
图 1.
含氟代表性药物
Figure 1.
Fluorine-containing drugs

有机小分子引入氟原子后往往表现出独特的物理、化学与生物特性, 因此含氟功能分子在医药、农药、生命科学和材料科学等领域中得到了广泛应用[1-4].尤其是在药物发展中起着重要作用, 含氟药物具有生物活性较高、稳定性较强和不易产生耐药性等优势, 因而向有机化合物分子中引入F原子或含氟砌块已经成为开发新的抗菌药物、抗肿瘤药物、抗病毒药物、消炎、中枢神经系统药物等的重要手段(图 1).因此, 发展新的含氟有机化合物合成方法也就更为迫切[5-6], 而对有机分子进行氟化和氟烷基化是合成含氟化合物的主要方法[4-5].
二氟烷基是一个重要的结构基元, 常见于医药、农药和功能材料中.尤其是在药物的设计和发现中具有重要应用和意义, 广泛地用于药物发展中.二氟甲基(CF2H)是羟甲基(CH2OH)、巯基(SH)等基团的生物电子等排体; 另外, 作为影响药物的药理活性和与靶点亲和力的重要因素, 药物分子与靶蛋白之间的氢键作用占有重要地位[7].而作为氢键供体, 二氟甲基(CF2H)相比于羟基(OH)和氨基(NH)等具有更好的亲脂性, 可以改善药物分子的脂溶性、膜透性、生物利用度及其他药代动力学特性[8].上市药物中有罗氟司特(Roflumilast)[9a]、泮托拉唑(Pantoprazole)[9b]、加雷沙星(Garenoxacin)[9c]和利奥地平(Riodipine)[9d]等(图 2), 还有一些用于儿童自闭症和抗革兰氏阴性菌感染的候选药物分子正处于临床试验阶段.发展稳定高效的二氟甲基试剂及相应二氟甲基化方法, 向药物分子中选择性地引入二氟甲基, 有助于研究药物结构与性质的相互关系, 发现新的具有特殊性质的先导化合物, 进而加快新药开发.所以, 二氟甲基的引入, 在药物设计和新药创制中占有非常重要的地位.
鉴于二氟甲基官能化的分子在创新药物发展中具有重要作用, 因此, 开发和发展二氟甲基试剂和二氟甲基化方法具有重要用途和意义.其中, 亲电型二氟甲基试剂可以通过直接亲核取代(+CF2H)、二氟卡宾(:CF2)和二氟甲基自由基(•CF2H)三种途径在分子合成的后期阶段便利地引入二氟甲基砌块, 引起了广泛的重视.早期的亲电二氟甲基试剂主要是一些作为亲电二氟卡宾源使用的试剂, 包括一些臭氧层消耗(ODS)试剂及其衍生物如HCF2Cl, CF3H, HCF2I, HCF2Br, CF2Br2, HCF2OTf, TMSCF2Br, TMSCF2Cl, FSO2CF2CO2H, FSO2CF2CO2Me, ISO2CF2CO2H, ClF2CCO2Na和ClCF2CO2Me等, 还有一些三氟甲基的金属化合物, 如Me3SnCF3、PhHgCF3, BrZnCF3和BrCdCF3也可作为二氟卡宾试剂使用.这些试剂存在诸如稳定性差、使用不便、效率低、环境污染严重、成本高、反应类型受限及官能团兼容性差底物受限等问题, 相关工作以前已有多篇文献进行了综述[10], 在本文中不再赘述.
本综述中, 我们将重点讨论2007年以来亲电型二氟甲基化试剂开发及其应用, 同时也对亲电型二氟烷基化试剂参与的亲电二氟烷基化反应进行了分析梳理.最后, 对亲电型二氟烷基化试剂及其应用作了展望.
亲电型二氟甲基试剂主要包括直接亲电型二氟甲基试剂和保护的亲电二氟甲基试剂.反应类型主要表现为亲电二氟甲基(+CF2H)直接对亲核剂的亲电反应和原位产生的二氟卡宾(:CF2)对亲核剂的二氟甲基化反应, 以及通过单电子(SET)转移产生的二氟甲基自由基(•CF2H)所发生的自由基二氟甲基反应.
近年来发展的稳定型直接亲电型二氟甲氟甲基试剂主要包括锍鎓盐型亲电二氟甲基试剂、苯基亚砜亚胺型二氟甲基试剂、硫叶立德型二氟甲基试剂和膦叶立德型二氟甲基试剂四类.
过去20多年中, 一系列亲电氟甲基化试剂被开发和合成出来, 基于这些试剂的氟甲基化新反应被不断地发展和报道出来, 尤其是亲电三氟甲基化试剂及其应用的开发得到了快速充分的发展.然而, 相比较于亲电三氟甲基试剂及其应用研究的成功, 无论是亲电的二氟甲基化试剂的开发, 还是其应用的发展都遇到了困难. Umemoto试剂[11]是非常著名的亲电型三氟甲基试剂, 阳离子锍鎓盐具有很好的电子分散性, 使得该试剂能够以固态形式稳定保存.而类似的锍鎓盐型亲电二氟甲基试剂的发展却艰难得多, 由于氟的超强电负性, 致使CF2H中的质子酸性太强, 对碱、光和热等外部条件较敏感, 致使试剂稳定性差, 二氟甲基化反应效率低下, 且可能产生背景反应.
基于三氟甲基二苯并噻吩锍鎓盐和三氟甲基二苯基锍鎓盐等亲电型三氟甲基试剂研究的工作基础[12], 2007年, Olah和Prakash等[13]报道了第一例非对称二芳基锍鎓盐型亲电型二氟甲基试剂1的合成(Scheme 1).并对其应用进行了研究, 该亲电型二氟甲基试剂能对磺酸盐的磺酸负离子发生亲电进攻得到磺酸二氟甲酯, 同时也能对三级胺、咪唑类化合物和膦中的N、P发生亲电二氟甲基化(Scheme 2), 但是试剂1即使低温冷冻保存也只是半固体状态, 其稳定性差且不易保存, 易于分解(-20 ℃低温保存三个月有13%的分解), 所以该试剂的合成收率和成本以及可适用的反应类型和效率, 与同类型的三氟甲基试剂相去甚远.

2016年, 肖吉昌小组[14]报道了一种非对称的二芳基锍鎓盐型亲电型二氟甲基试剂2对1, 3-二酮选择性O-二氟甲基化的方法, 该反应即使在没有碱参与的条件下也可低收率地得到目标产物, 是因为该试剂不稳定, 在常温下也能释放二氟卡宾参与反应, 以四氢呋喃作溶剂时会导致溶剂的聚合也证实了这一点.机理研究表明该反应中同时存在二氟甲基亲电过程和二氟卡宾过程(Scheme 3).

二氟甲基自由基反应是向有机小分子中引入二氟甲基的一种重要方法, 近年来发展迅速, 亲电型[15, 21, 35-38]和亲核型[16]的二氟甲基试剂都可在适宜的条件下作为二氟甲基自由基源发生二氟甲基自由基反应. 2017年, Koike和Akita等[15]对二芳基锍鎓盐型亲电二氟甲基试剂的结构进行了改良, 发展了一种稳定的锍鎓盐3 (Scheme 4).并将该试剂用于可见光催化的自由基二氟甲基化反应, 发展了一种在可见光照射无金属参与的条件下, 对芳基烯烃的区域选择性氨基-二氟甲基化的方法.激发态的苝能够发生单相电子转移, 二氟甲基试剂获得一个电子生成二氟甲基自由基, 随后与烯烃双键发生自由基反应后被苝自由基正离子氧化, 最后发生一步Ritter胺化得到目标产物, 利用该试剂能够从烯烃双键(C=C)一步构建C(sp3)—CF2H和C(sp3)—N键, 9, 10-双(二(对叔丁基苯基)氨基)蒽(tBu-BDA)也能作为有效的有机光催化剂实现这一反应(Scheme 4).虽然该试剂能在室温稳定保存三个月, 但是合成收率不高.

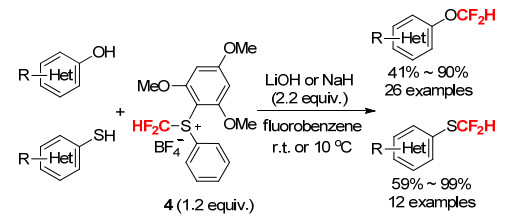
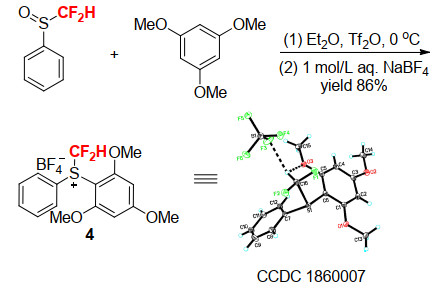
尽管上述工作推动了锍鎓盐型亲电二氟甲基试剂的发展, 但是, 试剂的稳定性、反应活性等方面存在的问题依然限制了其应用. 2018年, 我们小组在对芳基引入给电子基团的同时, 引入分子内氢键的设计, 从而达到调控+CF2H中质子的酸性和电正性的效果, 并通过X射线单晶衍射证实了二氟甲基与芳环邻位的甲氧基之间存在分子内氢键.该锍鎓盐型亲电二氟甲基试剂4能够在室温稳定存放六个月而不发生分解, 解决了试剂的稳定性问题, 而且其合成的收率非常高, 这也从侧面证明了其稳定性的大幅提高(Scheme 5).该试剂在对β-酮酸酯和丙二酸酯类化合物的亲电C-二氟甲基化上表现出高反应效率和高C/O选择性(Scheme 6)[17a].同时, 该试剂也能在有机碱的作用下对1, 3-二酮选择性地O-二氟甲基化(Scheme 7)[17b].


2019年, 我们小组[17c]对该试剂与脂肪醇的反应性进行了系统研究, 发现在温和条件下能高收率得到二氟甲基烷基醚类化合物, 控制实验和密度泛函理论(DFT)计算证实反应主要经历了一个二氟卡宾参与的五元环过渡态, 水在反应过渡态中起着重要作用(Scheme 8).

同年, 我们小组[17d]还研究了对苯酚和苯硫酚的反应, 合成了二氟甲氧基芳烃和二氟甲硫基芳烃, 反应同样经历一个二氟卡宾亲电过程, 系统的研究表明该条件下产生的二氟卡宾对O, S-亲核剂的反应活性为ArS->RS-, ArO->ROH>RO-, ArSH, RSH (Scheme 9).
此外, 使用其他原子代替锍鎓盐中二氟甲基的酸性氢原子, 是提高锍鎓盐型二氟甲基试剂稳定性的有效方法.所以, 去质子官能化的亲电二氟甲基化试剂可作为一种替代试剂.肖吉昌小组[18a]于2010年首次报道了一步合成稳定耐贮存的对称的溴二氟甲基二芳基锍鎓盐5的方法, 并发现该试剂能对C亲核剂如端炔和β-酮酸酯进行亲电溴二氟甲基化, 该试剂能对1, 3-二酮选择性地发生氧-二氟甲基化而不会得到溴二氟甲基化产物, 但因效率不高导致适用范围受限(Scheme 10).

2012年, Shibata等[18b]进一步发展了非对称双芳基硫鎓盐型亲电溴二氟甲基化试剂6.经由溴二氟甲基芳基亚砜和带有不同取代基的苯, 在三氟甲磺酸酐的作用下成功地合成了一系列非对称的双芳基硫鎓盐型的溴二氟甲基化试剂.通过芳基上不同取代基的电子及其空间效应的影响, 能够精细地调整和优化溴二氟甲基化试剂的稳定性及反应活性等, 可有效提高溴二氟甲基化试剂的反应效能.此外, 所得溴二氟甲基化产物经进一步转化能有效得到含二氟亚甲基的功能分子(Scheme 11).

同年, Shibata等[18c]对这类非对称溴二氟甲基二芳基锍鎓盐试剂的应用进行了拓展, 发现该试剂在有机碱的诱导下原位生成的二氟卡宾能够对sp3碳核发生亲电进攻, 完成了对1, 1-偕二氰基烯丙位碳和β-酮酸酯α位碳的sp3-C亲电二氟甲基化(Scheme 12), 值得一提的是, 对β-酮酸酯的亲电二氟甲基化存在C/O选择性问题, 通过改变锍鎓盐阴离子能够提高反应的C/O选择性比例, 同时也伴随一定量的α-溴代产物生成.

随后在2013年, Shibata等[18d]将该非对称的溴二氟甲基二芳基锍鎓盐应用于1, 3-二酮类化合物的选择性O-二氟甲基化(Scheme 13).

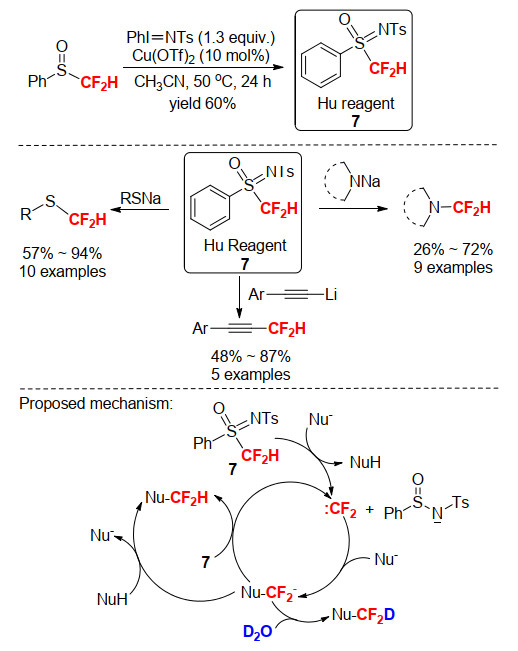
苯基亚砜亚胺类二氟甲基化试剂是过去10年中发展起来的一类重要二氟甲基化试剂, 能使某些类型的亲核剂发生高效的二氟甲基化反应, 如芳基硫醇类、咪唑类和磷亲核试剂等.然而, 也因为试剂稳定性和反应活性等方面存在的问题, 其主要用于N、P、S、O等杂原子亲核试剂的二氟甲基化反应, 对于重要的C亲核试剂的二氟甲基化效能不高, 其应用受到很大限制. 2009年, 胡金波小组[19a]将亲核性的二氟甲基苯基亚砜经过氧化亚胺化, 合成了一种新型的苯基亚砜亚胺型亲电二氟甲基试剂7 (Scheme 14), 并将该试剂作为二氟甲基阳离子(+CF2H)等效体, 用于对S-、N-和C-亲核剂的亲电二氟甲基化反应.但氘代实验证明反应经历的是一个二氟卡宾的过程, 首先, 试剂7经亲核剂作碱去质子化后迅速释放二氟卡宾物种(:CF2), 二氟卡宾与亲核剂(Nu-)反应生成二氟甲基负离子(Nu-CF2-), 随后经试剂7或亲核剂(NuH)质子化生成二氟甲基化产物, 在重水的存在下能得到氘代二氟甲基化产物(Scheme 14), 说明该试剂实际上是作为一个二氟卡宾前体试剂使用.
2015年, Magnier小组[19b]又开发了一种无金属参与的胡试剂7的合成方法, 苯基二氟甲基亚砜烯胺化后氧化得到试剂7和8 (Scheme 15a).但这个方法存在较大的缺点, 即中间体A不稳定, 限制了后续的试剂合成效率和收率.该小组初步研究了试剂7和8对β-酮酸酯的亲电二氟甲基化反应, 但是收率极低, 且C/O选择性也很差(Scheme 15b).

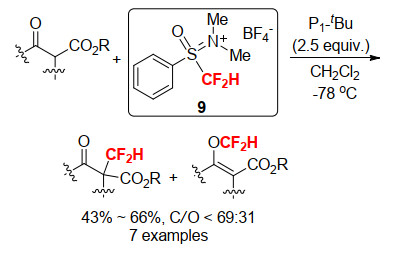
2011年Prakash和Olah等[20a]也报道了一种原位生成的N, N-二甲基苯基亚砜亚胺盐型亲电二氟甲基试剂9, 该试剂能与N、P、O、S亲核剂发生二氟甲基化反应分别得到二氟甲基季铵盐、二氟甲基季鏻盐、二氟甲基烷基醚和二氟甲硫基芳烃, 但是该反应条件苛刻, 需要严格的无水无氧操作.而且该类试剂稳定性差以至于只能原位生成使用, 可能由于其自身的不稳定而易于分解造成了反应效率的低下.机理研究证明该反应是一个直接的二氟甲基亲电过程而非常见的二氟卡宾过程(Scheme 16).

2012年, Shibata小组[20b]将N, N-二甲基苯基亚砜亚胺四氟硼酸盐9应用于β-酮酸酯的亲电二氟甲基化, 但是反应的C/O选择性不高, 实验结果和理论计算表明, 该反应可能包含二氟甲基阳离子亲电过程和自由基二氟甲基化过程两种途径, 直接的二氟甲基阳离子亲电过程得到的是C-选择性产物, 而自由基二氟甲基过程得到的是O-选择性产物(Scheme 17).
2016年, Koike和Akita等[21a-21b]将稳定、易合成的胡试剂首次用于光催化自由基二氟甲基化反应, 实现了末端和非末端烯烃双键(C=C)的一步区域选择性构建C(sp3)—CF2H和C(sp3)—O键, 合成了一系列的二氟甲基化的醇、醚和螺醚类化合物, 这一方法的构建为烯烃的双官能化和含氟多功能分子的构建提供了可靠的依据.作者提出的反应机理认为光催化剂IrⅢ经光照生成激发态的*IrⅢ, 试剂7与强还原性的*IrⅢ发生单电子转移产生二氟甲基自由基(•CF2H)和亚磺酰胺, 二氟甲基自由基(•CF2H)与烯烃反应生成自由基中间体C, C随后经强氧化性的IrⅣ单电子氧化生成阳离子中间体D, 光催化剂完成一个催化循环, 最后阳离子中间体D被亲核试剂ROH进攻得到氧-二氟甲基化产物(Scheme 18).

2019年, 该小组[21c]又将这一试剂用于可见光催化芳基烯烃的酮-二氟甲基双官能化, 制备了一系列α-二氟甲基化的酮类化合物, 同时该反应可使用连续流反应, 轻松的将反应规模放大到克级.二甲基亚砜(DMSO)在该反应过程中作为溶剂的同时也作为氧化剂, 循环伏安法测得该试剂7的还原电位为-2.26 V (vs. Cp2Fe in DMSO), 而激发态的fac-[Ir(ppy)3]为-2.14 V, 证明二氟甲基试剂7很可以被激发态的光催化剂Ir还原(Scheme 19).

2016年, 沈其龙小组[22a]报道了一个硫叶立德型亲电二氟甲基试剂10, 该试剂可用芳基二氟甲基硫醚与重氮化合物在铑催化下反应得到.在温和条件下, 并在路易斯酸或者布朗斯特酸作用下, 该试剂可以与醇反应, 制备烷基二氟甲基醚(Scheme 20).氘代实验证明该反应是一个直接亲电二氟甲基化过程, 没有二氟卡宾的参与.此外, 苯环上取代基的电性对该试剂的反应活性影响非常大, 降低试剂的电子云密度能够有效地提高试剂的反应活性.

2018年, 该小组[22b]又将试剂10应用于弱亲核性的碳亲核试剂如β-酮酸酯、丙二酸酯、氧化吲哚、苯并呋喃酮和乙烯酮甲硅烷基缩醛类化合物的亲电C-二氟甲基化(Scheme 21), 机理研究认为反应是一个二氟卡宾的过程.首先, 底物在无机碱Li2CO3的作用下失去一个质子形成碳负离子E, 试剂10在无机碱Li2CO3的作用下失去一个质子产生二氟卡宾(:CF2), 随后二氟卡宾(:CF2)对碳负离子E发生亲电进攻生成中间体F, 中间体F经质子化最终生成C-二氟甲基化产物, 并通过DFT计算证实了这一过程.

膦叶立德主要用于与醛、酮类化合物的Wittig反应构建碳-碳双键, 肖吉昌和卿凤翎等在二氟甲基膦叶立德前体试剂作为二氟卡宾前体的研究方面做了大量工作. 2013年, 肖吉昌小组[23]首次合成了膦叶立德型二氟甲基试剂Ph3P+CF2CO2- (PDFA, 11), 该试剂能够在无外加添加剂和相对较低的温度条件下转化为二氟卡宾, 并将其用于烯烃的二氟环丙烷化[24a](Scheme 22).

二氟甲基试剂11可用于对O、S、N-亲核试剂的二氟甲基化. 2015年, 肖吉昌小组[24b]报道了膦叶立德型二氟甲基试剂11 (Ph3P+CF2CO2-, PDFA)脱羧产生的二氟卡宾对O、S、N-亲核试剂的二氟甲基化(Scheme 23)以及对炔烃的谐二氟环丙烯化(Scheme 24).

2016年, 肖吉昌小组[24c]还通过二氟甲基试剂11 (Ph3P+CF2CO2-, PDFA)产生的二氟卡宾实现了在无碱参与的条件下对1, 3-二酮类化合物的选择性O-二氟甲基化, 合成了一系列二氟甲基烯醇醚(Scheme 25).同年, 朱卫华课题组[25]也报道了膦叶立德型二氟甲基试剂11 (Ph3P+CF2CO2-, PDFA)对2-羟基查尔酮的选择性O-二氟甲基化方面的工作(Scheme 26).


鉴于二氟甲基试剂11产生的二氟卡宾具有很好的亲电性, 肖吉昌小组[26]将二氟甲基试剂11用于对N-芳基磺酰基腙的亲电二氟甲基化反应, 合成了一系列的二氟甲基芳基砜类化合物, 反应机理认为底物先经CsHCO3去质子化生成负离子A, 随后被PDFA产生的二氟卡宾捕捉转化为中间体B, 中间体B经分子内环化转化为中间体C, 随后经开环得到D, D分解为E和F, 中间体F经质子化生成最终产物二氟甲基芳基砜(Scheme 27).

此外, 二氟甲基试剂PDFA经脱羧释放的二氟卡宾在三苯基膦和碱参与的条件下能与苄溴反应得到谐二氟芳基乙烯, 该反应实际上是一个二氟卡宾对膦叶立德的亲电过程.首先, 苄溴与三苯基膦反应生成季鏻盐, 在碱性条件下失去一个质子成为膦叶立德A, PDFA在加热条件下产生的二氟卡宾对膦叶立德发生亲电进攻生成1, 3-二极中间体B和膦杂环丙烷中间体C, B和C消除一分子三苯基膦都可得到最终产物. C经开环得到中间体D, 随后D消除一分子三苯基膦也能得到最终产物(Scheme 28)[27].

2015年, 肖吉昌小组[28]发现二氟甲基试剂PDFA也能用于对重氮类化合物的亲电二氟甲基化制备谐二氟苯乙烯类化合物.其中, 在与芳基重氮乙酸酯的卡宾交叉偶联合成谐二氟烯烃的反应中, 作者认为经历了一个铜二氟卡宾复合物的过程.首先, 铜卡宾A被二氟甲基试剂PDFA释放的三苯基膦捕捉生成中间体B, 随后中间体B与二氟卡宾生成铜二氟卡宾C, 中间体B (C)和叶立德H之间是可逆的, 随后中间体C的二氟卡宾经迁移插入Cu—C键生成中间体D, 最后经还原消除得到目标产物(Scheme 29, Proposed mechanism 1, Path I); 二氟卡宾也可能一开始与CuBr生成铜二氟卡宾中间体E, 随后与重氮化合物反应生成的中间体F对三苯基膦发生亲电进攻也能转化为中间体C (Scheme 29, Proposed mechanism 1, Path II).而二芳基重氮甲烷与二氟卡宾可在无Cu参与下直接发生偶联反应, 二芳基重氮甲烷脱除一分子氮气后产生的卡宾I被二氟甲基试剂PDFA释放的三苯基膦捕捉生成膦叶立德中间体K.另一种可能的途径是二芳基重氮甲烷先与三苯基膦反应生成的中间体J, J脱除一分子氮气后也能转化为膦叶立德中间体K.膦叶立德中间体K对二氟卡宾发生亲核进攻转化为中间体L, 最后发生β-PPh3消除转化为最终产物.二氟卡宾也可能直接对二芳基重氮甲烷的C-端发生亲电进攻生成中间体M, 最后脱除一分子氮气转化为最终产物(Scheme 29, Proposed mechanism 2).

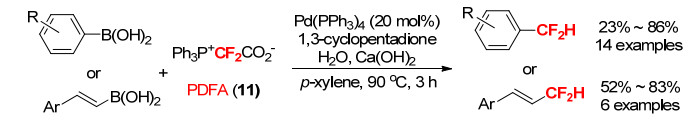
自1978年首例稳定的金属二氟卡宾物种被报道以来, 金属二氟卡宾参与的反应得到了一定的发展[29]. 2016年, 张新刚等[30]以BrCF2CO2Et为二氟卡宾源, 报道了首例钯-二氟卡宾参与的芳基硼酸的二氟甲基化反应, 其经过了一个二氟卡宾的迁移反应过程.同年, 肖吉昌小组[31]使用PDFA作为二氟卡宾源, 在钯催化下与芳基硼酸和烯基硼酸发生二氟卡宾迁移反应, 成功制备了二氟甲基芳烃、烯烃, 也证实了二氟卡宾迁移这一过程(Scheme 30).在这类金属二氟卡宾参与的反应中, 相关机理研究已经证实金属作为亲电位点, 二氟卡宾作为亲核位点(Scheme 31)[32], 这与正常的二氟卡宾亲电特性相反.

2015年, 肖吉昌小组[33a]发现以1, 5-二氮杂二环[5.4.0]十一烯-5 (DBU)作碱也能使PDFA试剂产生二氟卡宾, 经铜催化也能实现4-碘联苯的三氟甲基化, 其中另一分子的氟来源于试剂本身的分解.类似的, 以[Ph3PCF2H]+Br- (DFPB, 12a)代替PDFA试剂也能实现这一过程得到相同的产物, 作者提出的反应机理认为二氟卡宾被DBU捕捉生成中间体I, 中间体I经重排释放的F-离子被二氟卡宾亲电进攻生成CF3-离子, 随后经Cu(I)催化实现碘代芳烃的三氟甲基化(Scheme 32)[33b].

在分子硫的参与下, 试剂11也可用于对脂肪亲电体的三氟甲硫基化, 对亲电性的烷基卤发生亲核反应得到三氟甲硫基烷基化合物[34a]; 2017年, 肖吉昌课题组[34b-34c]在同样的反应条件下, 实现了溴代酮类化合物的α-碳上的三氟甲硫基化以及芳胺与二氟卡宾产生的硫代碳酰氟进行的N-三氟甲基化、串联二氟甲硫基化, 实验和理论计算都证实了硫代碳酰氟中间体的产生, 机理研究认为膦叶立德型二氟甲基试剂首先经脱羰基原位生成二氟卡宾, 随后与分子硫发生硫化转化为高亲电性的硫代碳酰氟(S=CF2), 并很快被亲核物种捕捉.基于分子硫参与下二氟卡宾能生成高亲电性的硫代碳酰氟(S=CF2), 随后发展了醇的脱羟基三氟甲硫基化反应[34d].同样地, 分子硒(Se)也能与二氟卡宾反应得到相应的亲电性硒代碳酰氟(Se=CF2), 首次实现了在外加氟化铯为氟源的条件下与苄基卤发生了三氟甲硒化反应(Scheme 33)[34e].

溴二氟甲基溴化季鏻盐[Ph3PCF2Br]+Br- (13)被认为是二氟卡宾前体试剂, 通过激发态光催化剂的单电子还原, 它能有效地转化为二氟甲基自由基参与反应. 2016年, 卿凤翎等[35a]报道了首例以[Ph3PCF2Br]+Br- (13)为自由基二氟甲基试剂, H2O和四氢呋喃(THF)为氢源, 可见光催化烯烃的氢-二氟甲基化.作者提出的反应机理认为[Ph3PCF2Br]+Br- (13)在水参与条件下释放的二氟卡宾在NaI和PPh3存在下能转化为二氟甲基季鏻盐, 季鏻盐能很容易地被激发态的光催化剂IrⅢ*单电子还原生成二氟甲基自由基(•CF2H), 随后二氟甲基自由基对烯烃进行自由基加成生成中间体A, 中间体A从THF攫取一个质子生成最终产物和中间体B, 中间体B能还原IrⅣ回到IrⅢ完成一个光催化循环(Scheme 34)[35a].

卿凤翎小组[35b-35d]发现[Ph3PCF2H]+Br- (DFPB)在不需要外加添加剂的条件下, 可以直接被激发态的光催化剂单电子还原产生二氟甲基自由基(•CF2H), 利用二氟甲基自由基(•CF2H)对烯烃的自由基加成, 发展了一系列对烯烃类化合物的氧-二氟甲基化、溴-二氟甲基化和氢-二氟甲基化反应方法(Scheme 35).

与[Ph3PCF2H]+Br- (DFPB, 12a)类似, [Ph3PCF2H]+ OTf- (12b)也可以作为自由基二氟甲基试剂, 2017年, 卿凤翎小组[36]又报道了对巯基化合物的自由基二氟甲基化反应合成二氟甲基硫醚化合物(Scheme 36).

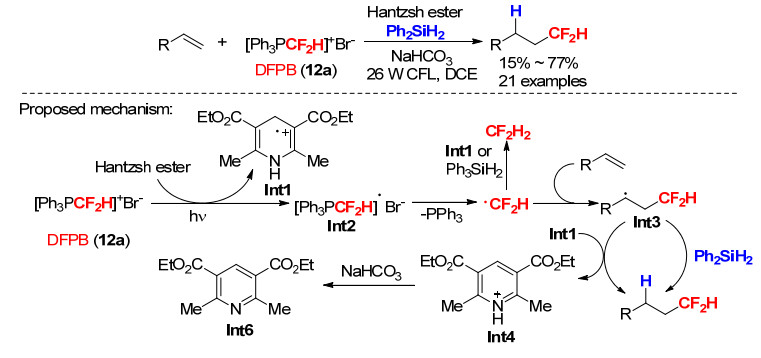
2019年, 肖吉昌等[37]报道了以DFPB为二氟甲基自由基源, 在无需金属光催化剂参与的条件下, 对烯烃的自由基氢-二氟甲基化方法(Scheme 37).在光照条件下经汉斯酯单电子还原, DFPB产生二氟甲基自由基•CF2H和中间体Int1, 自由基•CF2H对烯烃进行自由基加成后生成的自由基中间体Int3从Int1或Ph2SiH2攫取一个氢原子得到氢-二氟甲基化最终产物(Scheme 37).
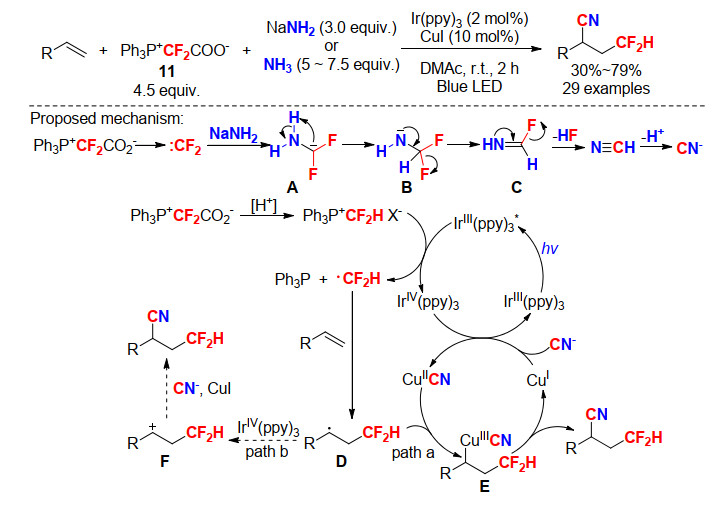
2019年, 肖吉昌小组[38]将PDFA (11)应用于光催化自由基二氟甲基化反应, 报道了首例原位生成氰基的氰-二氟甲基化反应. PDFA试剂在反应中作为二氟甲基和氰基源, 该反应避免了使用剧毒的氰化试剂.此外, Ph3P+CF2CO2-能攫取一个质子转化为Ph3P+CF2HX-, 循环伏安法实验证明Ph3P+CF2HX- (Epred=-0.959 V vs. SCE)能很容易被激发态的[Ir(ppy)3*] (E1/2red=-1.73 V vs. SCE)还原为二氟甲基自由基(•CF2H) (Scheme 38).
直接的亲电二氟甲基试剂, 由于CF2H中质子酸性强, 对碱、光、热等因素敏感, 造成试剂不稳定易于分解, 因此也往往影响了试剂的使用效率, 反应及底物类型受限, 这也是稳定高效的亲电二氟甲基试剂开发的困难所在.因此, 采取一种迂回策略, 在试剂设计中把CF2H中的质子酸用其它官能团取代, 这样就避免了亲电二氟甲基试剂的上述不稳定因素, 设计发展了稳定的官能团保护的亲电二氟甲基试剂.与亲电型直接的二氟甲基试剂相比, 官能化亲电型二氟烷基化试剂相对于直接的二氟甲基试剂来说不会受制于强酸性质子的不稳定因素影响, 相对更加稳定.而弊端则是需要多一步保护基的脱除才能转化为二氟甲基.另一方面, 含官能团化二氟烷基如溴二氟甲基能进一步地转化或与其他有机分子偶联, 更丰富的化学性质又是其优势所在.保护的亲电二氟甲基试剂主要有亲电溴二氟甲基试剂和苯磺酰基二氟甲基化试剂两类.
2010年, Magnier小组[39]开发了一类苯基硫亚胺型溴二氟甲基试剂14~17, 但是该试剂的亲电溴二氟甲基化性能很弱, 无实际应用价值, 作者也未对该试剂的可能应用做进一步研究(Scheme 39).

随后, 他们[39b]对该试剂进行氧化期望以升高硫的价态来提高试剂的反应活性和稳定性, 得到了苯基亚砜亚胺型亲电溴二氟甲基试剂18, 并将其用于炔烃的亲电溴二氟甲基化.作者认为, 该反应涉及二氟卡宾过程, 部分亲核性的炔基负离子需要先进攻试剂18产生二氟卡宾和炔基溴, 相当量的副产物炔基溴的产生导致目标产物溴二氟甲基炔的收率(Scheme 40).

2008年, 胡金波研究组[40a]发展了一种超价碘型亲电苯磺酰基二氟甲基试剂19, 该试剂能高效地与S、O亲核剂发生苯磺酰二氟甲基化反应.而且在铜盐催化下能与烯基和烯丙基羧酸发生脱羧苯磺酰二氟甲基化反应(Scheme 41), 铜盐作为路易斯酸在反应中起着加强试剂亲电性和促进α, β-不饱和羧酸脱羧的作用[40b-40c].苯磺酰二氟甲基可以经一步还原转化为二氟甲基或二氟亚甲基, 该试剂可作为一种间接亲电二氟甲基化的试剂.

另外, Shibata等[411]于2014年在报道了一种新的官能化的亲电二氟甲基化试剂S-(苯磺酰基二氟甲基)苯并噻吩鎓盐(20) (Scheme 42).该试剂能高效地对sp3-C亲核试剂进行苯磺酰二氟甲基官能化, 大量的β-酮酸酯和1, 1-偕二氰基丙烯在温和的反应条件下, 以较高收率选择性地生成相应的C-二氟甲基化产物, 特别是β-酮酸酯的反应中没有O-二氟甲基化产物的生成, 这对C亲核剂的二氟甲基反应的发展是一个突破.但该试剂对非环的β-酮脂和1, 3-二羰基底物氟化效率不高.

相对于早期的亲电型二氟甲基试剂, 近年来新发展的亲电型二氟甲基试剂有更加稳定、易保存和毒性小等特点, 能够对绝大多数杂原子亲核试剂和部分碳亲核试剂发生二氟甲基化.特别是新发展亲电型二氟甲基试剂更容易从激发态的光催化剂获得一个电子转化为二氟甲基自由基参与反应, 拓展了自由基二氟甲基化反应的应用.虽然大多数反应主要表现为二氟卡宾亲电过程, 直接亲电性的报道偏少, 特别是膦叶立德型二氟甲基试剂完全表现为二氟卡宾过程, 但是试剂的骨架结构修饰空间大, 反应性更加容易调控.虽然近年来亲电型二氟甲基试剂的研究取得了一些进展, 但与亲电型三氟甲基试剂以及亲核型的二氟甲基试剂相比, 仍相对滞后, 发展缓慢.试剂类型和数量都相对较少, 且在反应活性、普适性及试剂的制备便捷性等方面仍存在短板.而且, 本文涵盖的亲电二氟甲基试剂都存在副产物比重大及原子利用效率低的问题.因此, 对于亲电型二氟甲基试剂及其相应的二氟甲基化方法的开发, 进一步研究的空间和潜力仍较大.
基于近年来的研究工作基础和现状, 针对亲电型二氟甲基化试剂及其应用的发展, 将来需要引起关注和重点解决的问题有以下几方面: (1)发展更多结构新颖稳定的直接型亲电型二氟甲基化试剂, 丰富直接亲电型二氟甲基试剂库, 以拓展亲电二氟甲基化反应的应用范围, 拓宽反应类型和底物范围. (2)开发新的亲电型二氟甲基试剂的同时, 还需注重试剂的反应效率以及原子经济性, 尤其是发展更多普适性高、多功能的亲电二氟甲基试剂, 注重试剂的稳定、普适、高效、绿色、环保以及制备便捷性等. (3)鉴于含氟手性分子的重要性, 基于亲电二氟甲基试剂的不对称二氟甲基化反应的方法和策略至今仍是未解决的重大挑战.
(a) Smart, B. E. Chem. Rev. 1996, 96, 1555.
(b) Jeschke, P.; Baston, E.; Leroux, F. R. Mini-Rev. Med. Chem. 2007, 7, 1027 and references therein.
(c) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359.
(d) Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432.
(e) Bassetto, M.; Ferla, S.; Pertusati, F. Future Med. Chem. 2015, 7, 527.
(a) Goure, W. F.; Leschinsky, K. L.; Wratten, S. J.; Chupp, J. P. J. Agric. Food Chem. 1991, 39, 981.
(b) Pérez, R. A.; Sánchez-Brunete, C.; Miguel, E.; Tadeo, J. L. J. Agric. Food Chem. 1998, 46, 1864.
(c) Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed., Wiley-VCH, Weinheim, 2013.
(a) Kirsch, P.; Bremer, B. Angew. Chem., Int. Ed. 2000, 39, 4216.
(b) Tasaka, T.; Takenaka, S.; Kabu, K.; Morita, Y.; Okamoto, H. Ferroelectronics 2002, 276, 83.
(c) Boltalina, O. V.; Nakajima, T. New Fluorinated Carbons: Fundamentals and Applications, Elsevier, Amsterdam, 2016.
(a) Bégué, J. P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine, Wiley-VCH, Weinheim, 2008.
(b) Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, 2009.
(c) Gouverneur, V.; Muller, K. Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications, Imperial College Press, London, 2012.
(a) Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881.
(b) Purse, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320.
(c) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359.
(d) Kirk, K. L. Org. Process Res. Dev. 2008, 12, 305.
(e) Prakash, G. K. S.; Wang, F. Chim. Oggi 2012, 30, 30.
(f) Wang, J.; Liu, H. Chin. J. Org. Chem. 2011, 31, 1785(in Chinese).
(王江, 柳红, 有机化学, 2011, 31, 1785.)
For selected reviews on perfluoroalkylation reactions, see: (a) Lundgren, R. J.; Stradiotto, M. Angew. Chem., Int. Ed. 2010, 49, 9322.
(b) Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475.
(c) Wu, X.-F.; Neumann, H.; Beller, M. Chem.-Asian J. 2012, 7, 1744.
(d) Besset, T.; Schneider, C.; Cahard, D. Angew. Chem., Int. Ed. 2012, 51, 5048.
(e) Ye, Y.; Sanford, M. S. Synlett 2012, 2005.
(f) Qing, F.-L. Chin. J. Org. Chem. 2012, 32, 815(in Chinese).
(卿凤翎, 有机化学, 2012, 32, 815.)
(g) Pan, F.; Shi, Z. Acta Chim. Sinica, 2012, 70, 1679(in Chinese).
(潘菲, 施章杰, 化学学报, 2012, 70, 1679.)
(h) Wang, X.; Zhang, Y.; Wang, J. Sci. Sin. Chim. 2012, 42, 1417.
(王兮, 张艳, 王剑波, 中国科学: 化学, 2012, 42, 1417.)
(i) Chen, P.; Liu, G. Synthesis 2013, 45, 2929.
(j) Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214.
(k) Xu, J.; Liu, X.; Fu, Y. Tetrahedron Lett. 2014, 55, 585.
(l) Wang, G.; He, X.; Dai, J.; Xu, H. Chin. J. Org. Chem. 2014, 34, 837(in Chinese).
(王光祖, 赫侠平, 戴建军, 许华建, 有机化学, 2014, 34, 837.)
(m) Zhang, J.; Jin, C.; Zhang, Y. Chin. J. Org. Chem. 2014, 34, 662(in Chinese).
(张霁, 金传飞, 张英俊, 有机化学, 2014, 34, 662.)
(n) Merino, E.; Nevado, C. Chem. Soc. Rev. 2014, 43, 6598.
(o) Chu, L.; Qing, F.-L. Acc. Chem. Res. 2014, 47, 1513.
(a) Prakash, G. K. S.; Mandal, M.; Schweizer, S.; Petasis, N. A.; Olah, G. A. J. Org. Chem. 2002, 67, 3718.
(b) Narjes, F.; Koehler, K. F.; Koch, U.; Gerlach, B.; Colarusso, S.; Steinkhler, C.; Brunetti, M.; Altamura, S.; De Francesco, R.; Matassa, V. G. Bioorg. Med. Chem. Lett. 2002, 12, 701.
(c) Hu, J.; Zhang, W.; Wang. F. Chem. Commun. 2009, 7465.
(a) Li, Y.; Hu, J. Angew. Chem., Int. Ed. 2005, 44, 5882.
(b) Prakash, G. K. S.; Weber, C.; Chacko, S.; Olah, G. A. Org. Lett. 2007, 9, 1863.
(a) Cazzola, M.; Picciolo, S.; Matera, M. G. Expert Opin. Pharmacother. 2010, 11, 441.
(b) Kohl, B.; Sturm, E.; Rainer, G. US 4758579A, 1985.
(c) Gajjar, D. A.; Bello, A.; Ge, Z.; Christopher, L.; Grasela, D. M. Antimicrob. Agents Chemother. 2003, 47, 2256.
(d) Kastron, V. V.; Vitolin, R. O.; Fialkov, J. A.; Shelyazhenko, S. V. US 4219653, 1980.
(a) Brahms, D. L. S.; Dailey, W. P. Chem. Rev. 1996, 96, 1585.
(b) Dolbier, W. R. Jr.; Battiste, M. A. Chem. Rev. 2003, 103, 1071.
(c) Fedoryński, M. Chem. Rev. 2003, 103, 1099.
(d) Romanenko, V. D.; Kukhar, V. P. Chem. Rev. 2006, 106, 3868.
(e) Prakash, G. K. S.; Hu, J. Acc. Chem. Res. 2007, 40, 921.
(f) Hu, J.; Zhang, W.; Wang, F. Chem. Commun. 2009, 7465.
(g) Zhang, C.-P.; Chen, Q.-Y.; Guo, Y.; Xiao, J.-C.; Gu, Y.-C. Chem. Soc. Rev. 2012, 41, 4536.
(h) Bizet, V.; Kowalczyk, R.; Bolm, C. Chem. Soc. Rev. 2014, 43, 2426.
(i) Ni, C.; Hu, J. Synthesis 2014, 46, 0842.
(j) Shen, X.; Hu, J. Eur. J. Org. Chem. 2014, 4437.
(k) Belhomme, M.-C.; Besset, T.; Poisson, T.; Pannecoucke, X. Chem.-Eur. J. 2015, 21, 12836.
(l) Barata-Vallejo, S.; Bonesi, S. M.; Postigo, A. Org. Biomol. Chem. 2015, 13, 11153.
(m) Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765.
(n) Ni, C.; Zhu, L.; Hu, J. Acta Chim. Sinica 2015, 73, 90(in Chinese).
(倪传法, 朱林桂, 胡金波, 化学学报, 2015, 73, 90.)
(o) Pan, X.; Xia, H.; Wu, J. Org. Chem. Front. 2016, 3, 1163.
(p) Koike, T.; Akita, M. Chem 2018, 4, 409.
(q) Yerien, D. E.; Barata-Vallejo, S.; Postigo, A. Chem.-Eur. J. 2017, 23, 14676.
(r) Rong, J.; Ni, C.; Hu, J. Asian J. Org. Chem. 2017, 6, 139.
(s) Dilman, A. D.; Levin, V. V. Acc. Chem. Res. 2018, 51, 1272.
(t) Lemos, A.; Lemaire, C.; Luxen, A. Adv. Synth. Catal. 2019, 361, 1500.
(u) Koike, T.; Akita, M. Org. Biomol. Chem. 2019, 17, 5413.
(v) Wang, X.; Wang, X.; Wang, J. Tetrahedron 2019, 75, 949.
(w) Xie, Q.; Hu, J. Chin. J. Chem. 2020, 38, 202.
Umemoto, T. Chem. Rev. 1996, 96, 1757. doi: 10.1021/cr941149u
(a) Yagupol'skii, L. M.; Kondratenko, N. Y.; Timofeeva, G. N. Zh. Org. Khim. 1984, 20, 115.
(b) Umemoto, T.; Ishihara, S. J. Am. Chem. Soc. 1993, 115, 2156.
(c) Umemoto, T.; Ishihara, S.; Adachi, K. J. Fluorine Chem. 1995, 74, 77.
(d) Umemoto, T.; Ishihara, S. J. Fluorine Chem. 1999, 98, 75.
(e) Umemoto, T.; Adachi, K. J. Org. Chem. 1994, 59, 5692.
(f) Yang, J. J.; Kirchmeier, R. L.; Shreeve, J. M. J. Org. Chem. 1998, 63, 2656.
(g) Ma, J.-A.; Cahard, D. J. Org. Chem. 2003, 68, 8726.
(a) Prakash, G. K. S.; Weber, C.; Chacko, S.; Olah, G. A. Org. Lett. 2007, 9, 1863.
(b) Prakash, G. K. S.; Weber, C.; Chacko, S.; Olah, G. A. J. Comb. Chem. 2007, 9, 920.
Yue, C.-B.; Lin, J.-H.; Cai, J.; Zhang, C.-P.; Zhao, G.; Xiao, J.-C.; Li, H. RSC Adv. 2016, 6, 35705. doi: 10.1039/C6RA06338A
(a) Noto, N.; Koike, T.; Akita, M. Chem. Sci. 2017, 8, 6375.
(b) Noto, N.; Tanaka, Y.; Koike, T.; Akita, M. ACS Catal. 2018, 8, 9408.
(a) Tang, X.-J.; Thomoson, C. S.; Dolbier, W. R. Jr. Org. Lett. 2014, 16, 4594.
(b) Zhang, Z.; Tang, X.-J.; Thomoson, C. S.; Dolbier, W. R. Jr. Org. Lett. 2015, 17, 3528.
(c) Zhang, Z.; Tang, X.-J.; Dolbier, W. R. Jr. Org. Lett. 2015, 17, 4401.
(d) Rong, J.; Deng, L.; Tan, P.; Ni, C.; Gu, Y.; Hu, J. Angew. Chem., Int. Ed. 2016, 55, 2743.
(e) Fu, W.; Han, X.; Zhu, M.; Xu, C.; Wang, Z.; Ji, B.; Hao, X.-Q.; Song, M.-P. Chem. Commun. 2016, 52, 13413.
(f) Zhang, Z.; Tang, X.-J.; Dolbier, W. R. Jr. Org. Lett. 2016, 18, 1048.
(g) Miao, W.; Zhao, Y.; Ni, C.; Gao, B.; Zhang, W.; Hu, J. J. Am. Chem. Soc. 2018, 140, 880.
(h) Zhang, Z.; Martinez, H.; Dolbier, W. R. J. Org. Chem. 2017, 82, 2589.
(i) Zhu, M.; Fu, W.; Wang, Z.; Xu, C.; Ji, B. Org. Biomol. Chem. 2017, 15, 9057.
(j) Duchemin, N.; Buccafusca, R.; Daumas, M.; Ferey, V.; Arseniyadis, S. Org. Lett. 2019, 21, 8205.
(k) Wang, Z.-S.; Chen, Y.-B.; Zhang, H.-W.; Sun, Z.; Zhu, C.; Ye, L. J. Am. Chem. Soc. 2020, 142, 3636.
(a) Lu, S.-L.; Li, X.; Qin, W.-B.; Liu, J.-J.; Huang, Y.-Y.; Wong, H. N. C.; Liu, G.-K. Org. Lett. 2018, 20, 6925.
(b) Liu, G.-K.; Li, X.; Qin, W.-B.; Peng, X.-S.; Wong, H. N. C.; Zhang, L.; Zhang, X. Chem. Commun. 2019, 55, 7446.
(c) Liu, G.-K.; Qin, W.-B.; Li, X.; Lin, L.-T.; Wong, H. N. C. J. Org. Chem. 2019, 84, 15948.
(d) Liu, G.-K.; Li, X.; Qin, W.-B.; Lin, W.-F.; Lin, L.-T.; Chen, J.-Y.; Liu, J.-J. Chin. Chem. Lett. 2019, 30, 1515.
(a) Zhang, C.; Cao, H.; Wang, Z.; Zhang, C.; Chen, Q.; Xiao, J. Synlett 2010, 1089.
(b) Liu, G.; Mori, S.; Wang, X.; Noritake, S.; Tokunaga, E.; Shibata, N. New J. Chem. 2012, 36, 1769.
(c) Liu, G.; Wang, X.; Lu, X.; Xu, X.-H.; Tokunaga, E.; Shibata, N. ChemistryOpen 2012, 1, 227.
(d) Liu, G.; Wang, X.; Xu, X.-H.; Lu, X.; Tokunaga, E.; Tsuzuki, S.; Shibata, N. Org. Lett. 2013, 15, 1044.
(a) Zhang, W.; Wang, F.; Hu, J. Org. Lett. 2009, 11, 2109.
(b) Pégot, B.; Urban, C.; Bourne, A.; Le, T. N.; Bouvet, S.; Marrot, J.; Diter, P.; Magnier, E. Eur. J. Org. Chem. 2015, 3069.
(a) Prakash, G. K. S.; Zhang, Z.; Wang, F.; Ni, C.; Olah, G. A. J. Fluorine Chem. 2011, 132, 792.
(b) Yang, Y.; Lu, X.; Liu, G.; Tokunaga, E.; Tsuzuki, S.; Shibata, N. ChemistryOpen 2012, 1, 221.
(a) Arai, Y.; Tomita, R.; Ando, G.; Koike, T.; Akita, M. Chem.-Eur. J. 2016, 22, 1262.
(b) Noto, N.; Koike, T.; Akita, M. J. Org. Chem. 2016, 81, 7064.
(c) Nakayama, Y.; Ando, G.; Abe, M.; Koike, T.; Akita, M. ACS Catal. 2019, 9, 6555.
(a) Zhu, J.; Liu, Y.; Shen, Q. Angew. Chem., Int. Ed. 2016, 55, 9050.
(b) Zhu, J.; Zheng, H.; Xue, X.-S.; Xiao, Y.; Liu, Y.; Shen, Q. Chin. J. Chem. 2018, 36, 1069.
Zheng, J.; Cai, J.; Lin, J.-H.; Guo, Y.; Xiao, J.-C. Chem. Commun. 2013, 49, 7513. doi: 10.1039/c3cc44271c
(a) Zheng, J.; Lin, J.-H.; Cai, J.; Xiao, J.-C. Chem.-Eur. J. 2013, 19, 15261.
(b) Deng, X.-Y.; Lin, J.-H.; Zheng, J.; Xiao, J.-C. Chem. Commun. 2015, 51, 8805.
(c) Liu, C.; Deng, X.-Y.; Zeng, X.-L.; Zhao, G.; Lin, J.-H.; Wang, H.; Xiao, J.-C. J. Fluorine Chem. 2016, 192, 27.
Hua, M.-Q.; Wang, W.; Liu, W.-H.; Wang, T.; Zhang, Q.; Huang, Y.; Zhu, W.-H. J. Fluorine Chem. 2016, 181, 22. doi: 10.1016/j.jfluchem.2015.11.003
Zheng, Q.-T.; Wei, Y.; Zheng, J.; Duan, Y.-Y.; Zhao, G.; Wang, Z.-B.; Lin, J.-H.; Zheng, X.; Xiao, J.-C. RSC Adv. 2016, 6, 82298. doi: 10.1039/C6RA20629H
Deng, X.-Y.; Lin, J.-H.; Xiao, J.-C. J. Fluorine Chem. 2015, 179, 116. doi: 10.1016/j.jfluchem.2015.06.009
Zheng, J.; Lin, J.-H.; Yu, L.-Y.; Wei, Y.; Zheng, X.; Xiao, J.-C. Org. Lett. 2015, 17, 6150. doi: 10.1021/acs.orglett.5b03159
(a) Reger, D. L.; Dukes, M. D. J. Organomet. Chem. 1978, 153, 67.
(b) Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746.
Feng, Z.; Min, Q.-Q.; Zhang, X. Org. Lett. 2016, 18, 44. doi: 10.1021/acs.orglett.5b03206
Deng, X.-Y.; Lin, J.-H.; Xiao, J.-C. Org. Lett. 2016, 18, 4384. doi: 10.1021/acs.orglett.6b02141
Fu, X.-P.; Xue, X.-S.; Zhang, X.-Y.; Xiao, Y.-L.; Zhang, S.; Guo, Y.-L.; Leng, X.; Houk, K. N.; Zhang, X. Nat. Chem. 2019, 11, 948. doi: 10.1038/s41557-019-0331-9
(a) Zheng, J.; Lin, J.-H.; Deng, X.-Y.; Xiao, J.-C. Org. Lett. 2015, 17, 532.
(b) Wei, Y.; Yu, L.; Lin, J.; Zheng, X.; Xiao, J. Chin. J. Chem. 2016, 34, 481.
(a) Zheng, J.; Wang, L.; Lin, J.-H.; Xiao, J.-C.; Liang, S. H. Angew. Chem., Int. Ed. 2015, 54, 13236.
(b) Zheng, J.; Cheng, R.; Lin, J.-H.; Yu, D.-H.; Ma, L.; Jia, L.; Zhang, L.; Wang, L.; Xiao, J.-C.; Liang, S. H. Angew. Chem., Int. Ed. 2017, 56, 3196.
(c) Yu, J.; Lin, J.-H.; Xiao, J.-C. Angew. Chem., Int. Ed. 2017, 56, 16669.
(d) Luo, J.-J.; Zhang, M.; Lin, J.-H.; Xiao, J.-C. J. Org. Chem. 2017, 82, 11206.
(e) Chen, X.-L.; Zhou, S.-H.; Lin, J.-H.; Deng, Q.-H.; Xiao, J.-C. Chem. Commun. 2019, 55, 1410.
(a) Lin, Q.-Y.; Xu, X.-H.; Zhang, K.; Qing, F.-L. Angew. Chem., Int. Ed. 2016, 55, 1479.
(b) Ran, Y.; Lin, Q.-Y.; Xu, X.-H.; Qing, F.-L. J. Org. Chem. 2016, 81, 7001.
(c) Lin, Q.-Y.; Ran, Y.; Xu, X.-H.; Qing, F.-L. Org. Lett. 2016, 18, 2419.
(d) Hu, W.-Q.; Xu, X.-H.; Qing, F.-L. J. Fluorine Chem. 2018, 208, 73.
Ran, Y.; Lin, Q.-Y.; Xu, X.-H.; Qing, F.-L. J. Org. Chem. 2017, 82, 7373. doi: 10.1021/acs.joc.7b01041
Yu, J.; Lin, J.-H.; Cao, Y.-C.; Xiao, J.-C. Org. Chem. Front. 2019, 6, 3580. doi: 10.1039/C9QO00919A
Zhang, M.; Lin, J.-H.; Xiao, J.-C. Angew. Chem., Int. Ed. 2019, 58, 6079. doi: 10.1002/anie.201900466
(a) Urban, C.; Macé, Y.; Cadoret, F.; Blazejewski, J. C.; Magnier, E. Adv. Synth. Catal. 2010, 352, 2805.
(b) Urban, C.; Cadoret, F.; Blazejewski, J. C.; Magnier, E. Eur. J. Org. Chem. 2011, 4862.
(c) Macé, Y.; Magnier, E. Eur. J. Org. Chem. 2012, 2479.
(a) Zhang, W.; Zhu, J.; Hu, J. Tetrahedron Lett. 2008, 49, 5006.
(b) He, Z.; Luo, T.; Hu, M.; Cao, Y.; Hu, J. Angew. Chem., Int. Ed. 2012, 51, 3944.
(c) He, Z.; Hu, M.; Luo, T.; Li, L.; Hu, J. Angew. Chem., Int. Ed. 2012, 51, 11545.
Wang, X.; Liu, G.; Xu, X.-H.; Shibata, N.; Tokunaga, E.; Shibata, N. Angew. Chem., Int. Ed. 2014, 53, 1827. doi: 10.1002/anie.201309875

图式 2 二氟甲基二芳基锍鎓盐1对N, P, O的二氟甲基化反应
Scheme 2 N, P, O-Difluoromethylation with S-difluoromethyl-diarylsulfonium salt 1

图式 3 二氟甲基二芳基锍鎓盐2的合成及其对1, 3-二酮的O-二氟甲基化反应
Scheme 3 Synthesis of S-(difluoromethyl)diarylsulfonium salt 2 and O-difluoromethylation of 1, 3-diones

图式 4 锍鎓盐3的合成及其在可见光催化烯烃的氨化-二氟甲基化反应中的应用
Scheme 4 Synthesis of sulfonium salt 3 and its application in visible-light-induced aminodifluoromethylation of alkenes

图式 6 二氟甲基二芳基锍鎓盐4用于β-酮酸酯和丙二酸酯的选择性C-二氟甲基化反应
Scheme 6 Selective electrophilic C-difluoromethylation of β‑ketoesters and malonates with S-(difluoromethyl)-diaryl-sulfonium salt 4

图式 7 二氟甲基二芳基锍鎓盐4对1, 3-二酮的O-二氟甲基化反应
Scheme 7 O-Difluoromethylation of 1, 3-diones with S-(difluoro- methyl)-diarylsulfonium salt 4

图式 8 二氟甲基二芳基锍鎓盐4用于脂肪醇的二氟甲基化及其机理
Scheme 8 Difluoromethylation of aliphatic alcohols with S-(difluoromethyl)-diarylsulfonium salt 4 and its proposed mechanism

图式 9 亲电型二氟甲基试剂4对酚、硫酚的二氟甲基化反应
Scheme 9 Difluoromethylation of phenols and thiophenols with S-(difluoromethyl)-diarylsulfonium salt 4

图式 10 溴二氟甲基二芳基锍鎓盐5的合成及其用于亲电溴二氟甲基化反应
Scheme 10 Synthesis of S-bromodifluoromethyl-diarylsulfonium salt 5 and its application in electrophilic bromodifluoromethylation

图式 11 溴二氟甲基二芳基锍鎓盐6的合成及其对炔烃的亲电溴二氟甲基化反应
Scheme 11 Synthesis of S-bromodifluoromethyl-diarylsulfonium salt 6 and its electrophilic bromodifluoromethylation with alkynes

图式 12 溴二氟甲基二芳基锍鎓盐6用于选择性C-二氟甲基化反应
Scheme 12 C-Selective electrophilic difluoromethylation with S-bromodifluoromethyl-diarylsulfonium salt 6

图式 13 溴二氟甲基二芳基锍鎓盐6c用于1, 3-二酮的选择性O-二氟甲基化反应
Scheme 13 Selective O-difluoromethylation of 1, 3-diones with S-bromodifluoromethyl-diarylsulfonium salt 6c

图式 14 苯基亚砜亚胺型二氟甲基试剂7的合成及其在S, N, C-二氟甲基化中的应用和反应机理
Scheme 14 Synthesis of S-difluoromethyl-S-phenylsulfoximine reagent 7, its application in S, N, C-difluoromethylation and proposed mechanism

图式 15 苯基亚砜亚胺型二氟甲基试剂7和8的合成及其用于β-酮酸酯的二氟甲基化反应
Scheme 15 Synthesis of S-difluoromethyl-S-phenylsulfoximine reagents 7 and 8 and its application in difluoromethylation of β-ketoesters

图式 16 苯基亚砜亚胺型二氟甲基试剂9的合成及其用于N, O, S, P-二氟甲基化反应
Scheme 16 Synthesis of S-difluorome-thyl-S-phenylsulf- oximine reagent 9 and its N, O, S, P-difluoromethylation

图式 17 苯基亚砜亚胺型二氟甲基试剂9用于β-酮酸酯的亲电二氟甲基化反应
Scheme 17 Electrophilic difluoromethylation of β-ketoesters with S-difluoromethyl-S-phenylsulfoximine reagent 9

图式 18 可见光催化烯烃氧-二氟甲基化及其机理推测
Scheme 18 Photocatalytic oxydifluoromethylation of alkenes and its proposed mechanism

图式 19 可见光催化芳基烯烃的氧代-二氟甲基化
Scheme 19 Keto-difluoromethylation of aromatic alkenes by photoredox catalysis

图式 20 膦叶立德型二氟甲基试剂PDFA (11)的合成及其作为二氟卡宾前体用于芳基烯烃的二氟环丙烷化反应
Scheme 20 Preparation of difluoromethylated phosphonium ylide reagent PDFA (11) and its application as a difluorocarbene precuosor in difluorocyclopropanation of arylalkenes

图式 21 硫叶立德型二氟甲基试剂的亲电二氟甲基化反应及其机理推测
Scheme 21 Electrophilic difluoromethylation reaction with difluoromethylated sulfonium ylide reagent and its proposed mechanism

图式 22 PDFA用于芳基烯烃的二氟环丙烷化反应
Scheme 22 Difluorocyclopropanation of aryl alkenes with PDFA

图式 25 PDFA用于1, 3-二酮的O-二氟甲基化反应
Scheme 25 O-Difluoromethylation of 1, 3-diones with PDFA

图式 26 2-羟基查尔酮的选择性O-二氟甲基化反应
Scheme 26 Selective O-difluoromethylation of 2-hydroxy- chalcones

图式 27 PDFA用于对N-芳基磺酰基腙的亲电二氟甲基化及其机理推测
Scheme 27 Electrophilic difluoromethylation of N-arylsulfonyl hydrazones with PDFA and its proposed mechanism

图式 28 PDFA用于从苄溴合成谐二氟芳基乙烯及其机理推测
Scheme 28 Synthesis of gem-difluorostyrenes from benzyl bromide with PDFA and its proposed mechanism

图式 29 PDFA用于苄溴合成谐二氟芳基乙烯及其机理推测
Scheme 29 Synthesis of gem-difluorostyrenes from benzyl bromide with PDFA and its proposed mechanism

图式 31 钯催化二氟卡宾转移反应的可能机理
Scheme 31 A possible mechanism for controllable palladium-catalyzed difluorocarbene transfer

图式 32 膦叶立德型二氟甲基试剂DFPB (12a)的合成及其在铜催化碘代芳烃三氟甲基化反应中的应用和反应机理
Scheme 32 Synthesis of difluoromethylated phosphonium ylide reagent DFPB (12a) and its application in copper catalyzed trifluoromethylation of aromatic iodides and proposed mechanism

图式 33 分子硫、硒参与的氟烷基化反应
Scheme 33 Fluoroalkylation involving in elemental sulfur or selenium

图式 34 光催化烯烃的氢-二氟甲基化反应及其机理推测
Scheme 34 Photocatalytic hydrodifluoromethylation of alkenes and its proposed mechanism

图式 35 光催化烯烃的自由基二氟甲基化反应及其机理推测
Scheme 35 Photocatalytic radical difluoromethylation of alkenes and its proposed mechanism

图式 36 光催化巯基化合物的自由基二氟甲基化反应
Scheme 36 Photocatalytic radical difluoromethylation of thiols and thiophenols

图式 37 烯烃的自由基二氟甲基化反应及其反应机理
Scheme 37 Radical hydrodifluoromethylation of alkenes and its proposed mechanism

图式 38 光催化烯烃的氰-二氟甲基化及其机理推测
Scheme 38 Photocatalytic cyanodifluoromethylation of alkenes and its proposed mechanism

图式 39 苯基硫亚胺型溴二氟甲基试剂的合成
Scheme 39 Preparation of phenylsulfilimine bromodifluoromethylating reagents

图式 40 苯基亚砜亚胺型溴二氟甲基试剂18的合成及应用
Scheme 40 Preparation and application of phenylsulfoximine bromodifluoromethylating reagent 18

图式 41 超价碘型亲电苯磺酰基二氟甲基试剂19的合成及其应用
Scheme 41 Preparation and application of electrophilic hypervalent iodine(Ⅲ)-CF2SO2Ph reagent

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




 下载:
下载:







 下载:
下载: