
图式 1.
Kobayashi苯炔前体
Scheme 1.
Kobayashi benzyne precursor

含磷和含硫有机化合物被广泛应用于有机合成、功能材料和药物化学等领域中[1-2], 一直吸引着科学家们的目光.其中, 芳基膦配体因其独特的空间结构可与多种过渡金属形成均相催化剂, 在催化反应中发挥着越来越重要的作用[3].芳基硫化合物同样有着举足轻重的作用, 作为一类重要的结构单元广泛存在于天然产物和合成药物分子中[4].近年来, 磷和硫的芳基化反应成为了有机磷和有机硫化学研究的热点之一[5-6].传统芳基化主要利用有机磷或有机硫化合物与卤代芳烃的交叉偶联反应来实现, 但该方法通常依赖过渡金属催化剂, 且需要较高反应温度或较长反应时间.基于此, 发展绿色、温和、高效的磷和硫芳基化方法具有重要意义.
芳炔作为一种重要的有机反应活性中间体一直受到人们的普遍关注, 其参与的反应是构建碳碳键和碳杂键的重要方法之一, 在天然产物、生物碱和药物中间体的合成中得到了广泛的应用[7]. 1983年, Kobayashi课题组[8]发展了一种以邻三甲基硅苯基三氟磺酸酯为苯炔前体在氟离子存在下现场生成苯炔的温和方法(Scheme 1), 克服了苯炔反应受苛刻条件限制的缺点, 极大地推动了芳炔化学的快速发展.
具有高亲电特性的芳炔容易被亲核试剂进攻, 生成芳基负离子中间体.芳基负离子随后可作为新的亲核试剂进攻外部的亲电试剂, 生成邻二取代芳烃(Scheme 2).依据芳基负离子的淬灭方式, 可将芳炔与亲核试剂的反应分为三类[7]: (1)经质子化的直接芳基化反应(Scheme 2, Route a); (2)经分子间进攻亲电试剂的三组分反应(Scheme 2, Route b); (3)经分子内进攻亲电中心的σ-或π-键插入反应(Scheme 2, Route c).由于芳炔的反应可避免使用过渡金属催化剂, 条件温和且适用范围广, 芳炔参与有机磷和有机硫化合物的反应可以解决传统芳基化方法中存在的一些问题, 有效地促进了有机磷和有机硫化学的发展[9].本文拟结合本课题组在该领域的研究工作[10], 依据芳炔与亲核试剂的几种反应类型, 对近年来(2004年以来)芳炔参与的磷和硫芳基化反应研究进行综述.
传统的磷芳基化方法通常为过渡金属催化的亲核性含磷试剂与卤代芳烃等化合物的交叉偶联反应(Hirao反应)[5], 但反应条件较为苛刻, 具有一定的局限性.后来, 国内外有机化学家针对芳炔与有机磷化合物的反应展开了一系列研究.
2010年, Jugé课题组[11]报道了室温下芳炔参与的手性和非手性P(Ⅲ)化合物的直接芳基化反应(Eq. 1).该反应中, 含有孤对电子的磷原子亲核进攻芳炔, 产生的芳基负离子被溶剂中的质子淬灭后, 得到手性或非手性的芳基季鏻盐.此外, 三苯基胂与苯炔的反应同样以高产率得到苯基季鉮盐.
|
|
(1) |
2014年, Hosoya课题组[12]以1, 3-双(三氟甲磺酰氧基)-2-碘苯为芳炔前体, 在格氏试剂作用下可以现场生成3-三氟甲磺酰氧基苯炔.该反应中, 三苯基膦首先亲核进攻苯炔生成两性离子中间体.然后, 该中间体发生含硫的Fries重排, 导致三氟甲磺酰基迁移至芳环上, 最后得到芳基季鏻盐(Scheme 3).
2013年, Mhaske课题组[13]实现了芳炔参与亚磷酸三乙酯及其衍生物的直接芳基化(Scheme 4).该反应在温和条件下以良好的底物适应性得到芳基磷酸酯、芳基亚磷酸酯和芳基叔膦氧化物.该反应中, 亲核性的P(Ⅲ)化合物进攻芳炔后, 可能经两种反应途径得到目标产物.在途径a中, 反应经协同过程脱乙基化得到芳基化产物.途径b则与Michaelis-Arbuzov反应[14]类似, 乙基被体系中负离子攫取.同年, Hosoya课题组[15]报道了类似的反应方法, 反应以四丁基氟化铵(TBAF)为氟盐, 同样以高产率得到芳基磷酸酯类化合物.

2016年, 本课题组[10d]发展了芳炔参与亚磷酸二酯和仲膦氧化物的直接芳基化反应(Scheme 5).该反应在室温下以高产率得到芳基磷酸酯, 但在80 ℃下以高产率得到芳基叔膦氧化物.该反应中, 碳酸铯作为碱调节P(Ⅴ)与P(Ⅲ)的互变异构化平衡, 加快亲核性的P(Ⅲ)化合物进攻芳炔.如果没有添加碳酸铯, 该反应则不能得到目标产物.张万斌课题组[16]几乎同时报道了类似的反应方法, 但其反应需在100 ℃的较高温度下进行.考虑到仲膦氧化物的芳基化反应在较高温度下进行, 且需要添加过量的碳酸铯, 本课题组[10e]进一步发展了芳炔参与的铜催化仲膦氧化物的直接芳基化反应(Scheme 5).该反应无需加入额外碱, 在CuI催化下便可在室温下以高产率得到一系列芳基叔膦氧化物.该反应中, 仲膦氧化物与CuI作用生成P(O)Cu中间体, 然后进攻芳炔生成芳基铜中间体.最后, 芳基铜捕捉仲膦氧化物的质子得到目标产物, 同时P(O)Cu中间体再生.

σ-或π-键的芳炔插入反应是芳炔化学研究的热点之一, 其通过巧妙断裂化学键可制备结构多样化的邻二取代芳烃.含磷化学键需要具备电子极化的特征, 即需要有亲电中心和亲核中心, 才能顺利与芳炔发生插入反应.已报道可与芳炔发生插入反应的含磷化学键包括C—P键、C=P键、P—N键、P=N键、P—O键、P=O键和P—P键等.
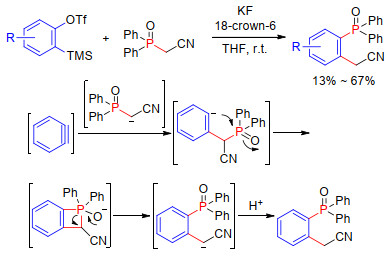
2005年, Yoshida和Kunai课题组[17]发展了磷酰基乙腈C—P键的芳炔插入反应(Scheme 6).该反应一步构建C—C键和C—P键, 以中等产率得到邻位取代的芳基膦.该反应中, 活泼亚甲基失去质子生成碳负离子, 然后亲核进攻芳炔, 生成芳基负离子中间体.芳基负离子进一步分子内亲核进攻磷原子, 生成不稳定的苯并四元环中间体.最后, 四元环开环导致C—P键断裂, 质子化后得到目标产物.
2011年, Alajarin、Lopez-Leonardo和Orenes课题组[18]报道了芳炔对P=N键的插入反应(Scheme 7).该反应中, 芳炔首先经[2+2]环加成对烯基膦亚胺P=N键插入, 形成的中间体开环后再经6π电环化生成苯并六元环中间体, 后者被溶剂乙腈质子化后得到季鏻盐产物.该方法同样适用于炔基膦亚胺和炔基膦硫化合物, 后者发生P=S键的芳炔插入反应. 2014年, 该课题组[19]进一步发展了烷基或芳基膦亚胺P=N键的芳炔插入反应(Scheme 7).与之前不同的是, 该反应没有经历6π电环化过程.两性离子中间体直接被溶剂质子化, 得到邻氨基苯基季鏻盐.此外, 产物可与过量苯炔进一步发生N-芳基化反应.该反应同样适用于烷基或芳基膦硫化合物, 发生P=S键的芳炔插入反应.
2013年, 张万斌课题组[20]实现了芳基磷酰胺P—N键的芳炔插入(Scheme 8).碳酸铯可加速磷酰胺的脱质子过程, 从而提高磷酰胺的亲核性.与C—P键的芳炔插入反应机理类似, 芳炔与磷酰胺的反应生成不稳定的苯并四元环中间体, 然后开环导致P—N键断裂, 最后质子化得到邻位芳胺基取代的芳基膦氧化物.值得一提的是, 产物经还原后可得到重要的双齿氮磷配体. 2016年, 郭键和贺耘课题组[21]报道了有机磷酸P—O键的芳炔插入反应(Scheme 8).与P—N的芳炔插入反应机理类似, 该反应经过苯并四元环中间体开环断裂P—O键, 得到邻位羟基取代的芳基膦氧化物.当芳炔过量时, 产物中的酚羟基进一步发生芳基化.
2019年, Gogoi课题组[22]发展了叔膦氧化物P=O键的芳炔插入反应(Scheme 9).该反应中, 芳炔首先插入稳定的P=O键形成苯并四元环中间体, 然后开环断裂P—O键生成1, 4-两性离子中间体.最后, 氧负离子与另一分子芳炔发生芳基化反应, 得到邻位芳氧基取代的芳基季鏻盐.
2018年, Hirano和Miura课题组[23]报道了四芳基二膦P—P键的芳炔插入反应(Scheme 10).该反应一步构建两个C—P键, 得到重要的双齿配体1, 2-双(二芳基膦基)苯类化合物.作者通过理论计算研究指出, 反应可能经过苯并四元环两性离子中间体, 再经开环断裂P—P键, 生成不稳定的三价双膦化合物.最后, P(Ⅲ)化合物经硫单质处理后分离得到稳定的硫代双膦氧化物.
芳炔、含磷有机化合物和亲电试剂的多组分反应较为罕见, 可能原因是磷原子与芳炔产生的两性离子中间体极易发生质子化, 导致芳基负离子难以进攻反应体系中的其它亲电试剂. 2013年, Hosoya课题组[15]报道了苯炔、甲氧基膦化合物和二氧化碳的三组分反应(Scheme 11).该反应中, 芳基负离子被二氧化碳捕捉, 生成苯甲酸根负离子.最后, 羧酸根负离子进攻分子内甲基, 得到邻位磷酰基取代的苯甲酸甲酯.由于作者仅给出了两个例子, 该反应的底物适用范围未知.

2014年, Biju课题组[24]报道了芳炔、叔膦和醛的三组分反应(Scheme 12).该反应中, 叔膦首先亲核进攻芳炔, 生成1, 3-两性离子中间体.然后, 中间体与醛发生[3+2]偶极环加成, 得到苯并氧杂磷杂茂类化合物.同年, 该课题组[25]进一步发展了芳炔、三芳基膦和活化酮的三组分反应.值得一提的是, 由该类方法得到的苯并氧杂磷杂茂类化合物在农药、材料化学与催化反应等领域有着潜在的应用.

2020年, Tobisu课题组[26]报道了芳炔引发的芳香亲核取代/脱芳基化的新型环合反应(Scheme 13).该反应中, 含磷底物为含氟三芳基膦类化合物. P(Ⅲ)化合物首先与芳炔产生芳基负离子, 然后发生分子内芳香亲核取代反应, 经C—F键断裂构建P—F键, 生成氟化磷中间体.最后, 该中间体与水作用发生脱芳基化, 得到二苯并膦杂茂类化合物.该方法适合制备多氟取代的二苯并膦杂茂, 产物可作为潜在的先进有机材料.此外, 作者尝试了无氟的三苯基膦与苯炔的反应, 同样得到二苯并膦杂茂, 但作者未说明原因.
硫芳基化通常由过渡金属催化的芳基卤化物和硫醇的交叉偶联反应实现[6], 但该类方法的反应条件较为苛刻.近年来, 芳炔参与的反应实现了温和条件下无过渡金属参与的硫芳基化.芳炔与含硫有机物的反应不仅可实现硫的直接芳基化, 而且可一步构建碳硫键和碳杂键等, 制备出结构多样化的芳基硫类化合物.
2006年, Larock课题组[27]发展了苯炔参与苯硫酚的直接苯基化反应(Eq. 2).该反应中, 苯硫酚与苯炔在室温下发生亲核加成, 以良好的产率得到二苯基硫醚.后来, Yoshida和Hosoya课题组[28]以邻碘芳基三氟磺酸酯为芳炔前体, 同样实现了硫醇的直接芳基化.
|
|
(2) |
2014年, Mhaske课题组[29]报道了芳炔参与亚磺酸钠类化合物的直接芳基化反应(Eq. 3).该反应中, 芳基或烷基亚磺酸钠中的硫原子亲核进攻芳炔, 在室温下以良好至优秀的产率得到系列芳基砜类化合物.同年, Singh课题组[30]报道了类似的反应方法, 但反应需在80 ℃的较高温度下进行.后来, Shibata课题组[31]进一步拓展了三氟甲基亚磺酸钠和芳炔的底物范围.
|
|
(3) |
2017年, 彭勃课题组[32]发现芳炔与二芳基硫醚反应可得到三芳基硫鎓盐(Eq. 4).由于二苯基硫醚的亲核性较弱, 苯环上的供电子取代基能有效提高反应效率.苯环上的大位阻取代基对反应起抑制作用, 导致产率显著降低, 说明位阻效应对反应有较大影响.值得一提的是, Hosoya课题组[12]以1, 3-双(三氟甲磺酰氧基)-2-碘苯为芳炔前体进行了类似反应研究, 实现了硫的直接芳基化, 其反应产物及机理与Scheme 3中所示的磷芳基化反应类似.
|
|
(4) |
2017年, 沈美华和徐华栋课题组[33]发展了芳炔参与2-噁唑烷硫酮的直接芳基化反应(Eq. 5).反应底物中存在硫原子和氮原子两个亲核中心, 该反应选择性地发生了硫芳基化, 得到芳硫基取代的二氢噁唑类化合物.
|
|
(5) |
由于硫原子能有效稳定α位碳负离子, 脂肪族硫醚与芳炔产生的芳基负离子容易发生分子内质子化, 生成芳基硫叶立德. 2017年, 郭键和贺耘课题组[34]发展了芳炔引发的α-苄硫基酮的Stevens重排反应(Scheme 14).几乎同时, Biju课题组[35]报道了芳炔引发的β-羰基烯丙硫醚的Stevens重排反应.这类反应中, 芳基负离子经分子内质子化生成硫叶立德, 随后发生Stevens重排, 得到β-羰基芳硫醚类化合物.底物中的羰基至关重要, 其增加了α位碳上氢原子的酸性, 从而使硫叶立德顺利生成.随后, 谭嘉靖和徐坤课题组[36]报道了类似的反应方法, 拓展了反应底物范围, 例如炔丙基硫醚和芳基烯丙基硫醚均适用于这类反应.
2018年, 谭嘉靖和徐坤课题组[37]报道了芳炔、环状硫醚与亲核试剂的三组分反应(Scheme 15).该反应中, 环状硫醚首先与芳炔生成硫鎓离子中间体.在亲核试剂进攻下, 环状硫鎓离子开环断裂C—S键, 得到结构多样化的芳基硫醚类化合物.该反应底物适用范围广泛, 以C、O、S、N和F为中心的亲核试剂均适用于该反应.随后, 该课题组[38]进一步对芳炔引发的开环氟代反应进行了详细研究, 反应在温和条件下得到多种高价值的氟代产物.同年, 何林课题组[39]报道了类似的三组分反应, 进一步扩大了底物范围.值得指出的是, Hoye课题组[40]在2016年报道了以三炔化合物为芳炔前体参与的类似反应.
2019年, Almqvist课题组[41]进一步发展了芳炔引发的硫叶立德反应(Eq. 6).该反应中, 芳炔与四氢噻唑底物中的硫原子生成两性离子中间体, 再经分子内1, 4-质子迁移生成硫叶立德中间体.最后, 经硫叶立德开环断裂C—S键得到N-烯基取代的吡啶酮类化合物.
|
|
(6) |
2011年, Greaney课题组[42]报道了硫脲C=S键的芳炔插入反应(Scheme 16).该反应中, 芳炔首先插入C=S键生成苯并四元环中间体, 然后开环断裂C—S键生成硫酚.最后, 硫酚与另一分子芳炔发生芳基化, 得到邻位芳硫基取代的芳基脒类化合物.该反应同时生成硫的直接芳基化副产物, 当硫脲上有吸电子基团时, 副产物的产率明显提升.
2015年, Werz课题组[43]发展了钯催化芳基硫氰酸酯C—S键的芳炔插入反应(Eq. 7).首先, 金属催化剂Pd(OAc)2和双膦配体Xantphos生成活性的[Pd(0)].然后, 零价钯与C—S键发生氧化加成, 生成[Pd(Ⅱ)]中间体.最后, 二价钯与芳炔配位后经还原消除过程一步构建C—C键和C—S键, 得到邻位硫取代的苯甲腈类化合物, 同时零价钯再生.由于芳炔具有高反应活性, 该反应容易生成副产物, 加入氧气可有效抑制副产物的生成.
|
|
(7) |
2016年, Studer课题组[44]报道了乙烯基硫醚C—S键的芳炔插入反应(Scheme 17).该反应利用乙烯基硫醚缺乏α-H的特性, 其与芳炔发生[3+2]环加成生成苯并五元环硫叶立德中间体, 再经质子转移和β-消除过程, 得到高立体选择性的三取代烯烃.当硫原子α位碳上有酰基时, 硫叶立德中间体则可与另一分子芳炔发生芳基化反应, 最后经β-消除过程得到高立体选择性的四取代烯烃.
2017年, 徐修华课题组[45]报道了三氟甲基砜类化合物C—S键的芳炔插入反应(Eq. 8).与C—P键的芳炔插入反应机理[17]类似, 三氟甲基砜的活泼亚甲基失去质子产生碳负离子, 然后亲核进攻芳炔, 生成芳基负离子中间体.最后, 芳基负离子分子内亲核进攻硫原子, 断裂C—S键后得到邻位取代的芳基砜类化合物.
|
|
(8) |
2018年, Mhaske课题组[46]进一步发展了硫叶立德C—S键的芳炔插入反应(Scheme 18).该反应中, 硫叶立德首先与芳炔生成苯并四元环中间体.然后, 溴负离子攫取该中间体的甲基, 并导致开环断裂C—S键, 最后得到邻位取代的苯甲硫醚类化合物.同年, 何林课题组[47]也报道了类似的反应方法, 底物范围得到了很大的拓展.
早在2005年, Larock课题组[48]报道了三氟甲基亚磺酰胺S—N键的芳炔插入反应(Eq. 9).该反应同样经历了苯并四元环中间体, 然后开环断裂S—N键得到芳烃邻位双官能团化产物.底物中的三氟甲基至关重要, 不仅增强了亚磺酰胺的酸性, 而且提升了硫原子的亲电性能.
|
|
(9) |
2016年, 李杨课题组[49]发展了三氟甲磺酰胺S—N键的芳炔插入反应(Scheme 19).该反应使用的芳炔前体为该课题组开发的含三个取代基的多米诺芳炔试剂(TPBT).首先, 三氟甲磺酰胺亲核进攻芳炔, 生成的中间体再次脱去一个OTf产生新芳炔.然后, 新芳炔对另一分子三氟甲磺酰胺S—N键进行插入反应, 最后得到三取代芳烃类化合物.值得指出的是, 该反应在苯环的1, 2, 3-三个位点上连续生成了C—N键、C—S键及C—N键.
2019年, Biju课题组[50]进一步发展了次磺酰胺S—N键的芳炔插入反应(Eq. 10).该反应中, 氮原子首先进攻芳炔, 产生两性离子中间体.然后, 芳基负离子分子内亲核进攻硫原子, 导致S—N键断裂, 最后得到邻位硫取代的苯胺类化合物.
|
|
(10) |
2015年, Hosoya课题组[51]报道了硫亚胺类化合物S=N键的芳炔插入反应(Scheme 20).该反应首先经过芳炔与S=N键的环加成, 生成苯并四元环中间体.然后, 四元环开环断裂S—N键得到两性离子中间体.最后, 氮负离子分子内进攻缺电子芳环(SNAr反应), 导致该芳环从硫原子迁移至氮原子上, 得到邻位硫取代的苯胺类化合物.值得注意的是, 当硫亚胺底物上有一个甲基取代基时, 反应则经过脱甲基化得到邻氨基芳硫醚.后来, 该课题组[52]进一步发展了邻溴芳基甲基硫亚胺S=N键的芳炔插入反应.同样, 该反应经脱甲基化得到邻氨基芳硫醚类化合物.由于产物的两个芳环上各带有氨基和溴, 产物可在金属钯催化下发生分子内偶联得到重要的吩噻嗪类化合物.
2017年, Yoshida和Hosoya课题组[53]进一步发展了亚砜亚胺类化合物S=N键的芳炔插入反应(Eq. 11).研究表明, 该反应受含硫底物上取代基的影响较大.当底物为二芳基亚砜亚胺时, 反应主要进行S=N键的芳炔插入, 以中等产率得到邻位亚磺酰基取代的苯胺类化合物.当底物为芳基甲基亚砜亚胺时, 反应则主要发生氮的直接芳基化.
|
|
(11) |
2015年, 王彬课题组[54a]报道了亚砜化合物S=O键的芳炔插入反应(Scheme 21).该反应中, 非对称亚砜与苯炔的反应得到两种芳基硫醚异构体[54a].作者给出了两种可能反应途径: (1)亚砜与两分子芳炔产生硫叶立德, 然后去甲基化得到产物; (2)亚砜与芳炔经[2+2]环加成生成苯并四元环中间体, 然后发生[1, 2]-甲基迁移得到产物.此外, 作者进行了靛红捕捉硫叶立德的实验, 证明了该反应存在硫叶立德中间体.后来, 该课题组[54b]利用这类现场生成的硫叶立德与醛或酮的反应实现了温和条件下无碱参与的羰基化合物的环氧化.
2017年, Studer课题组[55]进一步丰富了亚砜的底物范围, 发展了乙烯基亚砜S=O键的芳炔插入反应(Scheme 22).该反应同样经历了芳炔与亚砜的[2+2]环加成生成苯并四元环, 然后开环形成两性离子.最后, 乙烯基从硫原子迁移至氧原子上, 得到多取代的烯基醚类化合物.

2017年, 彭勃课题组[56]报道了二芳基亚砜S=O键的芳炔插入反应(Eq. 12).该反应中, 芳炔首先与S=O键作用生成两性离子中间体.然后, 氧负离子与另一分子芳炔发生氧芳基化, 最后得到邻位芳氧基取代的三芳基硫鎓盐类化合物.
|
|
(12) |
2017年, Yoshida和Hosoya课题组[57]进一步发展了二芳基亚砜S=O键的芳炔插入反应(Scheme 23).该反应中, 芳炔与二芳基亚砜生成两性离子中间体.然后, 氧负离子分子内亲核进攻缺电子芳环(SNAr反应), 生成Meisenheimer络合物, 最后得到邻位芳硫基取代的二芳基醚类化合物.值得注意的是, 该反应中的亚砜底物必须含有一个缺电子芳基.

2007年, Raminelli课题组[58]报道了二苯二硫醚S—S键的苯炔插入反应(Scheme 24).作者仅报道了一个例子, 反应得到1, 2-二(苯硫基)苯, 产率仅为29%.值得指出的是, 作者对Se—Se键的苯炔插入反应进行了较为细致的研究, 以中等收率得到相应的含硒产物. 2017年, Daugulis课题组[59]报道了双(三氟甲基)二硫醚S—S键的芳炔插入反应, 实现了芳环邻位双三氟甲基硫化(Scheme 24).值得一提的是, 三氟甲基芳基硫醚(ArSCF3)存在于许多生物活性分子结构中, 具有重要的医用价值.
2004年, Yoshida和Kunai课题组[60]报道了苯基三烷基锡硫醚S—Sn键的芳炔插入反应(Eq. 13).该反应中, 硫原子亲核进攻芳炔形成两性离子中间体.然后, 芳基负离子分子内亲核进攻锡原子导致S—Sn键断裂, 最后得到邻位芳硫基取代的芳基锡类化合物.此外, 作者进行了产物的应用研究, 以三丁基芳基锡为原料实现了Stille偶联等过渡金属催化的交叉偶联反应.
|
|
(13) |
2011年, Murafuji课题组[61]发展了芳基二芳基铋硫醚S—Bi键的苯炔插入反应(Eq. 14).该反应条件温和, 以较高产率得到邻位芳硫基取代的芳基铋类化合物.铋的传统芳基化方法为芳基格氏试剂或芳基锂试剂与铋(Ⅲ)卤代物的反应, 具有很大的局限性.此外, 作者还实现了S—Sb键的苯炔插入.
|
|
(14) |
含硫σ-或π-键的芳炔插入反应是实现芳烃邻位双官能团化的一种非常有效的策略, 但含硫底物通常需要预合成, 具有一定的局限性.芳炔参与的三组分反应可采用廉价易得的反应底物, 在邻位双官能化芳烃的合成上具有一定的优势.
2014年, 陈加荣和肖文精课题组[62]首次将二甲亚砜引入到芳炔和α-溴代羰基化合物的三组分反应中, 通过芳炔插入S=O键, 得到多取代芳基甲基硫醚类化合物(Scheme 25).该反应中, 二甲亚砜的氧原子亲核进攻芳炔产生两性离子中间体.然后, 芳基负离子分子内亲核进攻硫原子生成苯并四元环中间体.进一步, 四元环开环产生的羰基α位硫叶立德与α-溴代羰基化合物发生分子间亲核取代反应, 生成硫鎓盐中间体.最后, 溴负离子攫取甲基后得到目标产物.值得一提的是, 二甲亚砜在该反应中同时作为甲硫基化试剂和氧源.最近, Gogoi课题组[63]进一步拓宽了该反应的底物范围, 作者以亲电的丁炔二酸酯类化合物代替α-溴代羰基化合物, 反应同样能顺利进行.

由现有文献可知, 芳炔参与的反应通常得到芳烃邻位双官能团化产物.然而, 天然产物或药物分子的芳环上大多含有三个或以上的取代基.如果能打破传统苯炔化学只能邻位双官能化的限制, 实现一步构建多取代芳烃, 从而可快速合成药物分子或其它高价值化合物. 2016年, 李杨课题组[64]报道了芳炔、芳基烯丙基亚砜和溴代烷烃的三组分反应(Scheme 26).该反应实现了在芳环的三个连续位置上一步构建C—C键、C—S键和C—O键, 得到了1, 2, 3-三取代的芳烃官能化产物.该反应先后经历了烯丙基的分子内S→O迁移、克莱森重排、四元环开环以及氧负离子的亲核取代等过程.

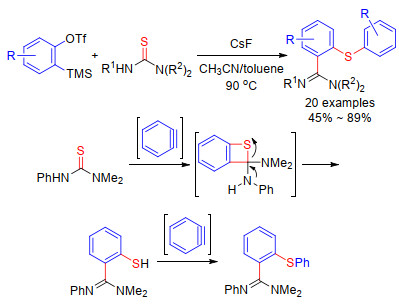
在Knochel等[65]的研究基础上, Greaney课题组[66]于2015年报道了芳炔、硫酚和O-苯甲酰基羟胺的三组分反应(Scheme 27).作者以邻碘苯基磺酸酯为芳炔前体, 在格氏试剂存在下可以原位生成芳炔.该反应中, 硫酚首先亲核进攻芳炔生成芳基氯化镁中间体.然后, 中间体在铜催化下发生亲电胺化反应, 最后得到邻位芳硫基取代的苯胺类化合物.

2016年, 胡金波课题组[67]报道了芳炔、三氟甲硫基银与2-碘苯乙炔的三组分反应(Scheme 28).该反应以稳定且易制备的三氟甲硫基银作为亲核的三氟甲硫基试剂, 以2-碘苯乙炔作为亲电的碘源.该反应一步构建C—S键和C—I键, 得到邻位三氟甲硫基取代的碘苯类化合物.此外, 反应产物和苯乙炔发生Sonogashira偶联反应后, 进一步在酸作用下发生分子内环合, 可制得重要的Yagupolskii-Umemoto-type三氟甲基化试剂.

2016年, 徐政虎课题组[68]发展了CuI催化的芳炔、硫代苯磺酸酯与端炔的三组分反应(Scheme 29).在碱存在下, CuI和端炔作用生成炔基铜中间体.然后, 炔基铜与苯炔生成芳基铜中间体.最后, 芳基铜被亲电的硫代苯磺酸酯捕捉, 得到邻位炔基取代的芳硫醚类化合物.由于活性中间体较多, 该反应体系中需加入分子筛和苄溴来抑制副产物的生成.

2019年, Kim课题组[69]报道了芳炔、仲胺与硫酰氟的三组分反应(Eq. 15).该反应中, 仲胺作为亲核试剂进攻芳炔, 生成两性离子中间体.然后, 中间体被亲电的硫酰氟淬灭, 一步构建C—S键和C—N键, 得到邻氨基芳基磺酰氟类化合物.
|
|
(15) |
2019年, Biju课题组[70]报道了苯炔、叔胺与2-苯硫基异吲哚啉-1, 3-二酮的三组分反应(Eq. 16).作者主要报道了含硒化合物与芳炔、叔胺的三组分反应, 而含硫化合物的反应仅有一例.该例反应中, 叔胺亲核进攻苯炔生成两性离子中间体.然后, 芳基负离子被亲电的苯硫基淬灭, 同时氮原子上的一个甲基被反应体系中的负离子攫取, 最后以中等产率得到邻氨基取代的苯硫醚.
|
|
(16) |
2007年, Larock课题组[71]报道了邻巯基苯甲酸甲酯与芳炔的环合反应(Eq. 17).该反应中, 硫原子首先亲核进攻芳炔, 产生的芳基负离子进一步分子内亲核进攻酯羰基, 最后得到噻吨酮类化合物.后来, Greaney课题组[72]报道了类似的环合反应.
|
|
(17) |
2015年, 李杨课题组[73]发展了硫代苯甲酰胺与芳炔的反应(Scheme 30).该反应的芳炔前体为多米诺芳炔试剂(TPBT), 硫原子首先亲核进攻芳炔, 生成的中间体再脱去一个OTf后产生新芳炔.然后, 氮原子分子内进攻新芳炔, 生成两性离子中间体.最后, 芳基负离子分子内亲核进攻羰基, 得到2, 4-二取代苯并噻唑.随后, Hoye课题组[74]以三炔化合物为芳炔前体对类似反应进行了研究.
2015年, Willis课题组[75]报道了1, 2, 5-噻二唑类化合物与芳炔的反应(Eq. 18).该反应中, 噻二唑失去甲腈后与芳炔发生环加成反应, 得到苯并异噻唑类化合物.
|
|
(18) |
2015年, Werz课题组[76]报道了苯并二硫代亚胺类化合物与芳炔的反应(Scheme 31).在碱作用下, 苯并二硫代亚胺的硫原子亲核进攻芳炔, 同时失去亚胺的质子以及开环断裂C—S键, 生成芳基负离子中间体.然后, 芳基负离子对分子内另一硫原子进行亲核取代, 最后得到重要的噻蒽类化合物.
2018年, Singh课题组[77]报道了酰基二硫缩烯酮类化合物与芳炔的反应(Eq. 19).该反应经历了底物与芳炔的[3+2]环加成反应, 生成的中间体中的巯基再与另一分子芳炔发生芳基化, 得到苯并噻吩类化合物.
|
|
(19) |
2019年, 孟祥太课题组[78]报道了硫代橙酮类化合物与芳炔的反应(Eq. 20).作者运用理论计算对反应机理进行了详细研究, 指出反应首先经历了硫代橙酮与芳炔的[3+2]环加成, 再经C—S键断裂扩环得到苯并八元硫杂环类化合物.
|
|
(20) |
总结了自2004年以来芳炔参与的磷和硫芳基化反应研究进展, 主要涉及直接芳基化反应、σ-或π-键的芳炔插入反应以及三组分反应.相对于传统芳基化方法, 芳炔参与的反应具有绿色、温和和高效等特点.近年来的发展已经实现了结构多样化的芳基膦和芳基硫化合物的合成, 特别是含磷和含硫化学键的芳炔插入反应得到了详细的研究.然而, 该领域仍然面临着机遇和挑战, 例如芳炔、有机磷化合物和亲电试剂的三组分反应研究十分罕见, 运用此策略快速合成重要的邻位取代的芳基膦配体具有很大的难度.因此, 依据该有机活性中间体的反应特性, 如何设计芳炔与含磷试剂及亲电试剂的多组分反应是该领域今后重点发展的方向.此外, 目前该类方法距离实现工业化仍然还有很大一段距离.
(a) Demmer, C. S.; Krogsgaard-Larsen, N.; Bunch, L. Chem. Rev. 2011, 111, 7981.
(b) Montchamp, J.-L. Acc. Chem. Res. 2014, 47, 77.
(c) Zhang, J.; Ding, D.; Wei, Y.; Xu, H. Chem. Sci. 2016, 7, 2870.
(d) Joachimiak, Ł.; Błażewska, K. M. J. Med. Chem. 2018, 61, 8536.
(a) Otzen, T.; Wempe, E. G.; Kunz, B.; Bartels, R.; Lehwark-Yvetot, G.; Hänsel, W.; Schaper, K. J.; Seydel, J. K. J. Med. Chem. 2004, 47, 240.
(b) Zhang, K.; Xu, X.; Qing, F. Chin. J. Org. Chem. 2015, 35, 556(in Chinese).
(张柯, 徐修华, 卿凤翎, 有机化学, 2015, 35, 556.)
(c) Lee, C.-F.; Liu, Y.-C.; Badsara, S. S. Chem.-Asian J. 2014, 9, 706.
(d) Chauhan, P.; Mahajan, S.; Enders, D. Chem. Rev. 2014, 114, 8807.
(a) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029.
(b) Martin, R.; Buchwald, S. L. Acc. Chem. Res. 2008, 41, 1461.
(a) Rueeger, H.; Lueoend, R.; Rogel, O.; Rondeau, J.-M.; Möbitz, H.; Machauer, R.; Jacobson, L.; Staufenbiel, M.; Desrayaud, S.; Neumann, U. J. Med. Chem. 2012, 55, 3364.
(b)Ilardi, E. A.; Vitaku, E.; Njardarson, J. T. J. Med. Chem. 2014, 57, 2832.
(a) Wang, Y.; Wu. M.; Ding, Y. Chin. J. Org. Chem. 2010, 30, 757(in Chinese).
(王勇, 吴梅, 丁贻祥, 有机化学, 2010, 30, 757.)
(b) Xu, Q.; Jia, X.; Li, X.; Sun, Q.; Zhou, Y.; Yin, S.; Han, L. Chin. J. Org. Chem. 2014, 34, 1340(in Chinese).
(徐清, 贾小娟, 李晓慧, 孙清, 周永波, 尹双凤, 韩立彪, 有机化学, 2014, 34, 1340.)
(c) Jablonkai, E.; Keglevich, G. Curr. Org. Synth. 2014, 11, 429.
(d) Jablonkai, E.; Keglevich, G. Org. Prep. Proced. Int. 2014, 46, 281.
(a) Yang, M.; Pei, J.; Yan, G.; Weng, Q. Chin. J. Org. Chem. 2013, 33, 343(in Chinese).
(杨明华, 裴吉, 严国兵, 翁秋月, 有机化学, 2013, 33, 343.)
(b) Umierski, N.; Manolikakes, G. Org. Lett. 2013, 15, 188.
(c) Johnson, M. W.; Bagley, S. W.; Mankad, N. P.; Bergman, R. G.; Mascitti, V.; Toste, F. D. Angew. Chem., Int. Ed. 2014, 53, 4404.
(d) Richards-Taylor, C. S.; Blakemore, D. C.; Willis, M. C. Chem. Sci. 2014, 52, 12679.
(a) Wenk, H. H.; Winkler, M.; Sander, W. Angew. Chem., Int. Ed. 2003, 42, 502.
(b) Okuma, K. Heterocycles 2012, 85, 515.
(c) Bhojgude, S. S.; Biju, A. T. Angew. Chem., Int. Ed. 2012, 51, 1520.
(d) Wu, C.; Shi, F. Asian J. Org. Chem. 2013, 2, 116.
(e) Modha, S. G.; Mehta, V. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2013, 42, 5042.
Himeshima, Y.; Sonoda, T.; Kobayashi, H. Chem. Lett. 1983, 12, 1211.
Matsuzawa, T.; Yoshida, S.; Hosoya, T. Tetrahedron Lett. 2018, 59, 4197.
(a) Chen, L.; Zhang, C.; Wen, C.; Zhang, K.; Liu, W.; Chen, Q. Catal. Commun. 2015, 65, 81.
(b) Chen, Q.; Zhang, C.; Chen, L.; Wen, C.; Du, Z.; Chen, H.; Zhang, K. Tetrahedron Lett. 2015, 56, 2094.
(c) Wen, C.; Chen, Q.; He, Z.; Yan, X.; Zhang, C.; Du, Z.; Zhang, K. Tetrahedron Lett. 2015, 56, 5470.
(d) Chen, Q.; Yan, X.; Du, Z.; Zhang, K.; Wen, C. J. Org. Chem. 2016, 81, 276.
(e) Chen, Q.; Yan, X.; Wen, C.; Zeng, J.; Huang, Y.; Liu, X.; Zhang, K. J. Org. Chem. 2016, 81, 9476.
Rémond, E.; Tessier, A.; Leroux, F. R.; Bayardon, J.; Jugé, S. Org. Lett. 2010, 12, 1568.
Yoshida, S.; Uchida, K.; Igawa, K.; Tomooka, K.; Hosoya, T. Chem. Commun. 2014, 50, 15059.
Dhokale, R. A.; Mhaske, S. B. Org. Lett. 2013, 15, 2218.
Arbuzov, B. A. Pure Appl. Chem. 1964, 9, 307.
Yoshida, S.; Hosoya, T. Chem. Lett. 2013, 42, 583.
Yang, G.; Shen, C.; Quan, M.; Zhang, W. Tetrahedron 2016, 72, 333.
Yoshida, H.; Watanabe, M.; Ohshita, J.; Kunai, A. Chem. Lett. 2005, 34, 1538.
Alajarin, M.; Lopez-Leonardo, C.; Raja, R.; Orenes, R. Org. Lett. 2011, 13, 5668.
Lopez-Leonardo, C.; Raja, R.; López-Ortiz, F.; del Águila-Sánchez, M. Á.; Alajarin, M. Eur. J. Org. Chem. 2014, 1084.
Shen, C.; Yang, G.; Zhang, W. Org. Lett. 2013, 15, 5722.
Qi, N.; Zhang, N.; Allu, S. R.; Gao, J.; Guo, J.; He, Y. Org. Lett. 2016, 18, 6204.
Neog, K.; Dutta, D.; Das, B.; Gogoi, P. Org. Biomol. Chem. 2019, 17, 6450.
Okugawa, Y.; Hayashi, Y.; Kawauchi, S.; Hirano, K.; Miura, M. Org. Lett. 2018, 20, 3670.
Bhunia, A.; Kaicharla, T.; Porwal, D.; Gonnade, R. G.; Biju, A. T. Chem. Commun. 2014, 50, 11389.
Bhunia, A.; Roy, T.; Gonnade, R. G.; Biju, A. T. Org. Lett. 2014, 16, 5132.
Fujimoto, H.; Kusano, M.; Kodama, T.; Tobisu, M. Org. Lett. 2020, 22, 2293.
Liu, Z.; Larock, R. C. J. Org. Chem. 2006, 71, 3198.
Yoshida, S.; Nagai, A.; Uchida, K.; Hosoya, T. Chem. Lett. 2017, 46, 733.
Pandya, V. G.; Mhaske, S. B. Org. Lett. 2014, 16, 3836.
Aithagani, S. K.; Yempalla, K. R.; Munagala, G.; Vishwakarma, R. A.; Singh, P. P. RSC Adv. 2014, 4, 50208.
Sumii, Y.; Sugita, Y.; Tokunaga, E.; Shibata, N. ChemistryOpen 2018, 7, 204.
Zhang, L.; Li, X.; Sun, Y.; Zhao, W.; Luo, F.; Huang, X.; Lin, L.; Yang, Y.; Peng, B. Org. Biomol. Chem. 2017, 15, 7181.
Sun, C.-H.; Lu, Y.; Zhang, Q.; Lu, R.; Bao, L.-Q.; Shen, M.-H.; Xu, H.-D. Org. Biomol. Chem. 2017, 15, 4058.
Xu, X.-B.; Lin, Z.-H.; Liu, Y.; Guo, J.; He, Y. Org. Biomol. Chem. 2017, 15, 2716.
Thangaraj, M.; Gaykar, R. N.; Roy, T.; Biju, A. T. J. Org. Chem. 2017, 82, 4470.
Tan, J.; Zheng, T.; Xu, K.; Liu, C. Org. Biomol. Chem. 2017, 15, 4946.
Zheng, T.; Tan, J.; Fan, R.; Su, S.; Liu, B.; Tan, C.; Xu, K. Chem. Commun. 2018, 54, 1303.
Fan, R.; Liu, B.; Zheng, T.; Xu, K.; Tan, C.; Zeng, T.; Su, S.; Tan, J. Chem. Commun. 2018, 54, 7081.
Jian, H.; Wang, Q.; Wang, W.-H.; Li, Z.-J.; Gu, C.-Z.; Dai, B.; He, L. Tetrahedron 2018, 74, 2876.
Chen, J.; Palani, V.; Hoye, T. R. J. Am. Chem. Soc. 2016, 138, 4318.
Singh, P.; Cairns, A. G.; Adolfsson, D. E.; Ådén, J.; Sauer, U. H.; Almqvist, F. Org. Lett. 2019, 21, 6946.
Biswas, K. Greaney, M. F. Org. Lett. 2011, 13, 4946.
Pawliczek, M.; Garve, L. K. B.; Werz, D. B. Org. Lett. 2015, 17, 1716.
Li, Y.; Mük-Lichtenfeld, C.; Studer, A. Angew. Chem., Int. Ed. 2016, 55, 14435.
Zhao, X.; Huang, Y.; Qing, F.-L.; Xu, X.-H. RSC Adv. 2017, 7, 47.
Ahire, M. M.; Thoke, M. B.; Mhaske, S. B. Org. Lett. 2018, 20, 848.
李志娟, 翦辉, 王伟华, 王强, 何林, 有机化学, 2018, 38, 2045. doi: 10.6023/cjoc201802003Li, Z.; Jian, H.; Wang, W.; Wang, Q.; He, L. Chin. J. Org. Chem. 2018, 38, 2045(in Chinese). doi: 10.6023/cjoc201802003
Liu, Z.; Larock, R. C. J. Am. Chem. Soc. 2005, 127, 13112.
Qiu, D.; He, J.; Yue, X.; Shi, J.; Li, Y. Org. Lett. 2016, 18, 3130.
Gaykar, R. N.; Bhattacharjee, S.; Biju, A. T. Org. Lett. 2019, 21, 737.
Yoshida, S.; Yano, T.; Misawa, Y.; Sugimura, Y.; Igawa, K.; Shimizu, S.; Tomooka, K.; Hosoya, T. J. Am. Chem. Soc. 2015, 137, 14071.
Matsuzawa, T.; Uchida, K.; Yoshida, S.; Hosoya, T. Chem. Lett. 2018, 47, 825.
Yoshida, S.; Nakajima, H.; Uchida, K.; Yano, T.; Kondo, M.; Matsushita, T.; Hosoya, T. Chem. Lett. 2017, 46, 77.
(a) Li, H.-Y.; Xing, L.-J.; Lou, M.-M.; Wang, H.; Liu, R.-H.; Wang, B. Org. Lett. 2015, 17, 1098.
(b) Lou, M.-M.; Wang, H.; Song, L.; Liu, H.-Y.; Li, Z.-Q.; Guo, X.-S.; Zhang, F.-G.; Wang, B. J. Org. Chem. 2016, 81, 5915.
Li, Y.; Studer, A. Org. Lett. 2017, 19, 666.
Li, X.; Sun, Y.; Huang, X.; Zhang, L.; Kong, L.; Peng, B. Org. Lett. 2017, 19, 838.
Matsuzawa, T.; Uchida, K.; Yoshida, S.; Hosoya, T. Org. Lett. 2017, 19, 5521.
Toledo, F. T.; Marques, H.; Comasseto, J. V.; Raminelli, C. Tetrahedron Lett. 2007, 48, 8125.
Mesgar, M.; Daugulis, O. Org. Lett. 2017, 19, 4247.
Yoshida, H.; Terayama, T.; Ohshita, J.; Kunai, A. Chem. Commun. 2004, 1980.
Chen, J.; Murafuji, T. Organometallics 2011, 30, 4532.
Liu, F.-L.; Chen, J.-R.; Zou, Y.-Q.; Wei, Q.; Xiao, W.-J. Org. Lett. 2014, 16, 3768.
Hazarika, H.; Neog, K.; Sharma, A.; Das, B.; Gogoi, P. J. Org. Chem. 2019, 84, 5846.
Li, Y.; Qiu, D.; Gu, R.; Wang, J.; Shi, J.; Li, Y. J. Am. Chem. Soc. 2016, 138, 10814.
Lin, W.; Sapountzis, I.; Knochel, P. Angew. Chem., Int. Ed. 2005, 44, 4258.
García-López, J.-A.; Çetin, M.; Greaney, M. F. Angew. Chem., Int. Ed. 2015, 54, 2156.
Zeng, Y.; Hu, J. Org. Lett. 2016, 18, 856.
Peng, X.; Ma, C.; Tung, C.-H.; Xu, Z. Org. Lett. 2016, 18, 4154.
Kwon, J.; Kim, B. M. Org. Lett. 2019, 21, 428.
Gaykar, R. N.; Guin, A.; Bhattacharjee, S.; Biju, A. T. Org. Lett. 2019, 21, 9613.
Zhao, J.; Larock, R. C. J. Org. Chem. 2007, 72, 583.
Hall, C.; Henderson, J. L.; Ernouf, G.; Greaney, M. F. Chem. Commun. 2013, 49, 7602.
Shi, J.; Qiu, D.; Wang, J.; Xu, H.; Li, Y. J. Am. Chem. Soc. 2015, 137, 5670.
Palani, V.; Chen, J.; Hoye, T. R. Org. Lett. 2016, 18, 6312.
Chen, Y.; Willis, M. C. Org. Lett. 2015, 17, 4786.
Pawliczek, M.; Garve, L. K. B.; Werz, D. B. Chem. Commun. 2015, 51, 9165.
Garg, P.; Singh, A. Org. Lett. 2018, 20, 1320.
Ding, W.; Yu, A.; Zhang, L.; Meng, X. Org. Lett. 2019, 21, 9014.

图式 4 芳炔参与亚磷酸三乙酯及其衍生物的直接芳基化
Scheme 4 Direct P-arylation of triethyl phosphite and its derivatives with arynes

图式 5 芳炔参与亚磷酸二酯和仲膦氧化物的直接芳基化
Scheme 5 Direct P-arylation of dialkyl phosphites and secondary phosphine oxides with arynes

图式 11 苯炔、三价磷化合物和二氧化碳的三组分反应
Scheme 11 Three component reactions of benzyne, P(Ⅲ) com- pounds and CO2

图式 12 芳炔、叔膦和醛的三组分反应
Scheme 12 Three component reactions of arynes, tertiary phos- phines and aldehydes

图式 22 乙烯基亚砜S=O键的芳炔插入反应
Scheme 22 Insertion of arynes into S=O bonds of vinyl sulfoxides

图式 23 二芳基亚砜S=O键的芳炔插入反应
Scheme 23 Insertion of arynes into S=O bonds of diaryl sulfoxides

图式 25 芳炔、二甲亚砜和α-溴代羰基化合物的三组分反应
Scheme 25 Three component reactions of arynes, dimethyl sul- foxide and α-bromo carbonyl compounds

图式 26 芳炔、烯丙基亚砜和溴代烷烃的三组分反应
Scheme 26 Three component reactions of arynes, allyl sulfoxide and bromoalkanes

图式 27 芳炔、硫酚和O-苯甲酰基羟胺的三组分反应
Scheme 27 Three component reactions of arynes, thiphenols and O-benzoylhydroxylamines

图式 28 芳炔、三氟甲硫基银和2-碘苯乙炔的三组分反应
Scheme 28 Three component reactions of arynes, trifluoromethylthiosilver and 2-iodophenylacetylene

图式 29 铜催化芳炔、硫代磺酸酯和端炔的三组分反应
Scheme 29 Copper-catalyzed three component reactions of arynes, thiosulfonates and terminal alkynes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:






































 下载:
下载:



















 下载:
下载: