20是一种含有4个三级C—H和5个亚甲基的三环结构的烃类化合物.他们巧妙地选择柏木烷为底物, 研究了四氧化钌催化的C—H氧化, 结果显示: C(8)位的三级C—H键被氧化为叔醇, 而C(2)位和C(5)位的C—H却没有发生氧化反应.这一结果正是由于C(2)位和C(5)位的邻位有更大的位阻所导致的. 2009年, Costas和Ribas等[11]在研究薄荷醇酯22的氧化时, 也发现了类似的结果.他们发现C—H羟化反应发生在位阻较小的C(5)位C—H而不是位阻较大的C(2)位异丙基的三级C—H.

2009年, Baran等[13]结合Eschenmoser和Schreiber在1955年提出的环张力释放理论和Houk等[12]的计算结果, 将甾醇氧化过程中产生选择性的原因——环张力释放——引入到C—H氧化反应中来, 解释了一些电性极其相似位阻也接近的多个C—H键在氧化反应中具有一定选择性的原因.他们考察了含有两个三级C—H的双环底物在氧化反应中的选择性(Scheme 7).通过核磁检测, 发现底物中异丙基中的三级碳的化学位移比与H2相连的碳的化学位移小δ 0.9, 这也表明异丙基中三级C—H1要比C—H2更富电性.因此, 如果仅仅考虑电性和位阻的话, C—H氧化反应位点更应倾向发生在H1的位置.然而, 实验结果却与之相反:他们发现即使使用不同的氧化剂, C—H氧化反应都发生在H2的位置.这一现象促使他们考虑是否存在其他因素主导了该类底物C—H氧化的位点选择性.随后, 他们合成了与化合物26结构极其相似的化合物27.在这两个化合物中H2的化学位移仅仅相差δ 0.1.当把等物质的量的26和27的混合物在同一反应体系下进行氧化时, 发现化合物26被氧化的量是27的3~4倍.这一结果进一步证实了:当底物中有相同电性和位阻的C—H时, 有利于1, 3轴向的环张力释放的C—H键氧化将优先发生.

在自然界中, 细胞色素P450参与了生命体中的甾醇类激素合成, 是一种末端单加氧酶.细胞色素P450的活性中心结构包含一个血红素铁中心, 在轴向上有一个半胱氨酸的巯基配体, 轴向的另外一端被水分子占据[14].细胞色素P450在催化氧化烷烃时, 烷烃先跟铁中心结合, 铁配合物即由低自旋态30转变成高自旋态31, 然后被还原为二价32, 接着被氧气氧化生成三价过氧自由基物种33, 进一步通过质子偶合电子转移过程生成三价Fe(III)-OOH物种34; 随后, Fe(III)-OOH与质子结合释放出一分子水后形成五价Fe(V)=O物种35, 35为真正的氧化性中间体.烷烃被氧化时, C—H键的断裂和C—O键的生成, 可能是协同过程, 也可能是摘氢-氧反馈的分步过程, 而五价Fe(V)=O物种最终变成三价Fe(III)物种进入下一次催化循环(Scheme 8).
师法自然, 化学家能够从生物酶催化的氧化反应受到启发而发展出高效的催化氧化反应新体系.目前, 针对C—H键不对称氧化反应发展的小分子金属配合物仿生催化剂主要有两大类:血红素酶模拟物卟啉金属配合物及基于非血红素金属蛋白酶的手性多齿氮配体的金属配合物[15].
1989年, Groves等[16]结合细胞色素P450模型化合物在烯烃不对称环氧化反应中的催化机制, 设计并合成了一种新型的四苯基卟啉配体, 并首次报道了卟啉Fe、Mn配合物催化C(sp3)—H不对称羟化反应.他们将手性联萘引入四苯基卟啉骨架, 用联萘作为桥联基团, 制备出了一种新型的手性卟啉配体并分别合成了相应的Fe、Mn配合物(Scheme 9).使用碘氧苯为氧化剂时, 他们分别考察了铁和锰配合物在乙苯C—H羟化反应中的催化性能, 发现二者的催化活性和选择性均不同:铁配合物催化时, 产率为40%, 对映选择性为40%, 产物的醇酮比为2.3:1;使用锰配合物催化该反应, 产率更高为77%, 对映选择性却更差(26% ee), 产物的醇酮比为1.3:1. 1999年, Gross等[17]利用缩酮修饰了四苯基卟啉骨架, 合成了手性卟啉钌配合物43.与之前工作不同的是, 他们采用2, 6-二氯吡啶氮氧化物42为氧化剂, 尝试了三级苄位C—H的羟化反应, 此反应优点是醇产物不会被进一步氧化为酮.他们发现, 在室温条件下2-苯基丁烷(40)以54%的产率和27%的ee被氧化成醇41.当降低反应温度到10 ℃时反应的产率降低到23%, 反应的对映选择性升高到38% (Scheme 10).同年, 支志明课题组[18]利用Halterman发展的氢化蒽骨架修饰的卟啉配体, 制备出相应的钌配合物45, 考察了其在苄位C—H氧化反应中的催化性能.他们同样使用2, 6-二氯吡啶氮氧化物为氧化剂.在二价钌配合物的催化下, 乙苯被以62%的产率转化为醇, 其对映选择性为72%, 而醇酮比为1.67:1 (Scheme 11).



2012年, Simonneaux等[19]进一步将Halterman卟啉配体进行优化, 引入了四个磺酸基, 合成了水溶性的卟啉锰配合物46 (Scheme 12).在以双氧水为氧化剂的条件下, 乙苯的苄位C—H氧化反应得以实现.产物中醇酮比例为1.3:1, 醇产物的ee为38%.随后, 他们使用二醋酸碘苯作为氧化剂, 产物的醇酮比最高为91:9且产物手性醇的对映选择性最高达到75% ee[20].

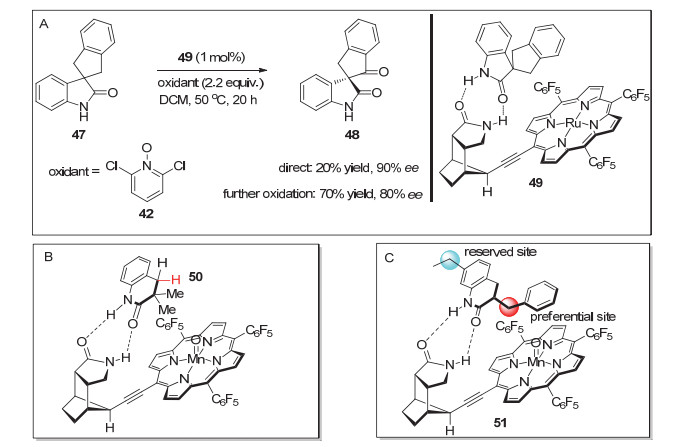
由于手性卟啉配体合成比较复杂, 随后的很多年此领域进展缓慢. 2015年, Bach课题组[21]将氢键导向的反应概念引入到螺环氧化吲哚的C—H氧化反应中.通过去对称化的策略, 实现了螺环氧化吲哚的对映选择性C—H氧化反应.在此之前, 他们[22]通过Sonogashira反应在卟啉配体中引入刚性大位阻手性酰胺骨架, 并以其新型的含酰胺导向基团的卟啉钌配合物成功实现了烯烃的不对称环氧化反应.作者发现, 此手性Ru催化剂49催化螺环氧化吲哚47的C—H键氧化的中间产物醇不稳定, 容易发生逆aldol反应, 导致反应的产率较低、对映选择性极不稳定.因此, 在完成C—H氧化反应后, 又将反应混合液置于Swern氧化或者氯铬酸吡啶(PCC)氧化的条件下继续氧化, 将羟基产物转化为酮.这一改进大大提高了产物手性酮48的收率, 且反应的对映选择性也基本保持(Scheme 13, A). 2018年, 该课题组[23]利用相似的策略, 实现了3, 3-二甲基-3, 4-二氢喹诺酮(50)的苄位C—H氧化反应(Scheme 13, B).与螺环氧化吲哚的苄位C—H氧化不同的是, 此反应使用碘氧苯为氧化剂, 且产物停留在醇产物.最近, 他们又将该反应体系应用到3-苯亚甲基喹诺酮(51)的苄位C—H不对称羟化反应中(Scheme 13, C), 该反应的对映选择性可高达99%[24].
手性salen配体是20世纪90年代发展起来的一类手性配体, 其在众多的不对称反应中得到应用, 被认为是一类优势配体[25]. Salen金属配合物为N2-O2型配体, 其金属配合物的N2-O2配位原子通常在同一平面, 通常被认为是一种类卟啉配体.由于salen金属配合物在烯烃的不对称环氧化反应中表现出的优异的催化性能, 自然也被考虑用于C—H不对称氧化反应.
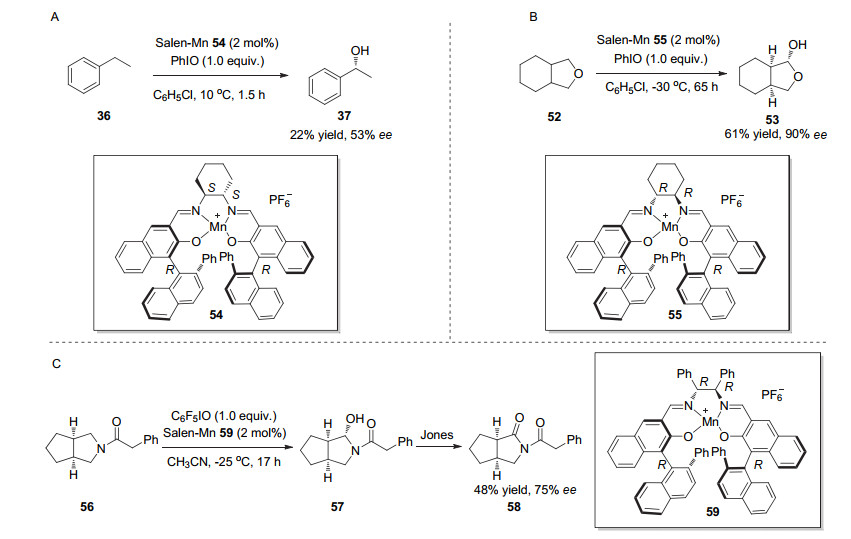
1996年, Katsuki课题组[26]将轴手性联萘骨架的salen-Mn配合物应用到苄位C—H羟化反应中.他们通过与Jacobsen催化剂作比较, 发现在Salen配体中引入的轴手性联萘骨架才是真正有利于不对称C—H羟化反应手性诱导的决定因素.同样的, 以乙苯的氧化为例, 在该体系的标准条件下, 乙苯以22%的产率和53% ee转化为1-苯基乙醇(Scheme 14, A). 1998年, 他们又通过对手性二胺和联萘骨架的手性匹配筛选, 调整了氧化剂、反应溶剂和温度, 最终实现了四氢呋喃类似物[27]和吡咯烷类底物[28]的去对称C—H羟化反应(Scheme 14, B, C).这是第一例金属配合物催化的内消旋底物的去对称C—H羟化反应研究.
Murahashi课题组[29]于2004年使用轴手性联萘骨架将salen配体桥连起来, 考察了其在茚满60的苄位C—H氧化反应中的催化性能(Scheme 15).作者还研究了反应体系中的添加剂对反应结果的影响.他们发现1, 5-二环己基咪唑(62)能提高反应的产率和对映选择性, 且该反应产物的醇酮比例也得到了一定程度的提高.此外, 该课题组[30]尝试了salen-Mn配合物在C(sp3)—H的去对称氧化反应中的催化性能.除了使用碘氧苯为氧化剂外, 在该体系中, 他们还添加了0.5 equiv.的4-苯基吡啶氮氧化物66来提高反应的活性和选择性.尽管通过去对称化的策略, 2-甲基-1, 3-二苯基丙烷(64)可以转化为手性酮65, 但是反应的产率和对映选择性都不能令人满意.

在过去几十年, 模拟氧化酶的活性中心结构, 设计、合成新型手性配体及其金属配合物, 实现化学选择、位点选择、对映选择地简单烃C—H的氧化反应一直是有机化学家追求的目标.除了卟啉骨架和类卟啉salen骨架, 选用不同的手性胺骨架以及氮供体, 调节配体的空间结构和电性, 进而控制金属配合物的催化活性和选择性是科学家们一致采用的策略.近十多以来, 基于手性二胺与吡啶等杂环构建的线性四氮配体所发展的铁、锰配合物, 作为非血红素酶的模型化合物, 在不对称氧化反应中表现出优秀的催化活性和对映选择性.
2017年, Costas课题组[31]将前期发展的新型配体——使用手性环己二胺为手性骨架, 三异丙基硅基修饰的吡啶为氮供体——应用到烷烃的不对称C—H氧化反应中, 获得了优异的反应结果.这一反应采用了去对称化的策略, 当使用酰胺为导向基团时, 环己烷的C—H氧化发生在C(3)位而不是C(4)位(Scheme 16, A).特别是当使用特戊酰胺为导向基团, 环丙烷甲酸为添加剂时, C(3)位氧化成酮的产率为90%, 对映选择性为90%.对于非血红素N4金属配合物/H2O2催化氧化反应体系中, 有机羧酸添加剂是决定反应活性和选择性的重要因素[32]. 2018年, 我们课题组[33]以环氧化反应为模板, 深入研究了酸添加剂在不对称氧化反应中的作用:双氧水氧化锰配合物形成可能的Mn-OOH物种, 然后有机羧酸辅助此物种的O—O键异裂, 进而脱去一分子水形成Mn=O活性中间体; 同时, 酸配位在金属中心, 有效提高中间体的不对称诱导能力, 大位阻的羧酸将有助于提高反应的对映选择性.

此外, Costas课题组[34]还尝试了1, 4-二取代的环己烷的C—H氧化.结果表明: 1, 4-顺式取代的底物71中一个酰胺基团被氧化为羰基, 而另一个酰胺基团保留; 1, 4-反式取代的底物72则不能发生反应(Scheme 16, B).这一结果提供了一种双取代环己烷的拆分策略.从另外一方面说, 也恰巧证明了环状结构烷烃C—H氧化反应中的1, 3-轴向的张力释放因素对C—H氧化反应位点的影响.
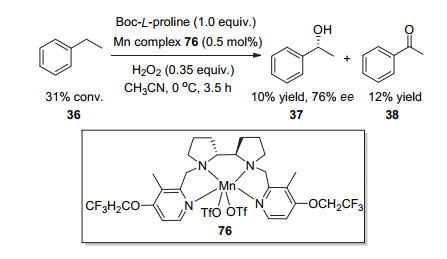
Bryliakov课题组[35]在同年报道了乙苯类苄位C—H氧化的反应结果.他们使用三氟乙氧基修饰的四齿氮配合物76为催化剂, 其中手性二胺骨架为联吡咯(Scheme 17).为了提高反应产物的醇酮比例, 他们使用了0.35 equiv.的H2O2作氧化剂, 使用Boc-L-脯氨酸为添加剂.然而反应的结果并不令人满意:乙苯的转化率仅仅为31%, 产物苯乙醇和苯乙酮的比例为1:1.2, 苯乙醇的对映选择性为76%.这一反应结果说明:在四齿氮金属配合物为催化剂, 双氧水为终端氧化剂, 酸为添加剂的反应体系中, C—H氧化反应想要停留在手性醇的阶段是非常难以控制的.
我们课题组[36]在过去的十多年中也一直致力于高效不对称氧化体系的构建, 在模拟非血红素蛋白酶活性中心结构的过程中, 发展了一系列易于制备、性能优异的四齿氮配体及其金属配合物.这些配合物在烯烃环氧化反应中展现出了较高的催化活性和不对称诱导能力[37].鉴于这些研究基础, 我们尝试挑战烷烃C—H键的不对称氧化反应.设计了一类具有螺环结构的化合物77, 经过催化剂的系统筛选, 发现L-脯氨酸衍生的手性配体的锰配合物82 (S-peb-Mn)在此类螺环底物C—H氧化反应中展现出优良的催化性能(Scheme 18).六元螺环化合物氧化产物的对映选择性可达94%, 五元螺环的C—H氧化产物的对映选择性可达98%.值得一提的是, 尽管该类底物中, 有多个苄位C—H, 但是该反应仍然给出了优异的位点选择性和对映选择性[38].此外, 该反应与之前报道的苄位C—H氧化反应的产率相比, 同样有很大程度的提高.随后, 我们还进一步研究了含氮杂环的螺环化合物的C—H氧化反应.比如, 我们研究了在药物活性分子和天然产物中较为常见的螺环氧化吲哚骨架和螺环喹啉酮骨架的C—H氧化[39].结果显示:不管是螺环氧化吲哚还是螺环喹啉酮底物, 都能被很好地转化为相应的手性酮; 当降低反应的温度和氧化剂的用量时, 该反应以22%~41%的产率获得了螺环喹啉酮的C—H羟化产物, 对映选择性高达99%.

金刚烷骨架中含有多个亚甲基以及三级C—H, 因此成为研究C—H氧化反应的模板底物之一.最近, Costas和Bietti等[40]利用羧酸作为导向基, 实现了金刚烷骨架的定点C—H氧化反应, 合成了内酯化产物84, 反应的产率可达88%, 对映选择性最高可以达到99% (Scheme 19).在该反应中, C—H氧化发生在羧基γ位, 这一位置远离拉电子基团, 适合形成五元环内酯产物.值得注意的是, 在羧基的诱导下更富电子的三级C—H并没有发生氧化反应, 且当羧基γ位附近有取代基形成位阻时, 可以通过催化剂结构的调控实现C—H氧化位点的控制.

C—H键广泛存在于有机分子中, 其氧化反应可直接向分子中引入氧或者羰基官能团, 或者在复杂分子全合成的后期实现直接官能化, 是现代有机合成中一类重要的反应.特别是一些简单烃类化合物的C—H不对称氧化反应将为生物活性分子的合成提供新的合成策略, 提高复杂分子全合成的效率.然而, 也正是由于同一有机分子中存在多个C—H键, 并且C—H之间的立体性差异较小, 难以识别, 给简单烷烃C—H的不对称氧化提出了更高的要求.本文综述了模拟生物酶发展的一系列新型手性卟啉、四氮配体金属配合物催化C—H键不对称氧化反应的研究进展, 并结合影响C(sp3)—H的氧化反应的因素、模拟酶金属配合物催化的C—H氧化反应的机理, 从配体类型的角度阐述了当前所实现的C—H键不对称氧化的一些实例.尽管目前这些研究仅仅是针对一些特殊底物取得了较高的催化活性和立体控制能力, 但是这些结果为人们发展更加高效和实用的不对称C—H氧化催化新体系奠定了坚实的基础, 提供了一些催化剂设计的思路.我们也期待, 随着对C—H不对称氧化反应的进一步深入研究和反应机制的全面认识, 底物普适性强、催化活性和选择性更高的小分子仿生催化剂将会出现.
(a) Katsuki, T. Comprehensive Asymmetric Catalysis I-Ⅲ: C—H Oxidation, Eds.: Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H., 1999, p. 791.
(b) Costas, M. Coord. Chem. Rev. 2011, 255, 2912.
(c) Newhouse, T.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 3362.
(d) Zhou, M.; Crabtree, R. H. Chem. Soc. Rev. 2011, 40, 1875.
(e) Qian, S.; Ma, Y.; Gao, S.; Luo, J. Chin. J. Org. Chem. 2018, 38, 1930 (in Chinese).
(钱少平, 马尧睿, 高姗姗, 骆钧飞, 有机化学, 2018, 38, 1930.)
(f) Wu, J.; Zhu, J.; Li, H.; Wu, C.; Shen, R.; Yu, L. Chin. J. Org. Chem. 2019, 39, 3328 (in Chinese).
(吴锦雯, 朱佳雯, 李慧, 吴春雷, 沈润溥, 余乐茂, 有机化学, 2019, 39, 3328.)
(a) Li, G.; Zhang, L. Chin. J. Org. Chem. 1985, 5, 27 (in Chinese).
(李广年, 张良辅, 有机化学, 1985, 5, 27.)
(b) Shen, D.; Miao, C.; Wang, S.; Xia, C.; Sun, W. Org. Lett. 2014, 16, 1108.
(c) Chen, J.; Lutz, M.; Milan, M.; Costas, M.; Otte, M.; Klein Gebbink, R. J. M. Adv. Synth. Catal. 2017, 359, 2590.
(d) Dantignana, V.; Milan, M.; Cusso, O.; Company, A.; Costas, M.; Bietti, M. ACS Cent. Sci. 2017, 3, 1350.
(e) Milan, M.; Costas, M.; Salamone, M.; Bietti, M. Acc. Chem. Res. 2018, 51, 1984.
(f) Wang, W.; Xu, D.; Sun, Q.; Sun, W. Chem.-Asian J. 2018, 13, 2458.
(g) White, M. C.; Zhao, J. J. Am. Chem. Soc. 2018, 140, 13988.
(a) Guo, M.; Corona, T.; Ray, K.; Nam, W. ACS Cent. Sci. 2019, 5, 13.
(b) Ray, K.; Pfaff, F. F.; Wang, B.; Nam, W. J. Am. Chem. Soc. 2014, 136, 13942.
(c) Yin, G. Acc. Chem. Res. 2013, 46, 483.
(d) Que, L. Jr.; Tolman, W. B. Nature 2008, 455, 333.
Luo, Y.-R. Handbook of Bond Dissociation Energies in Organic Compounds, CRC press, Boca Raton, 2003.
Asensio, G.; Castellano, G.; Mello, R.; González Núñez, M. E. J. Org. Chem. 1996, 61, 5564. doi: 10.1021/jo9604189
Chen, M. S.; White, M. C. Science 2007, 318, 783. doi: 10.1126/science.1148597
Asensio, G.; González Núñez, M. E.; Bernardini, C. B.; Mello, R.; Adam, W. J. Am. Chem. Soc. 1993, 115, 7250. doi: 10.1021/ja00069a025
Lee, M.; Sanford, M. S. J. Am. Chem. Soc. 2015, 137, 12796. doi: 10.1021/jacs.5b09099
罗飞华, 有机化学, 2019, 39, 3084. doi: 10.6023/cjoc201905027Luo, F. Chin. J. Org. Chem. 2019, 39, 3084 (in Chinese). doi: 10.6023/cjoc201905027
Tenaglia, A.; Terrenova, E.; Waegell, B. J. Org. Chem. 1992, 57, 5523. doi: 10.1021/jo00046a040
Gómez, L.; Garcia-Bosch, I.; Company, A.; Benet-Buchholz, J.; Polo, A.; Sala, X.; Ribas, X.; Costas, M. Angew. Chem., Int. Ed. 2009, 48, 5720. doi: 10.1002/anie.200901865
Chen, K.; Eschenmoser, A.; Baran, P. S. Angew. Chem., Int. Ed. 2009, 48, 9705. doi: 10.1002/anie.200904474
Du, X.; Houk, K. N. J. Org. Chem. 1998, 63, 6480. doi: 10.1021/jo9801519
(a) Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A. M.; Maves, S. A.; Benson, D. E.; Sweet, R. M.; Ringe, D.; Petsko, G. A.; Sligar, S. G. Science 2000, 287, 1615.
(b) Meunier, B.; de Visser, S. P.; Shaik, S. Chem. Rev. 2004, 104, 3947.
(c) Jiang, Y.; Li, S. Chin. J. Org. Chem. 2018, 38, 2307 (in Chinese).
(蒋媛媛, 李盛英, 有机化学, 2018, 38, 2307.)
(a) Ortiz de Montellano, P. R. Chem. Rev. 2010, 110, 932.
(b) Abu-Omar, M. M.; Loaiza, A.; Hontzeas, N. Chem. Rev. 2005, 105, 2227.
(a) Groves, J. T.; Viski, P. J. Am. Chem. Soc. 1989, 111, 8537.
(b) Groves, J. T.; Viski, P. J. Org. Chem. 1990, 55, 3628.
Gross, Z.; Ini, S. Org. Lett. 1999, 1, 2077. doi: 10.1021/ol991131b
Zhang, R.; Yu, W.-Y.; Lai, T.-S.; Che, C.-M. Chem. Commun. 1999, 2441.
Srour, H.; Le Maux, P.; Simonneaux, G. Inorg. Chem. 2012, 51, 5850. doi: 10.1021/ic300457z
Maux, P. L.; Srour, H. F.; Simonneaux, G. Tetrahedron 2012, 68, 5824. doi: 10.1016/j.tet.2012.05.014
Frost, J. R.; Huber, S. M.; Breitenlechner, S.; Bannwarth, C.; Bach, T. Angew. Chem., Int. Ed. 2015, 54, 691.
(a) Fackler, P.; Berthold, C.; Voss, F.; Bach, T. J. Am. Chem. Soc. 2010, 132, 15911.
(b) Fackler, P.; Huber, S. M.; Bach, T. J. Am. Chem. Soc. 2012, 134, 12869.
Burg, F.; Gicquel, M.; Breitenlechner, S.; Pöthig, A.; Bach, T. Angew. Chem., Int. Ed. 2018, 57, 2953. doi: 10.1002/anie.201712340
Burg, F.; Breitenlechner, S.; Jandl, C.; Bach, T. Chem. Sci. 2020, 11, 2121. doi: 10.1039/C9SC06089H
(a) Katsuki, T. Coord. Chem. Rev. 1995, 140, 189.
(b) Bhatia, B.; Punniyamurthy, T.; Iqbal, J. In Asymmetric Oxidation Reactions, Ed.: Katsuki, T., Oxford University Press, New York, 2001, p. 1~15.
(c) Matsumoto, K.; Saito, B.; Katsuki, T. Chem. Commun. 2007, 3619.
(d)Fan, Q.-H.; Ding, K. Top. Organomet. Chem. 2011, 36, 207.
(e) Shaw, S.; White, J. D. Chem. Rev. 2019, 119, 9381.
Hamachi, K.; Irie, I.; Katsuki, T. Tetrahedron Lett. 1996, 37, 4979. doi: 10.1016/0040-4039(96)00984-7
Miyafuji, A.; Katsuki, T. Tetrahedron 1998, 54, 10339. doi: 10.1016/S0040-4020(98)00489-X
Punniyamurthy, T.; Miyafuji, A.; Katsuki, T. Tetrahedron Lett. 1998, 39, 8295. doi: 10.1016/S0040-4039(98)80001-4
Murahashi, S. I.; Noji, S.; Komiya, N. Adv. Synth. Catal. 2004, 346, 195. doi: 10.1002/adsc.200303190
Murahashi, S.; Noji, S.; Hirabayashi, T.; Komiya, N. Tetrahedron: Asymmetry 2005, 16, 3527. doi: 10.1016/j.tetasy.2005.08.056
Milan, M.; Bietti, M.; Costas, M. ACS Cent. Sci. 2017, 3, 196. doi: 10.1021/acscentsci.6b00368
(a) Mas-Ballesté, R.; Que, L. Jr. J. Am. Chem. Soc. 2007, 129, 15964.
(b) Lyakin, O. Y.; Ottenbacher, R. V.; Bryliakov, K. P.; Talsi, E. P. ACS Catal. 2012, 2, 1196.
(a) Du, J.; Miao, C.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. ACS Catal. 2018, 8, 4528.
(b) Li, X.-X.; Guo, M.; Qiu, B.; Cho, K.-B.; Sun, W.; Nam, W. Inorg. Chem. 2019, 58, 14842.
Milan, M.; Bietti, M.; Costas, M. Org. Lett. 2018, 20, 2720. doi: 10.1021/acs.orglett.8b00929
Talsi, E. P.; Samsonenko, D. G.; Ottenbacher, R. V.; Bryliakov, K. P. ChemCatChem 2017, 9, 4580. doi: 10.1002/cctc.201701169
Sun, W.; Sun, Q. Acc. Chem. Res. 2019, 52, 2370. doi: 10.1021/acs.accounts.9b00285
(a) Wang, B.; Miao, C.; Wang, S.; Xia, C.; Sun, W. Chem.-Eur. J. 2012, 18, 6750.
(b) Wang, B.; Wang, S.; Xia, C.; Sun, W. Chem.-Eur. J. 2012, 18, 7332.
(c) Wu, M.; Wang, B.; Wang, S.; Xia, C.; Sun, W. Org. Lett. 2009, 11, 3622.
(d) Miao, C.; Wang, B.; Wang, Y.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. J. Am. Chem. Soc. 2016, 138, 936.
(e) Shen, D.; Qiu, B.; Xu, D.; Miao, C.; Xia, C.; Sun, W. Org. Lett. 2016, 18, 372.
(f) Wang, W.; Sun, Q.; Xia, C.; Sun, W. Chin. J. Catal. 2018, 39, 1463.
Qiu, B.; Xu, D.; Sun, Q.; Miao, C.; Lee, Y.-M.; Li, X.-X.; Nam, W.; Sun, W. ACS Catal. 2018, 8, 2479. doi: 10.1021/acscatal.7b03601
Qiu, B.; Xu, D.; Sun, Q.; Lin, J.; Sun, W. Org. Lett. 2019, 21, 618. doi: 10.1021/acs.orglett.8b03652
Cianfanelli, M.; Olivo, G.; Milan, M.; Klein Gebbink, R. J. M.; Ribas, X.; Bietti, M.; Costas, M. J. Am. Chem. Soc. 2020, 142, 1584. doi: 10.1021/jacs.9b12239

图式 1 C(sp3)—H的不对称氧化反应及面临的挑战
Scheme 1 Challenges associated with asymmetric C(sp3)—H oxidation

图式 2 不同C—H键的BDE与C—H氧化反应活性
Scheme 2 Relationship between BDE of C—H bonds and the reactivity of different C—H in oxidation

图式 3 诱导效应对C—H氧化反应位点的影响
Scheme 3 Site-selectivity of C—H oxidation influenced by inductive effect

图式 5 羧基作为导向基团对C—H氧化反应位点的影响
Scheme 5 Site-selectivity of C—H oxidation influenced by directing-group

图式 6 位阻影响的C—H氧化反应位点选择性
Scheme 6 Site-selectivity of C—H oxidation influenced by steric hindrance

图式 7 环张力释放影响的C—H氧化反应
Scheme 7 Site-selectivity of C—H oxidation influenced by strain release

图式 9 手性联萘桥连的卟啉配合物在乙苯C—H氧化反应中的催化性能
Scheme 9 Asymmetric C—H oxidation catalyzed by porphyrin complex modified using binaphthyl skeleton

图式 10 手性缩酮桥连的卟啉钌配合物催化的苄位C—H氧化反应
Scheme 10 Asymmetric C—H oxidation catalyzed by porphyrin complex bridged using ketal

图式 11 Halterman卟啉配体及其钌配合物催化的苄位C—H氧化反应
Scheme 11 Asymmetric benzylic C—H oxidation catalyzed by porphyrin-Ru complex

图式 12 水溶性卟啉锰配合物催化的乙苯C—H氧化反应
Scheme 12 Asymmetric benzylic C—H oxidation catalyzed by water-soluble porphyrin-Mn complex

图式 13 刚性酰胺骨架修饰的卟啉配合物催化的C—H氧化反应
Scheme 13 Asymmetric C—H oxidation catalyzed by porphyrin complex modified using amide

图式 14 联萘骨架修饰的Salen-Mn配合物催化的不对称C—H羟化反应
Scheme 14 Asymmetric C—H hydroxylation catalyzed by salen-Mn complex modified using binaphthyl skeleton

图式 15 联萘骨架桥连的salen-Mn配合物在不对称C—H氧化反应中的催化性能
Scheme 15 Asymmetric C—H oxidation catalyzed by salen-Mn complex bridged using binaphthol

图式 16 三异丙基硅基修饰的手性环己二胺骨架四齿氮配合物催化的C—H氧化反应
Scheme 16 Asymmetric C—H oxidation catalyzed by tetradentate nitrogen complex modified using tri-isopropylsilyl group

图式 17 三氟乙氧基修饰的手性联吡咯骨架四齿氮配合物催化的C—H氧化反应
Scheme 17 Asymmetric C—H oxidation catalyzed by tetradentate nitrogen complex modified using trifluoroethoxyl group

图式 18 脯氨酸衍生的S-peb-Mn配合物催化的C—H氧化反应
Scheme 18 Asymmetric C—H oxidation catalyzed by S-peb- Mn complex derived from L-proline

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:



 下载:
下载: