
图式 1.
烷基乙烯基碲化物的制备
Scheme 1.
Preparation of alkyl vinyl telluride

碲元素是在1782年由特兰西瓦尼亚化学家弗朗茨-约瑟夫·米勒·冯·赖兴施泰因在研究含金矿石时发现.虽然碲元素被发现的时间较早, 且在地壳中拥有相对来说较丰富的含量(约百万分之0.027, 与银和金等元素相当), 但它长期以来还是一种为人们所不熟知的元素, 在日常生活中的作用有限[1].近年来, 由于人们对碲元素的关注度越来越高.其在材料、生物和医药等方面的应用也越来越广泛.例如, 二维碲纳米片可以作为一种新型的光学材料, 被应用于非线性光学器件的制备.经碲纳米棒功能化处理后的钛涂层, 具有更强的抗菌性能.此外, 碲纳米线还可以作为无机纳米前药, 与细胞中的H2O2反应, 生成
碲单质作为最简单易得的碲源, 本身可以直接参与有机化学反应.与一些较活泼金属(如镁、锌)相似, 碲可与卤化物反应. 1988年, Amosova等[8]开发了利用强碱性还原体系KOH-SnC12-H2O制备烷基乙烯基碲化物2的方法.在该方法中, 碲单质、乙炔以及卤化物1直接反应, 直接合成烯基碲醚, 从而实现了将碲元素引入有机分子的合成目的(Scheme 1).该方法以水为溶剂, 在105~115 ℃下反应5 h, 即可以20%~70%的收率使底物转化为目标产物.与此同时, 还会产生少量的二乙烯基碲化物3以及二烷基碲化物4.该方法中各产物的收率与比例关系明显受到初始卤化物的种类与用量的影响, 其中, 伯烷基卤化物与仲烷基卤化物反应效果明显, 而叔烷基乙烯基碲化物却难以获得, 主要是由于叔烷基卤化物易在强碱性条件下脱去卤化氢而失去活性所导致.同时, 该方法的适用性按Br>I>Cl的顺序逐渐下降.随着卤化物用量的增加, 二乙烯基卤化物的产率也会随之上升, 几乎与烷基乙烯基碲化物产率一致.虽然该方法具有不少优点, 但过量KOH的使用以及Sn金属的残留都会对环境造成一定的危害.此外, 使用大量SnCl2以及繁琐的反应操作都使得该方法在大规模合成中的应用受到限制.
碲单质还可以与金属有机化合物反应. 2000年, Tanaka等[9]以有机膦作为催化剂催化碲单质插入Sn—Sn和Pb—Pb键, 从而构建新型含碲有机金属化合物.有机锡化合物5与碲在温度为80 ℃, 苯为溶剂的情况下反应, 以较好的收率(34%~100%)得到含碲有机锡化合物6 (Scheme 2, a).该方法不仅适用于芳香型有机锡化合物, 对烷基类有机锡化合物也有着较好的效果.在该反应中, 有机膦种类对产物的收率有着较大的影响.例如:三苯基膦催化三甲基二锡醚与碲单质反应时, 只能以45%的收率得到相应的含碲化合物, 而同等条件下三正丁基膦则能使产物的收率达到100% (Scheme 2, b).而且该方法相较于直接使用有机碲膦化合物(R'3P=Te)的方法而言, 效果相差不多.例如:三甲基二锡醚与三叔丁基碲膦反应1 h, 产物收率可以达到100%.与此同时, 使用催化量的三叔丁基膦催化三甲基二锡醚与碲单质反应3 h, 也能以100%收率得到产物.但该方法避免了使用有机碲膦化合物而产生等物质的量的R'3P的问题, 从而减轻了底物纯化的困难.不仅如此, 该方法还同时解决了有机碲膦化合物由于热不稳定性及高空气敏感性而难以制备的问题, 并可拓展到利用有机铅化合物7来制备含碲有机铅化合物8的过程中(Scheme 2, c).然而, 使用高毒性的苯作为反应溶剂, 使得该方法较易对环境造成污染, 同时也增加了后处理的成本.

从机理上来看, 有机膦化合物与碲单质发生加成反应, 得到相应的有机碲膦化合物9, 接着再与有机锡化合物反应, 生成含碲有机锡化合物10, 同时生成的有机膦化合物进入另一个催化循环体系继续参与反应.该方法实质上还是以有机碲膦化合物作为碲元素的转移基质, 但只需催化量的有机膦化合物就可以完成反应.同时也为后续的底物纯化带来了较大的便利(Scheme 3).

有机非金属盐也可与碲单质反应, 合成含碲化合物. 2019年, 谭启涛等[10]利用碲粉与易得的二芳基碘鎓盐11合成了嵌入碲的大环芳香族化合物12 (Scheme 4).该方法使用2-甲基吡啶作为添加剂, 二甲基亚砜(DMSO)作为溶剂来进行反应, 能够以较好的收率(35%~81%)得到目标产物.其中, 单取代的二芳基碘鎓盐和3, 7-二取代的二芳基碘鎓盐, 无论取代基团的电子效应如何, 其都能以良好的效率得到目标化合物.与此同时, 合成的含碲芳香族化合物表现出良好的空气/水分稳定性和高热稳定性, 故无需采取特殊的保护措施, 即可通过柱层析分离得到目标产物.令人惊讶的是, 该方法对含喹啉结构的二芳基碘鎓盐也可以起到相对良好的效果.而且在含有生物活性的类黄酮中也能成功地引入碲(图 1), 这对于研究含碲化合物潜在的生物性能起到了一定的作用.该方法还可以拓展到许多其他含碲共轭体系的杂环合成中.不仅如此, 合成的固态物质还具有有趣的室温可控磷光效应, 这也为新型室温可控磷光效应材料的研究提供了参考.


碲的金属性使其能够如金属一样催化有机化学反应.与一些贵金属催化剂(如钯、钌等)相似, 碲可催化插羰反应. Sonoda等[5b]发现, 在一氧化碳氛围中, 碲单质在催化胺类化合物13转化为尿素衍生物14以及甲酰胺类化合物15方面能够起到一定的作用(Scheme 5).虽然两种产物的收率较低, 但反应体系干净, 副产物仅为氢气.而氢气能够作为清洁能源被广泛应用, 所以该反应还是比较环保的.此外, 该反应的收率受反应时间的影响比较明显. 100 mmol的正叔丁基胺在1 mol%碲催化下反应10 h, 能够得到7.8 mol的N-二正丁基尿素, 7.2 mmol的氢气以及17.3 mmol的N-正丁基甲酰胺.而延长反应时间至72 h, 三种产物的产量均能够达到原来的3倍左右.与此同时, 提升反应温度以及增加一氧化碳压力都能使得三种产物的收率升高, 其中较高的反应温度以及较低的一氧化碳压力对于产物中尿素衍生物含量占比的提升有益.而选用硒来代替碲催化反应时, 尿素衍生物的产率就会明显下降, 可见在该方法中碲拥有比硒更好的催化活性.该方法对于脂肪族胺类化合物和芳香族胺类化合物均适用, 且两者的反应效果相差不大.
令人惊喜的是, 当向反应体系中添加10 mol%的硝基苯时, 能够显著地抑制氢气以及甲酰胺类化合物的生成, 同时不会对尿素的收率造成影响.从机理上可知, 碲在催化胺类化合物与一氧化碳反应后变成碲化氢16, 而此时硝基苯利用其氧化性, 将碲化氢氧化为碲单质, 自身变成苯胺(可被分离得到).而反应中使用硝基苯作为添加剂能够抑制甲酰胺类化合物的生成, 也说明了碲化氢在甲酰胺类化合物的产生过程中起到了重要的作用(Scheme 6).

一系列控制实验结果表明, 该方法中碲作为催化剂先催化胺类化合物和一氧化碳反应, 自身被还原为碲化氢16.接着H2Te脱氢分解为H2与碲单质.而碲单质作为催化剂继续参与下一个循环.另一方面, 一氧化碳与碲化氢发生插入反应得到碲代甲酸17, 碲代甲酸与胺类化合物反应得到甲酰胺类化合物与H2Te.而碲化氢则继续参与反应(Scheme 7).

与金属氧化物类似, 二氧化碲可催化各种C—H键的氧化反应, 从而实现其官能化. Bergman等[11]发现, 在含LiBr的醋酸溶液中, 二氧化碲能够将溶液中的乙酸缓慢氧化成乙酰氧基卡宾, 并与芳香族化合物18反应, 最终得到乙酰氧基甲基化产物19 (Scheme 8).该方法虽然耗时长, 产率较低(5%~30%).但反应体系干净, 副产物二芳基甲烷衍生物较少, 且没有出现溴化产物.例如:对二甲苯在该方法下能以30%的收率得到乙酸- 2, 5-二甲基苄酯, 并且没有副产物的产生.而在类似的反应环境下, 利用碲酸氧化甲苯衍生物20可以得到甲苯衍生物侧链乙酰氧基化的产物21, 且大幅度缩减了反应的时间(Scheme 9).但同时也产生了苯甲醛以及芳环溴化的副产物, 这使得反应的后处理变得更为繁琐.例如:邻二甲苯在此方法下能够以48%的收率得乙酸- 2-甲基苄酯, 同时会产生少量的二甲基苯甲醛以及2, 5-二甲基溴苯.此外, 对于苯环电子云密度较高的芳香族化合物, 芳环的溴化会成为主要反应, 例如4-甲氧基甲苯在此条件下反应时以68%的收率得到2-溴-4-甲氧基甲苯.该方法也体现了含碲化物在C—H键活化方面的有着一定的作用, 同时也展现了其优异的氧化性能, 但较高的反应温度与复杂的反应体系使得反应适用性受到影响.


一系列控制实验结果表明, TeO2先与溶液中的醋酸和溴化锂反应得到中间体22, 然后中间体22变成其共振中间体23, 中间体23在醋酸根离子的进攻下变成中间体24, 中间体24易发生解离得到二价碲化合物以及其他化合物.同时, 中间体24在醋酸根离子的作用下解离成二价碲化合物及乙酰氧基甲基卡宾.之后, 乙酰氧基甲基卡宾进攻芳香族化合物得到最终产物乙酰氧基甲基化芳香族化合物(Scheme 10).
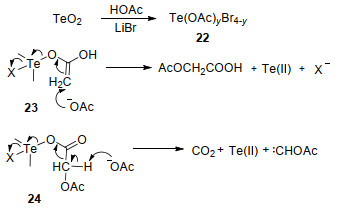
甲苯侧链乙酰氧基化反应中, Te(OH)6先将溶液中的溴离子氧化成溴单质, 而自身被还原为四价碲化合物.然后甲苯与溴单质反应得到溴代甲苯与溴化氢.其中, 溴代甲苯在四价碲化合物以及醋酸的作用下变成乙酰化甲苯与溴化氢, 溴化氢则继续参与反应循环(Scheme 11).

除氧化反应外, 碲还可以催化硫化反应. Fujiwan等[12]发现, 碲在由异氰化物25和硫单质合成异硫氰酸酯26的过程中有着较好的催化作用(Scheme 12).该反应中异氰化物与硫单质在三乙胺为添加剂, 四氢呋喃为溶剂的情况下能够以良好的收率(69%~85%)得到异硫氰酸酯.该方法不仅适用于芳香族化合物, 对于脂肪族异氰化物也有着一定的作用.例如:苄基异氰能够在该方法下以79%的收率得到苄基异硫氰酸酯, 而叔丁基异氰也能够以69%的中等收率得到叔丁基异硫氰酸酯.可见该方法适用性较广.不仅如此, 该反应中所需催化剂碲用量较少, 而且同等条件下碲的催化能力远高于硒的催化能力.例如:碲与硒用量均为0.5 mol%, 且其他条件都一样的情况下, 碲催化反应并使得产物收率达到100%所用的时间为3 h, 而相同时间内硒催化反应得到的产物收率不足40%(图 2).由于该反应的原子经济性较高, 避免了许多废弃物的产生, 所以反应的后处理也更为简便.但过量刺激性三乙胺的使用, 使得操作过程有潜在风险, 降低了反应的安全性.


与无机碲相比, 有机碲化合物结构更加复杂, 从而有着更加多样化的反应活性.有机碲参与的反应被广泛应用于含碲化合物的合成. 1989年, Comasseto课题组[13]利用含双键结构的端位炔烃27和有机碲化合物28反应得到全新的含碲烯烃化合物29和30 (Scheme 13, a).对于脂肪族炔烃和芳香族炔烃而言, 该反应的结果在产物的比例关系方面存在着一定的区别.其中, 有机碲化合物与芳香族炔烃反应, 单一地得到反马氏加成的含碲烯烃化合物29.有机碲化合物与脂肪族炔烃反应, 除反马氏加成的含碲烯烃主产物外, 还会有部分马氏加成的产物30生成.除较好的区域选择性外, 反应也具有较好的立体选择性, 所有产物中Z式构型的产物占比都超过E式构型的产物, 比例范围为2.57~8.09:1.不仅如此, 对于脂肪族和芳香族有机碲化合物而言, 该方法都具有良好的适用性.例如: (2Z)-3-甲基-2-烯-4-炔-1-戊醇与二十二烷基二碲醚反应, 能以80%的收率得到相应结构的化合物29.在此基础上, 还利用二乙烯基有机碲化合物32与苯乙炔衍生物31制备了全新的不对称二乙烯基有机碲化合物33 (Scheme 13, b).和之前端位炔烃与有机碲化合物反应的结果类似, 该方法得到的主要产物依旧是Z式构型的反马氏加成产物.此外还有少量的E式构型的反马氏加成产物.例如:苯乙炔与二乙烯基二碲醚反应, 能以76%的收率得到相应结构的化合物33.可见该方法适用性较广, 而且立体选择性良好.但2.5~5.0 equiv.炔烃的使用使得反应的原子经济性大大降低, 且易燃品NaBH4的使用, 也会导致反应的安全系数受到影响.
含碲化学键较弱, 容易发生均裂而产生碲自由基.该反应在可见光驱动下即可发生.例如, Sonoda等[14]发现, 将二苯基二碲醚35加入炔烃衍生物34中, 并在无溶剂条件下用光照射一段时间, 能够产生E式构型为主的含碲烯烃类化合物36 (Scheme 14). 此外炔烃中的R基团会对产物的收率造成较大的影响.当炔烃衍生物为脂肪族炔烃时, 由于产物在近紫外有吸收, 所以容易导致产物发生逆反应, 分解为炔烃衍生物和二苯基二碲醚.例如: 1-辛炔在波长为300 nm的光照射下, 仅有43%的核磁产率.而添加偏正光片过滤波长为300~400 nm的光之后, 产物的核磁收率为62%, 而分离收率则能够达到29%.而当炔烃上连有环己烯基等与叁键共轭的官能团时, 则能够使化合物的吸收峰偏移, 从而避免上述问题.例如丁炔二酸二乙酯和二苯基二碲醚反应, 能以90%的收率得到加成产物.并且仅需用波长为300 nm的光来照射反应体系即可.该方法中产物选择性良好, 而且反应体系干净.不仅如此, 该方法也充分体现了有机碲化合物具有光敏性的特点, 为后续研究有机碲化合物的光学性能提供了参考.
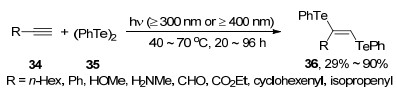
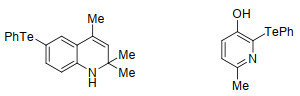
除了光照外, 一些过渡金属催化剂(如铜)也可促进碲键断裂. 2006年, Sangit等[15]在CuI/Bpy与过量镁粉的存在下, 利用微波加热使二苯基二碲醚35与卤代芳基37发生反应, 从而得到相应的单碲醚产物38 (Scheme 15).该方法大大缩减了利用卤代芳基和二苯基二碲醚来制备单碲醚的时间.除此以外, 该反应中无论是含杂环的卤代芳基还是大位阻的卤代芳基, 都能表现出良好的官能团容忍性.例如, 二苯基二碲醚与1-溴碘萘反应, 以68%的收率得到相应的单碲醚.而含有杂环的3-溴吡啶在该方法中以62%的收率得到相应的单碲醚.此外, 卤素的种类也会对产物的收率产生一定的影响.二苯基二碲醚与溴苯反应产率最高, 为65%;与碘苯反应的效果就稍差一点, 产物收率仅有60%;而与氯苯的反应效果最差, 在反应升温到200 ℃时, 产物收率依旧不太理想, 仅有40%.该反应可以用于制备不同的含碲抗氧化剂(图 3), 且对于单硫醚和单硒醚的制备也有着一定的作用.但是, 该反应也有着比较明显的缺陷, 较高的反应温度以及过量镁粉的使用都会使得该反应在应用方面受到较大的限制.

有机碲化合物的高反应活性使其在一些反应中亦可以充当催化剂. 2001年, Detty等[16]利用有机碲化合物催化烯烃的碘化(Scheme 16). 该方法利用无机碘化物作为碘源, 让底物39在磷酸盐缓冲溶液中进行反应, 以中等至良好的收率(40%~94%)得到碘代产物40.该反应通过有机碲化物对双氧水进行活化, 从而氧化无机碘化物并对底物进行碘化反应.从反应机理上可知, 首先无机碘化物与催化剂在双氧水的作用下发生加成反应, 同时催化剂上的氧原子对碲进行螯合配位, 防止另一个碘化物分子加成到碲原子上, 并最终形成中间体41(图 4).而中间体41的正电荷则激活碘配体, 从而完成对底物的亲核进攻(Scheme 17).而且使用pH为6的磷酸盐缓冲溶液作为溶剂也增加了酸性底物对反应体系的耐受性.不仅如此, 若底物具有一定的水溶性, 则可在不使用有机助溶剂的情形下进行反应.例如: 4-戊烯酸钠在不添加醚作为有机助溶剂的情况下, 即可以88%的收率得到碘代产物.而且该方法与直接使用碘单质来进行碘化反应得到结果几乎一致, 其中大部分底物的收率都有略微提升.但是对于一些未被活化的底物(例如苯和甲苯)而言, 该催化剂催化的效果就显得相对较差.进一步研究表明该方法对于利用无机溴化物来对底物进行溴化的效果并不突出, 所以其适用范围存在一定的限制.但该方法依旧具有很多优点, 例如:在易于底物纯化的同时, 也尽可能地避免了有机卤化物对环境的污染.而且此反应对底物的兼容性较好, 产物收率受底物空间位阻效应的影响也较小.但是过量强氧化剂的使用也对该反应的安全性造成了不可忽视的影响, 从而使得该方法在应用方面受到限制.
除卤化反应外, 碲化合物还可以催化氧化反应.在这些反应中, 碲作为氧转移催化剂, 起到运载氧的效果. 2005年, Parvez等[17]利用环状亚碲酸酯和螺二氧基碲烷等有机碲化合物催化硫酚42与过氧化物43反应, 得到相应的二硫化物44 (Scheme 18).该工作利用半衰期(t1/2)(将一半硫醇氧化成二硫醚所用时间)来表征各催化剂的催化活性.其中, 环状亚碲酸酯催化性能较环状亚硒酸酯而言显得比较突出.当使用56%过氧化叔丁醇作为过氧化物模拟物时, 环状亚硒酸酯催化底物所需半衰期为90 h, 而环状亚碲酸酯催化底物所需半衰期仅有0.05 h, 可以明显地看出环状亚碲酸酯的催化性能高于环状亚硒酸酯的催化性能.而且环状亚硒酸酯对于过氧化物种类以及浓度依赖过高.当过氧化叔丁醇用量为90%时, 环状亚硒酸酯催化底物所需半衰期为2.5 h, 而过氧化叔丁醇用量下降至56%时, 所需半衰期骤增到90 h.除了环状亚碲酸酯以外, 螺二氧基碲烷也展现出很好的催化能力, 其催化效果与环状亚碲酸酯的催化效果相近.而且此类化合物作为硒代谷胱甘肽过氧化物酶的模拟物, 还能够有效地减少有害的过氧化物来保护细胞免受氧化应激.所以有机碲化合物除了有较好的催化活性之外, 还有一定的生物活性.

肟类是易得的有机化合物, 而以肟为原料的反应, 有着很好的应用价值[18].其中, 脱肟反应被广泛应用于天然产物合成与药物合成中[19].受上述碲催化氧化反应的启发, 我们[20]最近以二苯基二碲醚作为催化剂来催化氧化脱肟反应(Scheme 19).该反应可在无溶剂条件下进行, 并使用氧气作为氧化剂.只需要使用2.5 mol%二苯基二碲醚作催化剂, 即可使得大部分肟(45)在60 ℃下以54%~94%的收率转化为酮(46).当芳环上连有给电子基团时, 产物收率会比连有吸电子基团的产物收率高.而且底物的位阻越大, 对底物收率造成的影响也就越大, 需要更高的温度才能完成反应.例如:苯丁酮肟在参与反应时, 在60 ℃下就能以54%的收率得到苯丁酮, 而苯戊酮肟在60 ℃时却难以发生反应, 需要将反应温度升至80 ℃才能得到苯戊酮.不仅如此, 该方法对脂肪族酮肟以及杂环酮肟也适用.
通过机理研究, 可推测出如下反应机理: (PhTe)2 (35)首先被氧化成次碲酸酐即PhTeOTePh (47), 随后裂解成中间体48与49, 中间体48可以被氧化成中间体49, 而中间体49会进一步被氧气氧化成中间体50, 中间体50与肟45加成得到中间体51, 这一步反应受底物的空间位阻影响较大, 有时需要更高的反应温度才能实现, 中间体51可分解为主要产物酮、副产物HNO以及中间体49, 而其中产生的中间体49则作为催化物质重新启动催化循环圈A.中间体49也可能直接与肟45加成得到中间体52, 并分解为副产物HNO、主要产物酮以及中间体48.中间体48会作为另一个催化物种重新启动催化循环圈B.该方法经历了一个自由基反应的历程, 从而与主要以离子型机理进行的有机硒催化氧化脱肟反应大不相同(Scheme 20)[21].

由上述可知, 碲元素独特的反应活性源自于其准金属特性.一方面, 碲作为非金属元素, 易于形成各种共价键, 从而便于与有机化合物反应; 另一方面, 碲元素的金属性使其能够表现出一些金属特性, 例如, 易与卤代烃反应生成含碲试剂, 易与一氧化碳发生作用从而可催化插羰反应, 易被氧化还原从而可催化氧化反应.与同族的硒元素相比, 含碲化学键更弱, 从而易发生均裂反应, 产生自由基.因此, 碲催化反应可通过自由基机理进行, 而与硒催化反应不同.与无机碲相比, 有机碲试剂结构更加复杂, 也可通过引入不同取代基来调控其反应性, 从而使其应用范围更加广阔.碲是较少见的准金属元素, 但其参与或催化的部分有机反应, 已经表现出一些绿色化学的特征, 符合有机合成化学的发展趋势[22].碲虽然比硒贵(约8元/g), 但与传统的贵金属如铂[23](约180元/g)、钯[24](约500元/g)、钌[25](约70元/g)、金[26](约360元/g)等相比, 价格更加便宜.因此, 碲催化同样有望被应用于工业合成领域.目前, 人们对于这方面的研究较少, 而该领域有待进一步深入研究.
(a) Gelbstein, Y.; Rosenberg, Y.; Sadia, Y.; Dariel, M. P. J. Phys. Chem. C 2010, 114, 13126.
(b) Cao, W.; Gu, Y.; Meineck, M.; Li, T.; Xu, H. J. Am. Chem. Soc. 2014, 136, 5132.
(c) Kaur, M.; Rob, A.; Williams, J. C.; Huang, Z. ACS Sym. Ser. 2013, 1152, 89.
(a) Wu, L.; Huang, W.; Wang, Y.; Zhao, J.; Ma, D.; Xiang, Y.; Li, J.; Ponraj, J.; Dhanabalan, S.; Zhang, H. Adv. Funct. Mater. 2019, 29, 1806346.
(b) Park, D.; Gautam, M.; Park, M.; Hwang, J.; Yong, C.; Kim, J.; Byeon, J. Environ. Sci.-Nano. 2019, 6, 2074.
(c) David, M.; María, U.; William, T.; Marcial, F.; Ada, V.; Iván, M.; Yves, H.; Thomas, J.; José, M. Nanomed-Nanotechnol 2019, 17, 36.
(d) Wu, Y.; Guo, T.; Qiu, Y.; Liu, Y.; Yao, Y.; Lian, W.; Lin, L.; Song, J.; Yang, H. Chem. Sci. 2019, 10, 7068.
(a) Cao, H.; Yang, Y.; Chen, X.; Liu, J.; Chen, C.; Yuan, S.; Yu, L. Chin. Chem. Lett. 2020, 31, 1887.
(b) Liu, Y.; Ling, H.; Chen, C.; Xu, Q.; Yu, L.; Jiang, X. Synlett 2019, 30, 1698.
(c) Cao, H.; Liu, M.; Qian, R.; Zhang, X.; Yu, L. Appl. Organomet. Chem. 2019, 33, e4599.
(d) Jing, X.; Chen, C.; Deng, X.; Zhang, X.; Wei, D.; Yu, L. Appl. Organomet. Chem. 2018, 32, e4332.
(e) Liu, M.; Li, Y.; Yu, L.; Xu, Q.; Jiang, X. Sci. China: Chem. 2018, 61, 294.
Chen, C.; Cao, Y.; Wu, X.; Cai, Y.; Liu, J.; Xu, L.; Ding, K.; Yu, L. Chin. Chem. Lett. 2020, 31, 1078. doi: 10.1016/j.cclet.2019.12.019
(a) Engman, L.; Cava, M. P. Tetrahedron Lett. 1981, 22, 5251.
(b) Kambe, N.; Hondo, K.; Ishii, H.; Sonoda, N. Bull. Chem. Soc. Jpn. 1981, 54, 1460.
(c) Engman, L.; Cava, M. P. J. Org. Chem. 1982, 47, 3946.
(a) Kumar, S.; Engman, L.; Valgimigli, L.; Amorati, R.; Fumo, M. G.; Pedulli, G. F. J. Org. Chem. 2007, 72, 6046.
(b) Kumar, S.; Johansson, H.; Kanda, T.; Engman, L.; Muller, T.; Bergenudd, H.; Jonsson, M.; Pedulli, G. F.; Amorati, R.; Valgimigli, L. J. Org. Chem. 2010, 75, 716.
(a) Liu, C.; Mao, J.; Zhang, X.; Yu, L. Catal. Commun. 2020, 133, 105828.
(b) Yu, L.; Cao, H.; Zhang, X.; Chen, Y.; Yu, L. Sustainable Energy Fuels 2020, 4, 730.
(c) Gao, G.; Han, J.; Yu, L.; Xu, Q. Synlett 2019, 30, 1703.
(d) Yang, Y.; Fan, X.; Cao, H.; Chu, S.; Zhang, X.; Xu, Q.; Yu, L. Catal. Sci. Technol. 2018, 8, 5017.
(e) Wang, T.; Jing, X.; Chen, C.; Yu, L. J. Org. Chem. 2017, 82, 9342.
Trofimov, B. A.; Gusarova, N. K.; Tatarinova, A. A.; Potapov, V. A.; Sinegovskaya, L. M.; Amosova, S. V.; Voronkov, M. G. Tetrahedron 1988, 44, 6739. doi: 10.1016/S0040-4020(01)90114-0
Han, L.; Mirzaei, F.; Tanaka, M. Organometallics 2000, 19, 722. doi: 10.1021/om990728s
Jiang, M.; Guo, J.; Liu, B.; Tan, Q.; Xu, B. Org. Lett. 2019, 21, 8328. doi: 10.1021/acs.orglett.9b03106
Bergman, J.; Engman, L. J. Org. Chem. 1982, 47, 5191. doi: 10.1021/jo00147a031
Fujiwara, S.; Tsutomu, S. Tetrahedron Lett. 1992, 33, 7021. doi: 10.1016/S0040-4039(00)60922-X
Barros, S. M.; Dabdoub, M. J.; Dabdoub, V. M. B.; Comasseto, J. V. Organometallics 1989, 8, 1661. doi: 10.1021/om00109a014
Ogawa, A.; Yokoyama, K.; Obayashi, R.; Han, L.; Kambe, N.; Sonoda, N. Tetrahedron 1993, 49, 1177. doi: 10.1016/S0040-4020(01)85809-9
Kumar, S.; Engman, L. J. Org. Chem. 2006, 71, 5400. doi: 10.1021/jo060690a
Higgs, D. E.; Nelen, M. I.; Detty, M. R. Org. Lett. 2001, 3, 349. doi: 10.1021/ol0068142
Back, T. G.; Kuzma, D.; Parvez, M. J. Org. Chem. 2005, 70, 9230. doi: 10.1021/jo0512711
(a) Yu, L.; Li, H.; Zhang, X.; Ye, J.; Liu, J.; Xu, Q.; Lautens, M. Org. Lett. 2014, 16, 1346.
(b) Zhang, X.; Sun, J.; Ding, Y.; Yu, L. Org. Lett. 2015, 17, 5840.
(c) Chen, R.; Qi, J.; Mao, Z.; Cui, S. Org. Biomol. Chem. 2016, 14, 6201.
(d) Wang, Y.; Wu, Z.; Li, Q.; Zhu, B.; Yu, L. Catal. Sci. Technol. 2017, 7, 3747.
(e) Jing, X.; Wang, T.; Ding, Y.; Yu, L. Appl. Catal. A-Gen. 2017, 541, 107.
(f) Zhang, D.; Huang, Y.; Zhang, E.; Yi, R.; Chen, C.; Yu, L.; Xu, Q. Adv. Synth. Catal. 2018, 360, 784.
(g) Chu, S.; Cao, H.; Chen, T.; Shi, Y.; Yu, L. Catal. Commun. 2019, 129, 105730.
(a) Corey, E. J.; Hopkins, P. B.; Kim, S.; Yoo, S. E.; Nambiar, K. P.; Falck, J. R. J. Am. Chem. Soc. 1979, 101, 7131.
(b) Grirrane, A.; Corma, A.; Garcia, H. J. Catal. 2009, 268, 350.
(c) Royals, E. E.; Horne Jr., S. E. J. Am. Chem. Soc. 1951, 73, 5856.
(d) Zheng, Y.; Wu, A.; Ke, Y.; Cao, H.; Yu, L. Chin. Chem. Lett. 2019, 30, 937.
Deng, X.; Chao, H.; Chen, C.; Zhou, H.; Yu, L. Sci. Bull. 2019, 64, 1280. doi: 10.1016/j.scib.2019.07.007
(a) Jing, X.; Yuan, D.; Yu, L. Adv. Synth. Catal. 2017, 359, 1194.
(b) Chen, C.; Zhang, X.; Cao, H.; Wang, F.; Yu, L.; Xu, Q. Adv. Synth. Catal. 2019, 361, 603.
(a) Ou, W.; Zou, R.; Han, M.; Yu, L.; Su, C. Chin. Chem. Lett. 2020, 31, 1899.
(b) Cao, H.; Qian, R.; Yu, L. Catal. Sci. Technol. 2020, 10, 3113.
(c) Bao, W.; He, M.; Wang, J.; Peng, X.; Sung, M.; Tang, Z.; Jiang, S.; Cao, Z.; He, W. J. Org. Chem. 2019, 84, 6065.
(d) Cao, Z.; Zhu, Q.; Lin, Y.; He, W. Chin. Chem. Lett. 2019, 30, 2132.
(e) Zhang, H.; Han, M.; Yang, C.; Yu, L.; Xu, Q. Chin. Chem. Lett. 2019, 30, 263.
(f) Yu, L.; Qian, R.; Deng, X.; Wang, F.; Xu, Q. Sci. Bull. 2018, 63, 1010.
(g) Cao, H.; Zhu, B.; Yang, Y.; Xu, L.; Yu, L.; Xu, Q. Chin. J. Catal. 2018, 39, 899.
(h) Fan, X.; Yi, R.; Wang, F.; Zhang, X.; Xu, Q.; Yu, L. Chin. J. Org. Chem. 2018, 38, 2736(in Chinese).
(范昕, 易容, 王芳, 张旭, 徐清, 俞磊, 有机化学, 2018, 38, 2736.)
(i) Yu, L.; Wang, J.; Cao, H.; Ding, K.; Xu, Q. Chin. J. Org. Chem. 2014, 34, 1986(in Chinese).
(俞磊, 王俊, 曹洪恩, 丁克鸿, 徐清, 有机化学, 2014, 34, 1986.
(a) Tong, Q.; Zhao, S.; Liu, Y.; Xu, B.; Yu, L.; Fan, Y. Appl. Organomet. Chem. 2020, 34, e5380.
(b) Zhao, S.; Xu, B.; Yu, L.; Fan, Y. Chin. Chem. Lett. 2018, 29, 884.
(c) Zhao, S.; Xu, B.; Yu, L.; Fan, Y. Chin. Chem. Lett. 2018, 29, 475.
(d) Wang, F.; Xu, L.; Sun, C.; Yu, L.; Xu, Q. Appl. Organomet. Chem. 2018, 32, e4505.
(a) Zhu, Z.; Wang, W.; Zeng, L.; Zhang, F.; Liu, J. Catal. Commun. 2020, 142, 106031.
(b) Yang, Y.; Xu, B.; He, J.; Shi, J.; Yu, L.; Fan, Y. Appl. Organomet. Chem. 2019, 33, e5204.
(c) Yang, Y.; Li, M.; Cao, H.; Zhang, X.; Yu, L. Mol. Catal. 2019, 474, 110450.
(d) Fan, X.; Yao, Y.; Xu, Y.; Yu, L.; Qiu, C. ChemCatChem 2019, 11, 2596.
(e) Wang, Q.; Jing, X.; Han, J.; Yu, L.; Xu, Q. Mater. Lett. 2018, 215, 65.
(f) Zhang, D.; Wei, Z.; Yu, L. Sci. Bull. 2017, 62, 1325.
(a) Liu, Z.; Zha, M.; Wang, Q.; Hu, G.; Feng, L. Chem. Commun. 2020, 56, 2352.
(b) Zhang, D.; Deng, X.; Zhang, Q.; Han, J.; Yu, L. Mater. Lett. 2019, 234, 216.
曹小卫, 陈帅, 鲍敏, 史宏灿, 李巍, 化学进展, 2018, 30, 1380. https://www.cnki.com.cn/Article/CJFDTOTAL-QHBH201805012.htmCao, X.; Chen, S.; Bao, M.; Shi, H.; Li, W. Prog. Chem. 2018, 30, 1380 (in Chinese). https://www.cnki.com.cn/Article/CJFDTOTAL-QHBH201805012.htm

图式 2 含碲有机金属化合物的制备
Scheme 2 Preparation of organometallic compounds containing tellurium

图式 3 有机膦催化的Te插入Sn—Sn键的可能机理
Scheme 3 Possible mechanism for the organophosphine-catalyzed insertion of tellurium into Sn—Sn bond

图式 4 含碲大环芳香族化合物的制备
Scheme 4 Preparation of large cyclic aromatic compounds containing tellurium

图 1 含喹啉、类黄酮结构的代表性化合物
Figure 1 Representative compounds containing quinoline and flavonoid structures

图式 6 硝基苯存在下Te催化胺与一氧化碳羰基化反应的机理
Scheme 6 Mechanism of Te-catalyzed carbonylation of amine with carbon monoxide in the presence of nitrobenzene

图式 7 碲催化胺与一氧化碳羰基化反应的可能机理
Scheme 7 Possible mechanism of Te-catalyzed carbonylation of amines with carbon monoxide

图式 8 二氧化碲催化的芳香族化合物的乙酰氧基甲基化反应
Scheme 8 Acetyloxymethylation of aromatic compounds catalyzed by tellurium dioxide

图式 9 碲酸催化的甲苯侧链的乙酰氧基化反应
Scheme 9 Acetylation of toluene side chains catalyzed by telluric acid

图式 10 芳香族化合物乙酰氧基甲基化反应的可能机理
Scheme 10 Possible mechanisms of acetyloxymethylation of aromatic compounds

图式 11 甲苯侧链乙酰氧基化反应的可能机理
Scheme 11 Possible mechanisms of acetylation of toluene side chains

图式 12 碲单质催化的异硫氰酸酯的合成
Scheme 12 Synthesis of isothiocyanates catalyzed by tellurium elements

图式 14 二苯基二碲醚与炔烃化合物的光化学反应
Scheme 14 Photochemical reaction of diphenyl ditelluride with alkynes

图式 15 卤代芳烃与二碲醚的反应
Scheme 15 Reaction of halogenated aromatic hydrocarbons with tellurium ethers

图 3 卤代芳烃与二碲醚反应制备的含碲抗氧化剂
Figure 3 Tellurium-containing antioxidant prepared by reaction of halogenated aromatic hydrocarbon and ditellurium ether

图式 18 有机碲催化硫酚与过氧化物的反应
Scheme 18 Organic tellurium catalyzed reaction of thiophenol with peroxides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:







 下载:
下载:








 下载:
下载: