
图式 1.
过渡金属催化烯丙基取代反应的区域选择性
Scheme 1.
Regioselectivity of transition metal-catalyzed allylic substitution reactions

烯丙基取代反应是指亲核试剂对烯丙基亲电试剂发生形式上的SN2或者SN2'的取代反应, 而这些反应通常是由钯[1-4]、铱[5-7]、钼[8-16]、钨[17]、钌[18-23]、铑[24-30]、镍[31-33]、铜[34-42]等过渡金属来催化完成的. 1965年, Tsuji等[43]报道了等物质的量的π-烯丙基氯化钯与丙二酸二乙酯钠盐发生的亲核取代反应. 1977年, Trost课题组[44]使用钯/手性磷配体催化体系首次实现了催化不对称烯丙基取代反应.随后, 经过数十年的发展, 过渡金属催化的不对称烯丙基取代反应已经成为有机合成中构建碳碳键和碳杂键最重要的方法之一, 在手性药物和天然产物的不对称合成有着广泛的应用[45-47].
在众多过渡金属催化的烯丙基取代反应中, 金属铱、钼、钨、钌、铑等催化的烯丙基取代反应有着不同于钯的特殊的支链区域选择性(Scheme 1), 其中金属铱在该类反应中表现出优异的对映选择性, 在近年来越来越多地受到化学家们的关注. 1997年, Takeuchi课题组[48]报道了金属铱催化的烯丙基取代反应.同年, Helmechen课题组[49]报道了第一例金属铱催化的不对称烯丙基取代反应, 通过使用PHOX配体可以取得99/1的区域选择性和95% ee的对映选择性.此后, 国内外Helm- chen、Hartwig、Carreira、Alexakis、游书力等多个课题组[50-54]均在该领域进行了深入研究, 各种各样的亲核试剂被成功运用到金属铱催化的不对称烯丙基取代反应中, 以非常优秀的区域选择性和对映选择性得到了结构多样的含烯丙基模块的手性产物.尽管如此, 由于铱催化的烯丙基取代反应独特的支链选择性, 其产物通常含有多个手性中心(两个及以上), 如何实现高效的立体发散性合成依然是该领域的重要挑战.
两种或者多种催化剂参与的协同催化模式[55-63], 因其具有简便、高效等优点而引起了化学家们的极大兴趣(Scheme 2).在这种催化模式中, 多种催化剂共存于同一反应体系中, 催化剂之间互相补充、协同作用, 彼此间不干扰, 每种催化剂选择性地平行作用于相应的反应底物或者催化循环, 进而顺利实现了各个反应底物的有序组装, 为许多以前无法实现的转化提供了新的解决方案.体系中的任意一种催化剂都可发挥手性诱导作用, 因此当手性催化剂的不对称诱导相互匹配时,有利于提高反应的对映选择性, 并且能够实现连续多个手性中心的立体化学控制, 对手性化合物的立体发散合成具有重要意义.
鉴于协同催化模式的独特优势, 将这种催化模式应用到金属铱催化的不对称烯丙基化反应中, 能够大大丰富亲核试剂的类型, 拓宽烯丙基取代反应的应用范围.更为重要的是, 当两种催化剂都具有手性环境时, 则有可能实现多手性中心产物的立体发散性合成.
本综述将总结协同催化模式在金属铱催化不对称烯丙基取代反应中的应用研究进展, 并根据协同催化剂类型的不同, 将按照以下几个方面进行展开: (1)金属铱与有机胺协同催化, (2)金属铱与相转移催化剂协同催化, (3)金属铱与布朗斯特酸协同催化, (4)金属铱与路易斯碱协同催化, (5)金属铱与其他过渡金属协同催化.
有机胺催化是有机小分子催化领域的一类重要分支[64-66], 包括伯胺、仲胺和叔胺催化三种类型, 伯胺和仲胺主要通过与醛或酮类底物形成烯胺或亚胺中间体参与反应, 而叔胺在催化反应时常作为路易斯碱发挥作用.近年来, 化学家们发现有机胺与过渡金属在反应体系中可以共存, 且分别发挥活化作用, 据此报道了一系列过渡金属与有机胺协同催化的不对称反应, 是协同催化领域发展最为迅速的一个研究方向[67-68].
2006年, Córdova课题组[69]报道了首例过渡金属与有机胺协同催化的例子.他们选用钯和吡咯烷作催化剂, 实现了醛、酮分子间羰基α位的烯丙基化反应.在此之后过渡金属与有机胺的协同催化模式得到了迅速的发展.
2013年, Carreira课题组[70]报道了首例金属铱与手性胺协同催化反应, 通过联合使用手性铱催化剂(采用自主设计的磷-烯配体)和金鸡纳碱衍生的伯胺催化剂, 成功实现了支链醛类化合物α位的不对称烯丙基取代反应.在此反应中, 通过改变金属铱催化剂配体和伯胺催化剂的立体构型, 能够以良好的收率、完美的对映选择性及非对映选择性获得目标产物的四种全部立体异构体(Scheme 3).控制实验表明, 金属铱络合物活化支链烯丙醇生成π-烯丙基铱中间体, 伯胺催化剂活化醛生成烯胺活性中间体, 随后两类中间体发生烯丙基取代反应得到目标产物, 并且铱催化剂与伯胺催化剂分别控制产物α和β位的立体构型(Scheme 4).

在上述研究工作的基础上, 该课题组后续利用类似的金属铱与有机胺的协同催化体系, 实现了不同类型醛类化合物的不对称烯丙基取代反应, 并成功应用于多种生物活性分子和药物的不对称合成中. 2014年, Carreira课题组[71]尝试利用相同的催化策略来实现直链烷基醛的α位不对称烯丙基取代反应, 但该反应存在一个不容忽视的挑战:目标产物的一个手性中心含有易于差向异构化的羰基α位氢原子.通过系统性的条件优化, 作者发现脯氨酸衍生的手性仲胺催化剂可以高效地活化直链烷基醛, 实现铱和有机胺协同催化的不对称烯丙基取代反应.同样, 通过改变手性配体和仲胺催化剂的立体构型, 反应也能够以良好的收率、优秀的立体选择性得到目标产物的全部立体异构体.此外, 作者还将该方法应用于抗抑郁药物帕罗西汀的不对称合成中(Scheme 5).

同年, Carreira课题组[72]通过对反应底物的设计, 成功的将该协同催化策略应用于天然产物△9-tetrahydro-cannabinols的不对称合成中.需要指出的是, 通过改变手性催化剂的立体构型, 可以方便的得到该天然产物的所有立体异构体(Scheme 6).

2015年, Carreira课题组[73]再次利用金属铱与手性仲胺协同催化体系实现了α-氨基乙醛和α-羟基乙醛的不对称烯丙基取代反应, 以良好的收率、优秀的立体选择性合成了一系列α位杂原子(氮、氧)取代的γ, δ-不饱和醛类化合物, 并且通过手性配体与仲胺催化剂不同绝对构型的组合能够实现γ, δ-不饱和醛16的全部4种立体异构体发散性合成(Scheme 7).

自然界中存在许多不稳定或者易挥发的亲核试剂, 如肼、羟胺、叔丁基过氧化物及小分子醛类化合物等, 此类化合物由于其不稳定性, 常储存于水溶液中.但过量的水(具有亲核性)会淬灭过渡金属活性中间体[74], 从而限制了此类化合物在有机合成中的应用.因此, 若要实现此类化合物的烯丙基取代反应, 就必须保证水对π-烯丙基金属中间体的亲核加成反应是非动力学优势的或可逆的. 2019年, Carreira课题组[75]利用金属铱与仲胺协同催化体系成功实现了α-氯乙醛、戊二醛等的不对称烯丙基取代反应, 并以良好的收率、优秀的立体选择性合成了一系列烯丙基化产物.近期, 该课题组利用类似的协同催化策略, 实现了乙醛(水溶液)的不对称烯丙基取代反应, 并以此为关键步骤实现了4种倍半萜烯类天然产物heliannuols C (E)和heliespirones A (C)的不对称合成(Scheme 8)[76].

除了以上介绍的Carreira课题组报道的一系列出色研究成果外, 其他课题组在此领域同样有着重要贡献. 2015年, Jørgensen课题组[77]利用过渡金属与手性仲胺协同催化体系, 实现了α, β-不饱和醛γ位的不对称烯丙基取代反应:以支链烯丙醇23为底物, 在金属铱催化下专一性地得到支链烯丙基化产物24, 而以肉桂醇羧酸酯25为底物, 在金属钯作用下则专一性地得到了直链烯丙基化产物26, 两种反应体系均能取得较好的收率和优异的对映选择性(Scheme 9).

杨玉荣课题组以吲哚基支链烯丙醇为原料, 利用醛类化合物α位不对称烯丙基取代反应为关键步骤, 高效地合成了多种多环吲哚生物碱. 2018年, 该课题组[78]以2-吲哚基支链烯丙醇为底物, 在金属铱与手性仲胺协同催化下, 实现了脂肪醛α位不对称烯丙基取代反应, 并高效地合成了具有羧肽酶抑制活性的天然产物(-)-acti- nophyllic acid (Scheme 10). 2019年, 该课题组[79]以3-吲哚基支链烯丙醇为原料, 通过类似的不对称烯丙基取代反应, 实现了天然产物(-)-alstoscholarine的不对称合成(Scheme 11).

2019年, 肖文精、陆良秋等[80]运用金属铱与有机伯胺协同催化策略, 实现了乙烯基氨基醇33与醛或β, γ-不饱和酮的不对称[4+2]环加成反应.醛在伯胺催化下形成的烯胺中间体与金属铱活化的乙烯基氨基醇发生的不对称[4+2]环加成反应, 能以良好的收率、良好至优秀的对映选择性和非对映选择性得到一系列光学纯的二氢喹啉酮(Scheme 12).需要指出的是, 该反应的关键中间体半缩醛36a通过“一锅法”能以良好的收率和优秀的对映选择性转化为多种不同官能团化的手性四氢喹啉衍生物.而β, γ-不饱和酮在手性伯胺A7催化下形成二烯胺中间体与金属铱活化的乙烯基氨基醇发生不对称[4+2]环加成反应, 通过改变铱配体的绝对构型, 能以良好的收率、优秀的立体选择性实现四氢喹啉45的非对映选择性发散合成(Scheme 13).

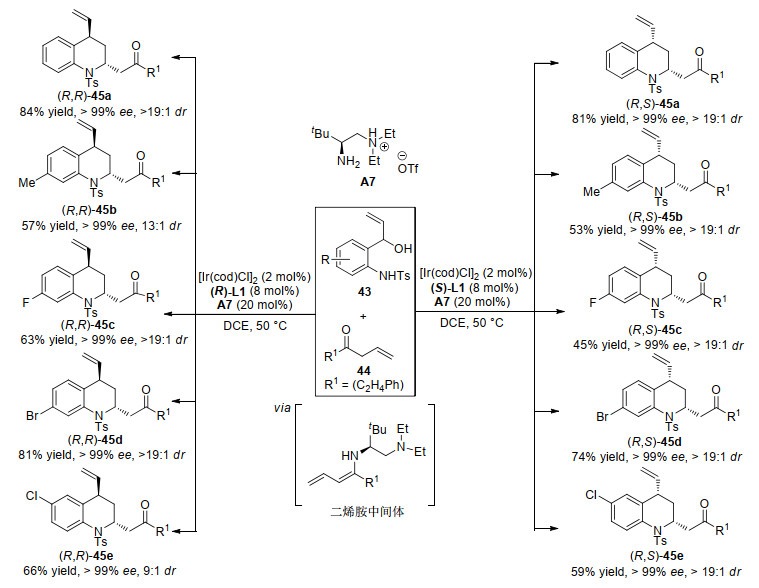
作者认为乙烯基氨基醇与醛的不对称[4+2]环加成反应机理为:首先, 金属铱络合物活化乙烯基氨基醇生成π-烯丙基铱金属中间体46, 与此同时醛与伯胺催化剂结合生成烯胺活性中间体47.中间体46和中间体47之间发生不对称烯丙基取代反应得到中间体48, 然后48经过分子内环化、水解得到半缩醛中间体39, 随后通过一系列的转化得到各种取代的二氢喹啉酮和四氢喹啉化合物(Scheme 14).

与伯胺或仲胺催化剂的活化模式不同, 叔胺催化剂作为一类具有弱亲核性的Lewis碱, 常被应用于活化Morita-Baylis-Hillman (MBH)碳酸酯, 实现不对称官能团化反应[81-83]. 2019年, 陈应春课题组[84]利用金属铱与叔胺协同催化策略, 实现了靛红衍生的MBH碳酸酯与烯丙基碳酸酯的不对称[4+3]环化反应.该反应的实现需要克服以下挑战: (1)亲核性的叔胺可作为过渡金属的配体, 因此叔胺催化剂与过渡金属催化剂存在兼容性问题. (2)两类底物均具有烯丙醇结构模块, 要实现叔胺高化学选择性地活化MBH衍生物, 而过渡金属高化学选择性地活化另外一个底物是具有挑战的. (3)所产生的两类偶极中间体均具有较高的反应活性, 这可能会存在较为严重的背景反应.通过系统性的条件优化, 作者发现叔胺1, 4-二氮杂二环[2.2.2]辛烷(DABCO)与预制备的金属铱/配体络合物组成的协同催化体系, 能够催化目标反应的发生, 以良好的收率、优秀的立体选择性合成了一系列吲哚酮螺氮杂䓬化合物(Scheme 15).

此外, 作者利用类似的叔胺与铱络合物协同催化策略, 成功实现了靛红衍生的MBH碳酸酯与乙烯基氮杂环丙烷的不对称[3+3]环化反应.该反应具有较为广泛的底物适用范围, 各种取代的靛红MBH碳酸酯均能以良好的收率、优异的立体选择性得到吲哚酮螺哌啶衍生物(Scheme 16).

2020年, 陈应春课题组[85]再次利用叔胺与金属铱协同催化策略, 实现了靛红衍生的MBH碳酸酯与烯丙基羧酸酯的不对称1, 3-氧烯丙基化/Cope重排反应, 并以良好的收率、优秀的对映选择性获得一系列含四取代烯烃结构的目标化合物(Scheme 17).此外, 作者通过选择不同类型的手性铱络合物及对底物的微小调整, 实现对目标产物的所有异构体(烯烃E/Z构型改变和中心手性改变)的立体发散性合成(Scheme 18).


通过控制实验, 作者认为可能的机理为:首先, DABCO和手性铱络合物分别活化MBH碳酸酯和烯丙醇酯形成中间体63和64, 接着通过协同催化过程发生γ-区域选择性的烯丙基烷基化反应形成中间体65, 该中间体进一步被苯甲酸根负离子进攻, 进而消除DABCO生成了1, 3-氧烯丙基化产物.在这步反应中产生的非对映选择性导致接下来协同Cope重排过程控制Z-式和E-式两种异构体的生成(Scheme 19).

相转移催化剂(PTC)是指可以帮助反应物从一相转移到能够发生反应的另外一相中, 从而加快异相系统反应速率的一类催化剂, 目前已经广泛应用于多种不同类型的有机反应中[86].
2001年, 龚流柱课题组[87]和Takemoto课题组[88]通过使用非手性钯催化剂与手性相转移催化剂组成的协同催化体系, 实现了甘氨酸酯席夫碱的不对称烯丙基取代反应.这是第一例过渡金属与有机小分子协同催化不对称反应的报道.
相转移催化剂与金属铱协同催化的不对称烯丙基取代反应由Takemoto课题组[89]在2003年报道, 在甘氨酸酯席夫碱与支链烯丙基醋酸酯的反应中, 作者首次尝试了PTC与铱组成的协同催化体系, 虽然反应的对映选择性和非对映选择性均不够理想, 但是这一初步结果也证实了该协同催化模式的可行性(Scheme 20).

2013年, Carreira课题组报道了支链烯丙醇的不对称烯基化[90]与炔基化[91]反应.此反应表现出了很高的区域选择性和立体选择性, 催化剂对水和空气稳定, 易于操作, 适用于芳基或者杂环芳基烯丙醇.相转移催化剂nBu4NHSO4和nBu4NBr的使用有效地解决了烯基三氟硼酸钾和炔基三氟硼酸钾在有机溶剂中溶解性不佳的问题(Scheme 21).

2017年, 韩志勇等[92]利用铱/相转移协同催化体系, 实现了α-亚胺羧酸酯和烯丙基醋酸酯的不对称烯丙基化反应.该反应过程中, 亚胺发生了极性反转(由亲电性变为亲核性), 以优异的产率和立体选择性得到了含有两相邻手性中心的α-季碳氨基酸衍生物, 通过简单的衍生反应可以方便地将产物转化为手性氨基酸盐和含有多个手性中心的α-季碳脯氨酸衍生物(Scheme 22).

作者提出了如下反应机理(Scheme 23):在氢氧化钾和相转移催化剂nBu4NBr作用下, α-亚胺羧酸酯81去质子化得到2-氮杂烯丙基负离子.同时烯丙基醋酸酯与金属铱络合物发生氧化加成得到π-烯丙基铱中间体86. 2-氮杂烯丙基负离子进攻π-烯丙基铱中间体, 完成立体选择性的烯丙基化反应, 得到中间体87, 经水解后得到α-季碳氨基酸酯产物83.在该反应过程中, 立体选择性主要由手性配体控制, 但相转移催化剂在反应中也起到重要作用, 不仅对反应收率有重要影响, 还作为2-氮杂烯丙基负离子的抗衡阳离子影响烯丙基化反应过程, 进而提高反应的立体选择性.

2017年, Hartwig课题组[93]报道了预制备的铱与手性配体的络合物与相转移催化剂协同催化的不对称烯丙基取代反应(Scheme 24).作者用3~4 mol%的nBu4NOBz作相转移催化剂, 实现了肉桂醇羧酸酯与硅烷基烯酮缩醛的取代反应, 以优秀的收率和立体选择性得到了脂肪烃取代的羧酸酯, 产物在温和的条件下就能转化为伯醇、羧酸、酰胺等衍生物.在此反应中, 相转移催化剂的存在至关重要, 缺少相转移催化剂反应将完全无法进行.

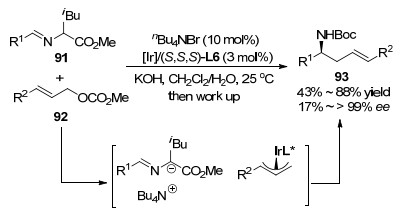
2020年, 王春江课题组[94]在报道了铱/铜协同催化的不对称烯丙基取代与2-氮杂-Cope重排串联反应后, 巧妙地用相转移催化剂nBu4NBr替代传统的铜催化剂, 对α位含大位阻基团的席夫碱活化, 得到了与铱/铜双金属体系相当的反应结果, 合成了一系列手性高烯丙基胺衍生物.该金属铱与相转移协同催化体系相较于双金属催化体系更为绿色环保且操作简单, 但反应对席夫碱底物的邻位取代基仅报道了iBu, 且产物取代基的多样性略逊于双金属体系(Scheme 25).
相转移催化剂的运用, 使得铱催化的不对称烯丙基取代反应得以在非均相体系中顺利进行, 推动了铱催化的不对称烯丙基取代的进一步发展.然而目前相关研究主要集中于非手性的相转移催化剂, 手性相转移催化剂在此体系中的应用并不理想, 两种催化剂间的手性匹配问题仍然有待深入研究.
2004年, Akiyama课题组[95]和Terada课题组[96]分别独立报道了联萘酚骨架的手性磷酸催化的不对称Mannich反应, 由此拉开了手性布朗斯特酸催化不对称反应研究的序幕.通常, 布朗斯特酸催化有机反应是通过质子化作用形成离子对或者氢键, 也可以用作双功能催化剂.手性磷酸作为布朗斯特酸的重要组成部分, 具有种类众多、结构可调节性大等特点.手性磷酸与金属铱协同催化结合了两类催化剂的优点, 为铱催化的不对称烯丙基取代反应提供了新的途径.
2017年, 钟国富课题组[97]利用手性铱络合物与磷酸协同催化体系, 实现了2-萘酚的不对称烯丙基取代去芳构化反应(Scheme 26).该反应以简便易得的支链烯丙醇为底物, 以良好的收率和优秀的对映选择性合成了一系列手性β-萘酮衍生物.控制实验结果表明, 手性铱络合物对反应的立体诱导起到关键作用, 而添加磷酸有助于烯丙醇脱水以降低反应能垒, 并且在立体控制步骤中大位阻的磷酸有助于提高反应的对映选择性.

结合控制实验和文献, 作者提出了如下可能的机理(Scheme 27):烯丙醇底物在手性铱络合物和布朗斯特酸共同作用下, 失去一分子的水, 生成π-烯丙基铱中间体97, 此时布朗斯特酸脱去质子成为相应的共轭碱.此共轭碱与萘酚羟基的氢原子产生氢键作用, 弱化了氢氧键, 活化的萘酚与中间体97络合产生了关键中间体98.随后发生分子内亲核加成得到目标产物, 同时脱去铱络合物和磷酸完成催化循环.在这个催化循环中, 大空间位阻的磷酸与手性铱络合物协同作用显著提高了反应的对映选择性, 而手性与消旋的磷酸在反应中都能得到优异的产率和对映选择性, 使用消旋配体衍生的金属铱络合物与手性磷酸组成的协同催化体系则只能得到消旋的产物.这表明手性铱络合物对产物的对映选择性控制起到决定作用.

路易斯碱是指在反应中能提供电子云的分子或者原子团, 包括有机磷、卡宾、三级胺等, 在催化反应时往往对底物进行亲核进攻生成鏻盐和铵盐、烯醇负离子等活泼中间体.
2003年, Krische课题组[98]报道的三叔丁基膦作为路易斯碱与四三苯基膦钯协同催化的分子内烯丙基取代反应是最早关于路易斯碱与过渡金属协同催化的报道, 但文中并没有涉及到不对称诱导效应的研究.
金属铱与路易斯碱协同催化的不对称烯丙基取代反应于2017年才开始被逐渐报道, 其中已经被证明能与金属铱兼容并实现协同催化的路易斯碱有三类:异硫脲类催化剂、氮杂卡宾催化剂以及在第二部分中介绍过的叔胺催化剂.
异硫脲类催化剂于20世纪初由Birman课题组[99]设计合成, 最初适用于催化醇的不对称动力学拆分反应. 2008年, Romo课题组[100]首次将异硫脲类催化剂应用于烯胺醇负离子中间体的生成中, 实现了酮酸的不对称分子内环化反应, 大大地拓展了这类催化剂的应用范围. 2016年, Snaddon课题组[101]实现了金属钯催化剂与异硫脲催化剂协同催化的芳基乙酸酯的不对称α位苄基化反应, 这是过渡金属与该类催化剂协同催化的首例成功报道.
2017年, Hartwig课题组[102]实现了金属铱与异硫脲催化剂协同催化芳基乙酸酯的不对称支链烯丙基取代反应(Scheme 28).在反应中, 当异硫脲类催化剂(BTM)作为路易斯碱与铱的亚磷酰胺配体络合物协同作用, 可以以优秀的立体选择性得到目标产物, 同时改变两种催化剂的立体构型, 可以成功得到所有立体构型的产物.需要指出的是, 五氟苯基酯与叔丁氧羰基酯都是该反应的关键, 其他的芳基衍生物与离去基团都会显著降低产率和立体选择性.

氮杂环卡宾是一类重要的路易斯碱催化剂, 它催化的有机反应具有操作简单、选择性好、无污染等优点而引起了化学家们的广泛关注.但是由于其较强的亲核性, 可作为配体与过渡金属形成稳定的金属配合物, 容易降低过渡金属催化剂活性, 这一性质在一定程度上阻碍了过渡金属与氮杂环卡宾协同催化反应的发展. 2016年, 游书力课题组[103]报道了氮杂环卡宾作为手性配体在金属铱催化分子内不对称烯丙基取代反应中的应用, 作者通过单晶X射线衍射证实了氮杂环卡宾与金属铱络合物的存在, 并证明了其催化活性.
2019年, Glorius课题组[104]报道了金属铱与氮杂环卡宾协同催化的不对称烯丙基取代环化反应(Scheme 29), 证实了这两种催化剂的兼容性与匹配性.作者从烯丙基环碳酸酯104和106出发, 与肉桂醛衍生物及α-氯代醛以优秀的收率和立体选择性得到了环化产物.但在对其进行发散性合成时, 作者发现原先的体系并不能得到良好的结果, 通过对条件的再次筛选, 作者发现氮杂卡宾催化剂N2更有利于发散合成, 但是对于反式内酯的产物也未能得到与顺式内酯相当的非对映选择性.为了克服这一缺点, 作者利用反式异构体在热力学和动力学上更受青睐的特点, 通过二异丙基氨基锂(LDA)处理顺式产物(R, S)-105a, 成功地得到了具有高非对映选择性的反式内酯(S, S)-105a, 为合成具有高非对映选择性的反式内酯提供了一条替代路线.

作者提出了如下反应机理(Scheme 30), 氮杂环卡宾游离后与α, β-不饱和醛形成反式烯醇中间体, 然后通过β质子化(或者由α氯代醛通过碱介导发生氯化物消除).卡宾上的取代基屏蔽了烯醇化物中间体的背面.在金属的催化循环中, 碳酸乙烯基亚乙酯在金属铱作用下形成了π-烯丙基铱中间体(该中间体可以通过质谱捕获得到), 理论计算表明, 中间体的背面被屏蔽.最后烯醇中间体的Si面与π-烯丙基铱中间体的Si面靠近, 发生分子间烯丙基取代反应, 然后经过内酯化得到产物, 并游离出催化剂完成催化循环.

除了上述介绍的金属铱与有机小分子催化剂协同催化的不对称烯丙基取代反应外, 金属铱与其他过渡金属协同催化的不对称烯丙基取代反应在最近几年也取得了重要进展, 另一种过渡金属的引入大大拓展了亲核试剂的范围, 同时通过调整手性配体的构型也可以实现立体发散性合成.
2016年, 张万斌课题组[105]报道了在温和条件下, 铱/锌双金属协同催化的α-羟基酮的α-烯丙基化反应(Scheme 31).衍生于亚磷酰胺配体的手性铱络合物和手性Zn-Pro苯酚络合物的协同作用, 使反应具有很高的活性和优秀的对映选择性及非对映选择性, 反应的关键中间体就是锌与α-羟基苯乙酮形成的五元环过渡态.该反应通过不同绝对构型配体的组合可以得到四种立体异构体, 实现立体发散性合成.

2017年, 张万斌课题组[106]又采用同样的体系实现了非保护的α-羟基茚酮的α-烯丙基化反应(Scheme 32).该反应克服了α-羟基茚酮反应位点多、立体位阻大和烯醇化困难等困难, 在温和条件下高效、高立体选择性地得到具有连续三取代/四取代手性中心的环状α-羟基茚酮衍生物, 并成功实现了产物立体发散性合成.

2018年, 张万斌课题组[107]开发了可用于立体发散性合成的铱/铜双金属协同催化体系, 并将其用于非天然α-氨基酸的立体选择性合成.该反应底物适用性广泛, 一系列的烯丙基底物和醛亚胺酯都可以顺利用于该催化体系, 取得了良好的收率及优秀的对映选择性和非对映选择性.此外, 反应产物通过简单的转化即可用于天冬氨酸类似物的立体发散性合成, 可作为一种谷氨酸转运体的亚型选择性阻断剂(Scheme 33a).值得注意的是, 几乎同时, 王春江课题组[108]报道了类似的铱/铜协同催化反应(Scheme 33b), 并且也成功地以优秀的收率和立体选择性构建了邻位双取代氨基酸衍生物, 不同构型配体的组合也能以优秀的结果得到四种立体异构体.此外, 两种金属与两种配体同时加入同一容器, 分别配位再混合得到了相当的结果,两种活性催化剂可以通过“金属/配体自分类”方式获得, 简化了操作步骤.

同年, 张万斌课题组[109]利用相似的铱/铜协同催化体系, 进一步实现了酮亚胺酯与烯丙基碳酸酯的不对称取代反应(Scheme 34).该反应进一步拓展了席夫碱的范围, 合成了邻位单取代氨基酸衍生物, 通过简单的后处理即可得到伯胺产物.此外, 配体改变获得四种立体异构体的优势在此报道中也得以体现.

2018年, Hartwig课题组[110]报道了铱/铜协同催化的氮杂芳基乙酰胺的不对称烯丙基取代反应(Scheme 35), 该反应实现了手性含氮杂芳基化合物的立体发散性合成, 且反应具有较高的对映选择性和非对映选择性.各种含吡啶基、苯并噻唑基、苯并噁唑基、吡嗪基、喹啉基和异喹啉基的氮芳基乙酰胺都表现出了良好的活性.

2019年, 王春江课题组[111]应用铱/铜协同催化体系, 从简单易得的醛亚胺酯和吲哚基烯丙基碳酸酯出发构建了含有多手性中心的四氢-γ-咔啉衍生物.在该反应中, 吲哚基烯丙基碳酸酯的烯丙基邻位空间结构较大, 使得传统的Feringa类型配体(L3、L6等)不能取得良好的结果, 而游书力课题组发展的亚磷酰胺配体L13, 由于其与铱配位后在空间上不拥挤, 可以兼容位阻较大的底物[112], 在反应中取得了优秀的立体选择性控制结果.该体系实现了四氢-γ-咔啉衍生物的立体发散合成, 通过改变配体构型可以获得四种光学异构产物.作者通过控制实验证实, 立体发散的烯丙基取代反应和随后发生的立体专一性的iso-Pictet-Spengler环化是反应顺利进行的关键(Scheme 36).

王春江课题组[113]在铱/铜协同催化席夫碱的不对称烯丙基取代反应的研究中发现, 亚甲胺叶立德发生烯丙基化反应后的产物具有特殊的烯丙基-氮杂烯丙基结构, 巧妙地通过增大亚甲胺叶立德α位取代基的位阻, 迫使中间体通过重排反应得到了更稳定的高烯丙胺衍生物, 成功地实现了不对称烯丙基取代反应与2-氮杂-Cope重排的串联反应(Scheme 37), 以优秀的立体选择性和收率获得了一系列1, 3-和1, 4-取代高烯丙胺化合物.
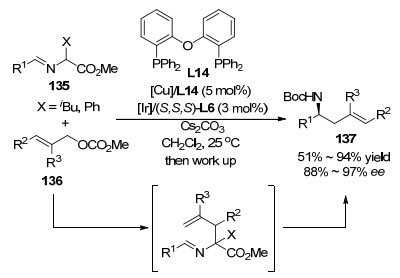
2016年, 钮大文课题组[114]以铱/银协同催化体系实现了合成有机硼类化合物的新策略, 直接向目标分子中立体选择性地引入了“CH2B(OR)2”基团, 这种策略较容易地合成了在硼原子的β位具有手性中心的化合物, 从谐二硼甲烷和烯丙基叔丁氧羰基酯出发合成了一系列支链高烯丙基硼酸酯(Scheme 38).值得指出的是, 针对不同类型底物, 他们使用了不同类型的手性配体, 其中对邻位有大位阻取代基团的底物, 游书力课题组发展的亚磷酰胺配体L13相较于L6也表现出了独特的优势.对于缺电子类型底物, 使用了Carreira课题组发展的磷-烯类型配体.该反应具有较高的收率、较广的底物范围、以及优秀的对映选择性.

综上所述, 近十几年来金属铱与其他催化剂协同催化的不对称烯丙基取代反应已经取得了重要进展, 协同催化策略的引入进一步丰富了该反应中亲核试剂的类型(如醛类化合物、α-羟基酮、氨基酸酯席夫碱等), 实现了多种金属铱单独催化作用下难以实现的不对称烯丙基取代反应.更为重要的是, 通过不同绝对构型的手性催化剂的组合能够实现目标分子所有立体异构体的发散性合成.此外, 该类不对称烯丙基取代反应在△9-te- trahydrocannabinols、(-)-actinophyllic acid、(-)-alsto- scholarine、heliannuols C (E)和heliespirones A (C)等多种天然产物和抗抑郁药物帕罗西汀的不对称合成中展现出其重要应用价值.尽管如此, 除金属铱与有机胺协同催化体系应用较为全面之外, 金属铱与相转移催化剂、布朗斯特酸、路易斯碱或其他过渡金属的协同催化体系仅有零星报道, 因此开发金属铱参与的新型协同催化体系仍然是一个重要的研究方向.此外, 目前的研究主要集中于协同催化体系在单一不对称烯丙基取代反应中的应用, 设计开发基于不对称烯丙基取代的串联反应, 从而实现环状产物所有立体异构体的发散性合成, 也是将来值得研究的课题之一.尤其需要指出的是,该领域研究的进展也将为一些重要的多手性中心活性药物分子的合成及相应的手性药物发现提供新的合成方法和策略.
Trost, B. M.; van Vranken, D. L. Chem. Rev. 1996, 96, 395. doi: 10.1021/cr9409804
Trost, B. M. Chem. Pharm. Bull. 2002, 50, 1. doi: 10.1248/cpb.50.1
Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921 doi: 10.1021/cr020027w
Milhau, L.; Guiry, P. J. Top. Organomet. Chem. 2011, 38, 95.
Takeuchi, R.; Kashio, M. J. Am. Chem. Soc. 1998, 120, 8647. doi: 10.1021/ja981560p
Takeuchi, R.; Ue, N.; Tanabe, K.; Yamashita, K.; Shiga, N. J. Am. Chem. Soc. 2001, 123, 9525. doi: 10.1021/ja0112036
Takeuchi, R. Synlett 2002, 1954.
Belda, O.; Moberg, C. Acc. Chem. Res. 2004, 37, 159. doi: 10.1021/ar030239v
Trost, B. M. Org. Process Res. Dev. 2012, 16, 185. doi: 10.1021/op200294r
Moberg, C. Org. React. 2014, 84, 1.
Trost, B. M.; Hachiya, I. J. Am. Chem. Soc. 1998, 120, 1104. doi: 10.1021/ja973298a
Malkov, A. V.; Gouriou, L.; Lloyd-Jones, G. C.; Starý, I.; Langer, V.; Spoor, P.; Vinader, V.; Kočovský, P. Chem.-Eur. J. 2006, 12, 6910. doi: 10.1002/chem.200501574
Trost, B. M.; Zhang, Y. J. Am. Chem. Soc. 2007, 129, 14548. doi: 10.1021/ja0755717
Trost, B. M.; Zhang, Y. Chem.-Eur. J. 2010, 16, 296. doi: 10.1002/chem.200902770
Trost, B. M.; Zhang, Y. Chem.-Eur. J. 2011, 17, 2916. doi: 10.1002/chem.201002569
Ozkal, E.; Pericas, M. A. Adv. Synth. Catal. 2014, 356, 711. doi: 10.1002/adsc.201300967
Lloyd-Jones, G. C.; Pfaltz, A. Angew. Chem., Int. Ed. Engl. 1995, 34, 462. doi: 10.1002/anie.199504621
Matsushima, Y.; Onitsuka, K.; Kondo, T.; Mitsudo, T.; Takahashi, S. J. Am. Chem. Soc. 2001, 123, 10405. doi: 10.1021/ja016334l
Onitsuka, K.; Matsushima, Y.; Takahashi, S. Organometallics 2005, 24, 6472. doi: 10.1021/om050739n
Onitsuka, K.; Okuda, H.; Sasai, H. Angew. Chem., Int. Ed. 2008, 47, 1454. doi: 10.1002/anie.200704457
Trost, B. M.; Rao, M.; Dieskau, A. P. J. Am. Chem. Soc. 2013, 135, 1869.
Kawatsura, M.; Uchida, K.; Terasaki, S.; Tsuji, H.; Minakawa, M.; Itoh, T. Org. Lett. 2014, 16, 1470. doi: 10.1021/ol5002768
Kanbayashi, N.; Hosoda, K.; Kato, M.; Takii, K.; Okamura, T.; Onitsuka, K. Chem. Commun. 2015, 51, 10895. doi: 10.1039/C5CC02414E
Leahy, D. K.; Evans, P. A. In Modern Rhodium-Catalyzed Organic Reactions, Ed.: Evans, P. A., John Wiley & Sons, Inc., New York, 2005; p. 191.
Evans, P. A.; Nelson, J. D. J. Am. Chem. Soc. 1998, 120, 5581. doi: 10.1021/ja980030q
Hayashi, T.; Okada, A.; Suzuka, T.; Kawatsura, M. Org. Lett. 2003, 5, 1713. doi: 10.1021/ol0343562
Kazmaier, U.; Stolz, D. Angew. Chem., Int. Ed. 2006, 45, 3072. doi: 10.1002/anie.200600100
Sidera, M.; Fletcher, S. P. Nat. Chem. 2015, 7, 935. doi: 10.1038/nchem.2360
Li, C.; Breit, B. Chem.-Eur. J. 2016, 22, 14655. doi: 10.1002/chem.201603532
Parveen, S.; Li, C.; Hassan, A.; Breit, B. Org. Lett. 2017, 19, 2326. doi: 10.1021/acs.orglett.7b00718
Didiuk, M. T.; Morken, J. P.; Hoveyda, A. H. J. Am. Chem. Soc. 1995, 117, 7273. doi: 10.1021/ja00132a039
Chung, K.-G.; Miyake, Y.; Uemura, S. J. Chem. Soc., Perkin Trans. 1 2000, 15.
Kita, Y.; Kavthe, R. D.; Oda, H.; Mashima, K. Angew. Chem., Int. Ed. 2016, 55, 1098. doi: 10.1002/anie.201508757
Langlois, J. B.; Alexakis, A. Organomet. Chem. 2011, 38, 235.
Malda, H.; van Zijl, A. W.; Arnold, L. A.; Feringa, B. L. Org. Lett. 2001, 3, 1169. doi: 10.1021/ol0156289
Van Veldhuizen, J. J.; Campbell, J. E.; Giudici, R. E.; Hoveyda, A. H. J. Am. Chem. Soc. 2005, 127, 6877. doi: 10.1021/ja050179j
Yoshikai, N.; Zhang, S.-L.; Nakamura, E. J. Am. Chem. Soc. 2008, 130, 12862. doi: 10.1021/ja804682r
Selim, K. B.; Matsumoto, Y.; Yamada, K.; Tomioka, K. Angew. Chem., Int. Ed. 2009, 48, 8733. doi: 10.1002/anie.200904676
Langlois, J.-B.; Alexakis, A. Adv. Synth. Catal. 2010, 352, 447. doi: 10.1002/adsc.200900790
Shi, Y.; Jung, B.; Torker, S.; Hoveyda, A. H. J. Am. Chem. Soc. 2015, 137, 8948. doi: 10.1021/jacs.5b05805
You, H.; Rideau, E.; Sidera, M.; Fletcher, S. P. Nature 2015, 517, 351. doi: 10.1038/nature14089
Rideau, E.; You, H.; Sidera, M.; Claridge, T. D.W.; Fletcher, S. P. J. Am. Chem. Soc. 2017, 139, 5614. doi: 10.1021/jacs.7b02440
Tsuji, J.; Takahashi, H.; Morikawa, M. Tetrahedron Lett. 1965, 4387.
Trost, B. M.; Strege, P. E. J. Am. Chem. Soc. 1977, 99, 1649. doi: 10.1021/ja00447a064
Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 396.
Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921. doi: 10.1021/cr020027w
Lu, Z.; Ma, S. Angew. Chem., Int. Ed. 2008, 47, 258. doi: 10.1002/anie.200605113
Takeuchi, R.; Kashio, M. Angew. Chem., Int. Ed. 1997, 36, 263. doi: 10.1002/anie.199702631
Janssen, J. P.; Helmchen, G. Tetrahedron Lett. 1997, 38, 8025. doi: 10.1016/S0040-4039(97)10220-9
Hartwig, J. F.; Stanley, L. M. Acc. Chem. Res. 2010, 43, 1461.
Liu, W.-B.; Xia, J.-B.; You, S.-L. Top. Organomet. Chem. 2011, 38, 155.
Qu, J.; Helmchen, G. Acc. Chem. Res. 2017, 50, 2539. doi: 10.1021/acs.accounts.7b00300
Cheng, Q.; Tu, H.; Zheng, C.; Qu J.; Helmchen, G.; You, S.-L. Chem. Rev. 2019, 119, 1855. doi: 10.1021/acs.chemrev.8b00506
邓颖颍, 杨文, 杨新, 杨定乔, 有机化学, 2017, 37, 3039. doi: 10.6023/cjoc201604029Deng, Y.; Yang, W.; Yang, X.; Yang, D. Chin. J. Org. Chem. 2017, 37, 3039(in Chinese). doi: 10.6023/cjoc201604029
Shao, Z.; Zhang, H. Chem. Soc. Rev. 2009, 38, 2745. doi: 10.1039/b901258n
Zhong, C.; Shi, X. Eur. J. Org. Chem. 2010, 2010, 2999. doi: 10.1002/ejoc.201000004
Zhou, J. Chem. Asian J. 2010, 5, 422. doi: 10.1002/asia.200900458
Allen, A. E.; MacMillan, D. W. C. Chem. Sci. 2012, 3, 633. doi: 10.1039/c2sc00907b
Du, Z.; Shao, Z. Chem. Soc. Rev. 2013, 42, 1337. doi: 10.1039/C2CS35258C
Chen, D.-F.; Han, Z.-Y.; Zhou, X.-L.; Gong, L.-Z. Acc. Chem. Res. 2014, 47, 2365. doi: 10.1021/ar500101a
Inamdar, S. M.; Shinde, V.S.; Patil, N. T. Org. Biomol. Chem. 2015, 13, 8116. doi: 10.1039/C5OB00986C
Afewerki, S.; Córdova, A. Chem. Rev. 2016, 116, 13512. doi: 10.1021/acs.chemrev.6b00226
张毛毛, 骆元元, 陆良秋, 肖文精, 化学学报, 2018, 76, 838.Zhang, M.-M.; Luo, Y.-L.; Lu, L.-Q.; Xiao, W.-J. Acta Chim. Sinica 2018, 76, 838(in Chinese).
Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471. doi: 10.1021/cr0684016
Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Angew. Chem., Int. Ed. 2008, 47, 6138. doi: 10.1002/anie.200705523
Xu, L.-W.; Luo, J.; Lu, Y. Chem. Commun. 2009, 1807.
Gualandi, A.; Mengozzi, L.; Wilson, C. M.; Cozzi, P. G. Chem. Asian J. 2014, 9, 984. doi: 10.1002/asia.201301549
Afewerki, S.; Córdova, A. Chem. Rev. 2016, 116, 13512. doi: 10.1021/acs.chemrev.6b00226
Ibrahem, I.; Córdova, A. Angew. Chem., Int. Ed. 2006, 45, 1952. doi: 10.1002/anie.200504021
Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065. doi: 10.1126/science.1237068
Krautwald, S.; Schafroth, M. A.; Sarlah, D.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 3020. doi: 10.1021/ja5003247
Schafroth, M. A.; Zuccarello, G.; Krautwald, S.; Sarlah, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2014, 53, 13898. doi: 10.1002/anie.201408380
Sandmeier, T.; Krautwald, S.; Zipfel, H. F.; Carreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 14363. doi: 10.1002/anie.201506933
For an example of water as a nucleophile in allylic substitution, see: Lüssem, B. J.; Gais, H.-J. J. Am. Chem. Soc. 2003, 125, 6066.
Sandmeier, T.; Goetzke, F. W.; Krautwald, S.; Carreira, E. M. J. Am. Chem. Soc. 2019, 141, 12212. doi: 10.1021/jacs.9b05830
Sandmeier, T.; Carreira, E. M. Org. Lett. 2020, 22, 1135. doi: 10.1021/acs.orglett.9b04658
Næsborg, L.; Halskov, K. S.; Tur, F.; Mønsted, S. M. N.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2015, 54, 10193. doi: 10.1002/anie.201504749
Liang, X.; Zhang, T.-Y.; Meng, C.-Y.; Li, X.-D.; Wei, K.; Yang, Y.-R. Org. Lett. 2018, 20, 4575. doi: 10.1021/acs.orglett.8b01861
Yao, J.-N.; Liang, X.; Wei, K.; Yang, Y.-R. Org. Lett. 2019, 21, 8485. doi: 10.1021/acs.orglett.9b03319
Zhang, M.-M.; Wang, Y.-N.; Wang, B.-C.; Chen, X.-W.; Lu, L.-Q.; Xiao, W.-J. Nat. Commun. 2019, 10, 2716. doi: 10.1038/s41467-019-10674-3
Liu, T.-Y.; Xie, M.; Chen, Y.-C. Chem. Soc. Rev. 2012, 41, 4101. doi: 10.1039/c2cs35017c
Wei, Y.; Shi, M. Chem. Rev. 2013, 113, 6659. doi: 10.1021/cr300192h
Pellissier, H. Tetrahedron 2017, 73, 2831. doi: 10.1016/j.tet.2017.04.008
Chen, Z.-C.; Chen, Z.; Yang, Z.-H.; Guo, L.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2019, 58, 15021. doi: 10.1002/anie.201907797
Chen, P.; Li, Y.; Chen, Z.-C.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2020, 59, 7083. doi: 10.1002/anie.202000044
Shirakawa, S.; Maruoka, K. Angew. Chem., Int. Ed. 2013, 52, 4312. doi: 10.1002/anie.201206835
Chen, G.; Deng, Y.; Gong, L.-Z; Mi, A.; Cui, X.; Jiang, Y.; Choi, M. C.; Chan, A. S. Tetrahedron: Asymmetry 2001, 21, 1567.
Nakoji, M.; Kanayama, T.; Okino, T.; Takemoto, Y. Org. Lett. 2001, 3, 3329. doi: 10.1021/ol016567h
Kanayama, T.; Yoshida, K.; Miyabe, H.; Kimachi, H.; Takemoto, Y. J. Org. Chem. 2003, 68, 6197. doi: 10.1021/jo034638f
Hamilton, J. Y.; Sarlah, D.; Carreira, E. M. J. Am. Chem. Soc. 2013, 135, 3, 994.
Hamilton, J. Y.; Sarlah, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2013, 52, 7532. doi: 10.1002/anie.201302731
Su, Y.-L; Li, Y.-H.; Chen, Y.-G.; Han, Z.-Y. Chem. Commun. 2017, 53, 1985. doi: 10.1039/C6CC09654A
Jiang, X.; Hartwig, J. F. Angew. Chem., Int. Ed. 2017, 56, 8887. doi: 10.1002/anie.201704354
Wei, L.; Xiao, L.; Wang, Z.-F.; Tao, H.-Y.; Wang, C.-J. Chin. J. Chem. 2020, 38, 82. doi: 10.1002/cjoc.201900391
Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem., Int. Ed. 2004, 43, 1566. doi: 10.1002/anie.200353240
Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356. doi: 10.1021/ja0491533
Shen, D.; Chen, Q.; Yan, P.; Zeng, X.; Zhong, G. Angew. Chem., Int. Ed. 2017, 56, 3242. doi: 10.1002/anie.201609693
Jellerichs, B. G.; Kong, J. R.; Krische, M. J. J. Am. Chem. Soc. 2003, 125, 7758. doi: 10.1021/ja0301469
Birman, V. B.; Ulffman, E. W.; Jiang, H.; Li, X.; Kilbane, C. J. J. Am. Chem. Soc. 2004, 126, 12226. doi: 10.1021/ja0491477
Purohit, V. C.; Matla, A. S.; Romo, D. J. Am. Chem. Soc. 2008, 130, 10478. doi: 10.1021/ja803579z
Schwarz, K. J.; Amos, J. L.; Klein, J. C.; Do, D. T.; Snaddon, T. N. J. Am. Chem. Soc. 2016, 138, 5214. doi: 10.1021/jacs.6b01694
Jiang, X.; Beiger, J. J.; Hartwig, J. F. J. Am. Chem. Soc. 2017, 139, 87. doi: 10.1021/jacs.6b11692
Ye, K.-Y.; Cheng, Q.; Zhuo, C.-X.; Dai, L.-X.; You, S.-L. Angew. Chem., Int. Ed. 2016, 55, 8113. doi: 10.1002/anie.201603266
Singha, S.; Serrano, E.; Mondal, S.; Daniliuc, C. G.; Glorius, F. Nat. Catal. 2020, 3, 48. doi: 10.1038/s41929-019-0387-3
Huo, X.; He, R.; Zhang, X.; Zhang, W. J. Am. Chem. Soc. 2016, 138, 11093. doi: 10.1021/jacs.6b06156
He, R.; Liu, P.; Huo, X.; Zhang, W. Org. Lett. 2017, 19, 5513. doi: 10.1021/acs.orglett.7b02577
Huo, X.; Zhang, J.; Fu, J.; He, R.; Zhang, W. J. Am. Chem. Soc. 2018, 140, 2080. doi: 10.1021/jacs.8b00187
Wei, L.; Zhu, Q.; Xu, S.-M.; Chang, X.; Wang, C.-J. J. Am. Chem. Soc. 2018, 140, 1508. doi: 10.1021/jacs.7b12174
Zhang, J.; Huo, X.; Li, B.; Chen, Z.; Zou, Y.; Sun, Z.; Zhang, W. Adv. Synth. Catal. 2019, 361, 1130. doi: 10.1002/adsc.201801148
Jiang, X.; Boehm, P.; Hartwig, J. F. J. Am. Chem. Soc. 2018, 140, 1239. doi: 10.1021/jacs.7b12824
Xu, S.-M.; Wei, L.; Shen, C.; Xiao, L.; Tao, H.-Y.; Wang, C.-J. Nat. Commun. 2019, 10, 5553. doi: 10.1038/s41467-019-13529-z
Liu, W.-B.; Zheng, C.; Zhuo, C.-X.; Dai, L.-X.; You, S.-L. J. Am. Chem. Soc. 2012, 134, 4812. doi: 10.1021/ja210923k
Wei, L.; Zhu, Q.; Xiao, L.; Tao, H.-Y.; Wang, C.-J. Nat. Commun. 2019, 10, 1594. doi: 10.1038/s41467-019-09563-6
Zhan, M.; Li, R.-Z.; Mou, Z.-D.; Cao, C.-G.; Liu, J.; Chen, Y.-W.; Niu, D. ACS Catal. 2016, 6, 3381. doi: 10.1021/acscatal.6b00719

图式 1 过渡金属催化烯丙基取代反应的区域选择性
Scheme 1 Regioselectivity of transition metal-catalyzed allylic substitution reactions

图式 3 铱/伯胺协同催化:支链醛的不对称α-烯丙基化
Scheme 3 Synergetic Ir/primary amine catalysis: asymmetric α-allylations of branched aldehydes

图式 5 铱/仲胺协同催化:直链醛的不对称α-烯丙基化
Scheme 5 Synergetic Ir/secondary amine catalysis: asymmetric α-allylations of linear aldehydes

图式 6 Δ9-tetrahydrocannabinol所有异构体的立体发散性合成
Scheme 6 Stereodivergent preparation of all stereoisomers of Δ9-tetrahydrocannabinol

图式 7 铱/仲胺协同催化: α-氨基(羟基)乙醛的不对称烯丙基化
Scheme 7 Synergetic Ir/secondary amine catalysis: asymmetric α-allylations of α-amino- and α-hydroxyacetaldehydes

图式 8 铱/仲胺协同催化:小分子醛的不对称烯丙基化
Scheme 8 Synergetic Ir/secondary amine catalysis: asymmetric allylation of small molecule aldehydes

图式 9 铱(钯)/仲胺协同催化: α, β-不饱和醛的γ位不对称烯丙基化
Scheme 9 Synergetic Ir(Pd)/secondary amine catalysis: asymmetric γ-allylations of α, β-unsaturated aldehydes

图式 10 吲哚烯丙基醇的双催化烯丙基化
Scheme 10 Dual catalytic allylation of indole-based allylic alcohol

图式 12 乙烯基氨基醇和醛类化合物的不对称[4+2]环加成反应
Scheme 12 Asymmetric [4+2] cycloadditions of vinyl aminoalcohols and aldehydes

图式 13 乙烯基氨基醇和醛或β, γ-不饱和酮的不对称[4+2]环加成反应
Scheme 13 Asymmetric [4+2] cycloadditions of vinyl aminoalcohols and aldehydes or β, γ-unsaturated ketones

图式 14 铱/胺催化剂协同催化对映选择性合成氢化喹啉化合物的机理
Scheme 14 Proposed mechanism of synergetic iridium/amine catalysis for the enantioconvergent synthesis of hydroquinolines

图式 15 铱/三级胺协同催化不对称[4+3]环化反应
Scheme 15 Synergetic iridium/tertiary amine catalysis for the asymmetric [4+3] annulations

图式 16 铱/三级胺协同催化不对称[3+3]环化反应
Scheme 16 Synergetic iridium/tertiary amine catalysis for the asymmetric [3+3] annulations

图式 17 铱/三级胺协同催化:不对称1, 3-氧烯丙基化/Cope重排串联反应
Scheme 17 Synergetic iridium/tertiary amine catalysis: the cascade 1, 3-oxo-allylation and Cope rearrangement process

图式 18 四取代烯烃的四种立体异构体的发散性合成
Scheme 18 Stereodivergent preparation of all stereoisomers of tetrasubstituted alkenes

图式 19 不对称1, 3-氧烯丙基化/Cope重排串联反应的机理研究
Scheme 19 Mechanism study for the cascade 1, 3-oxo-allylation and Cope rearrangement process

图式 20 铱/相转移协同催化体系: β-取代的α-氨基酸的合成
Scheme 20 Synergetic Ir/PTC catalysis: synthesis of β-substi- tuted α-amino acids

图式 21 铱/相转移协同催化体系:烯丙醇与烯基三氟硼酸钾及炔基三氟硼酸钾的不对称取代反应
Scheme 21 Synergetic Ir/PTC catalysis: enantioselective substitution of allylic alcohols with potassium alkenyltrifluoroborates and alkynyltrifluoroborates

图式 22 铱/相转移协同催化体系: α-亚胺羧酸酯的不对称极性反转烯丙基化
Scheme 22 Synergetic Ir/PTC catalysis: asymmetric umpolung allylation of α-imino ester

图式 23 铱/相转移协同催化α-亚胺羧酸酯的不对称烯丙基取代中可能的机理
Scheme 23 Proposed mechanism of Ir/PTC synergetic catalyzed asymmetric allylation of α-imino ester

图式 24 铱/相转移协同催化体系:硅烷基烯酮缩醛的不对称烯丙基取代
Scheme 24 Synergetic Ir/PTC catalysis: enantioselective allylic substitution of silyl ketene acetals

图式 25 铱/相转移协同催化体系:亚甲胺叶立德参与的烯丙基化/2-氮杂-Cope重排反应
Scheme 25 Synergetic Ir/PTC catalysis: allylic alkylation/ 2-aza-Cope rearrangement of azomethine ylides

图式 26 铱/磷酸协同催化体系:萘酚衍生物和烯丙基醇的不对称烯丙基去芳构反应
Scheme 26 Synergetic Ir/phosphoric catalysis: the asymmetric allylic dearomatization of naphthols with secondary allylic alcohols

图式 27 萘酚衍生物和烯丙基醇的不对称烯丙基去芳构反应机理
Scheme 27 A proposed mechanism of the asymmetric allylic dearomatization reaction of naphthols with secondary allylic alcohols

图式 28 铱/BTM协同催化体系:芳基乙酸酯的不对称烯丙基化
Scheme 28 Synergetic Ir/BTM catalysis: enantioselective allylations of aryl acetic acid esters

图式 29 铱/氮杂环卡宾协同催化体系:不对称[3+2]和[4+2]环加成反应
Scheme 29 Synergetic Ir/NHC catalysis: asymmetric [3+2] and [4+2] cycloadditions

图式 30 协同的铱/氮杂环卡宾催化:不对称[3+2]环加成反应机理
Scheme 30 Synergetic Ir/NHC catalysis: a proposed mechanism of asymmetric [3+2] cycloadditions

图式 31 铱/锌协同催化: α-羟基酮的不对称烯丙基化反应
Scheme 31 Synergetic Ir/Zn catalysis: asymmetric allylation α-allylation of α-hydroxyketones

图式 32 铱/锌协同催化: α-羟基茚酮的不对称烯丙基化反应
Scheme 32 Synergetic Ir/Zn catalysis: asymmetric allylation α-allylation of α-hydroxy indanones

图式 33 铱/铜协同催化:邻位双取代氨基酸的立体发散合成
Scheme 33 Synergetic Ir/Cu catalysis: stereodivergent synthesis of α, α-disubstituted α-amino acids

图式 34 铱/铜协同催化:甘氨酸席夫碱的不对称烯丙基化
Scheme 34 Synergetic Ir/Cu catalysis: asymmetric allylation of the glycine-based ketimine esters

图式 35 铱/铜协同催化:氮杂芳基乙酰胺的不对称烯丙基化
Scheme 35 Synergetic Ir/Cu catalysis: asymmetric allylation of azaaryl acetamides

图式 36 铱/铜协同催化:四氢-γ-咔啉衍生物的合成
Scheme 36 Synergetic Ir/Cu catalysis: synthesis of tetrahydro-γ-carboline derivatives

图式 37 铱/铜协同催化:亚甲胺叶立德参与的烯丙基化/2-氮杂-Cope重排反应
Scheme 37 Synergetic Ir/Cu catalysis:: allylic alkylation/2-aza- Cope rearrangement of azomethine ylides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



 下载:
下载:




 下载:
下载: