
图式 1.
可见光促进的过渡金属催化的有机反应
Scheme 1.
Organic reaction by visible-light-driven transition- metal catalysis

钯催化的有机转化是构建碳-碳键和碳-杂键最有效的方法之一.由于具有反应选择性好、底物适用范围广、官能团兼容性好以及反应条件温和等优点, 钯催化的有机转化已被广泛应用于合成化学、材料科学和生物医药等领域[1].传统的钯化学主要涉及双电子转移过程, 包括Pd0, PdII, PdIV等偶数变价的催化循环.最近, 人们发现钯催化剂也可以参与到诸多单电子转移反应之中, 越来越多的涉及PdI, PdIII等奇数变价的钯催化循环被发现, 扩展了钯催化有机反应的应用范围[2].
可见光催化以可见光为能量来源, 通过产生高活性的反应中间体, 实现了许多常规热反应不能实现的化学转化[3].近年来, 可见光催化领域的蓬勃发展, 也给传统金属有机化学带来了新的契机.通过结合可见光催化与传统过渡金属催化, 可以实现许多新颖的化学转化.目前这一领域发展了两种典型的模式: (1)可见光诱导过渡金属催化体系[4].该催化模式充分利用钯络合物能够吸收部分可见光的性质, 使其不仅作为传统催化剂直接与反应底物作用, 还作为光敏剂发生敏化作用(Scheme 1, a). (2)可见光与过渡金属协同催化体系[5].该策略通过可见光催化与传统过渡金属催化的有机地结合(通过电子转移或能量转移来活化有机分子, 并通过光敏剂改变过渡金属催化剂的价态), 使之具备单一光催化或过渡金属催化不具备的催化活性(Scheme 1, b).
鉴于钯催化的重要性, 近年来可见光与钯协同催化引起了众多化学家们的关注.本文主要介绍近年来可见光与钯协同催化在有机合成领域应用的研究进展, 将从反应类型的角度对可见光与钯协同催化的化学转化进行分类总结, 主要包括芳基化、烷基化、羰基化、酰基化、羧基化、以及碳杂键构建等反应.
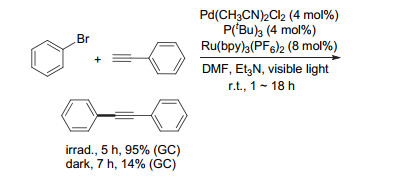
2007年初, Akita等[6]通过结合可见光与钯催化, 实现了光敏剂代替铜盐的Sonogashira反应(Scheme 2).实验表明, 在可见光照射条件下, 溴苯与苯乙炔的反应能在5 h内以95%的产率得到相应的二苯乙炔; 而暗反应条件下, 7 h后的产率仅为14%.基于此, 作者认为光催化剂和光照对该反应有至关重要的作用.遗憾的是当时并不清楚光催化剂在催化循环中的具体作用, 但由于能够检测到电子给体三乙胺的降解产物, 作者认为光敏剂在该催化循环应该具有不可忽视的作用.尽管作者在文中提到可能是经由能量转移的机制, 但随着近几年可见光/金属协同催化的发展, 该反应经由电子转移的可能性更大.
2011年, Sanford等[7a]通过结合可见光与钯催化, 实现了温和条件下导向基团(directed group, DG)导向的芳基C—H键芳基化反应(Scheme 3).相比于作者之前在高温(100 ℃)下实现的氧化性的二芳碘鎓盐参与的C(sp2)—H键的芳基化反应[7b], 在新发展的协同催化体系中, 光敏剂的使用避免了强氧化性前体的使用, 同时使得反应能够在室温条件下得以进行.新方法显示出较好的普适性, 吡啶、酰胺、吡唑、嘧啶和肟醚, 甚至是游离的肟, 都可以作为导向基引入产物之中.作者认为该转化涉及到RuII/*RuII/RuIII和PdII/PdIII/PdIV的循环:首先, 激发的*RuII催化剂还原芳基重氮盐以产生芳基自由基并得到氧化态的RuIII; 该芳基自由基被经C—H活化产生的环钯物种捕获得到PdIII中间体, 氧化态的RuIII经单电子转移(single electron transfer, SET)将该PdIII中间体氧化成PdIV中间体, 并再生RuII完成光敏剂的催化循环; 最后, PdIV中间体经还原消除得目标产物, 并再生PdII催化剂完成钯催化循环.文章首次明确光敏剂在该类反应之中起到了双重作用:充当还原剂促使产生芳基自由基; 充当氧化剂促使高价钯中间体的生成.该模式为后续研究提供了有力的借鉴, 在此之后, 一系列报道随之出现.

随后, Sanford等[8]将该可见光与钯协同催化体系扩展到了以二芳基碘鎓盐作为自由基源的导向C(sp2)—H芳基化之中(Scheme 4).与之前的报道相似, 吡啶、肟醚以及各类酰胺等都可以作为导向基, 以较好的收率得到相应的邻位芳基化产物.此外, 该自由基型芳基化反应还具有底物范围广和官能团耐受性好等特点.
2017年, 郭海明等[9]利用这一催化体系, 以嘌呤作为导向基团, 成功实现了6-芳基嘌呤核苷类化合物中C(sp2)—H键的选择性单芳基化(Scheme 5).该反应官能团兼容性好, 反应条件温和, 室温条件下, 家用蓝光LED灯照射反应2~4 h, 即能以较高的收率得到具有潜在药用价值的嘌呤核苷类衍生物.
同年, Spencer等[10]将这一光与钯协同催化体系扩展到苯二氮䓬的C(sp2)—H键芳基化之中, 高产率地合成一系列苯二氮䓬类衍生物(Scheme 6).作者通过密度泛函理论(DFT)计算比较了传统单一钯催化途径与Sanford等所提出的光钯协同催化途径, 发现在协同催化机制之中, 光敏剂的存在会使反应倾向于以能垒更低的SET的形式进行, 这对PdIII和PdIV中间体的形成十分有利, 进而加速反应的进行.
此外, 有机光敏剂也被用来代替昂贵的钌、铱金属配合物. 2017年, 许华建等[11]使用廉价易得的9, 10-二氢吖啶类有机光敏剂(AcrH2), 实现了钯催化芳基重氮盐参与的酰胺导向的C(sp2)—H键芳基化(Scheme 7, a).反应官能团兼容性好, 卤素、酯基、酰胺、醚、酰基等官能团都能够兼容.初步实验结果表明该反应机理与Sanford提出的机理类似.同年, Balaraman等[12]使用有机光敏剂Eosin Y也实现了类似的转化(Scheme 7, b).
2019年, 王磊等[13]利用可见光与钯协同催化体系, 在氧气气氛下实现了芳炔与苯甲酰胺的C(sp2)—H键芳基化环化反应(Scheme 8).机理实验表明氧气在该双催化体系中起着至关重要的作用.该反应的历程为:首先是PdII与底物中的氮原子配位, 经历C—H键活化得到五元环钯物种, 再与原位产生的苯炔物种经配位插入、还原消除得到目标产物, 同时激发态的光敏剂氧化Pd0生成PdII物种, 分子氧氧化还原态的光敏剂完成催化循环.此外, 该反应中不能排除由分子氧生成的超氧自由基负离子氧化Pd0到PdII的过程.

2016年, 黄学良等[14]在室温和无外加配体的条件下实现了光与钯协同催化碘代芳烃的Heck反应(Scheme 9).该反应对丙烯酸酯有较好的立体选择性, 选择性地合成单一的E-肉桂酸酯类化合物; 当反应底物为苯乙烯时, 主要得到Z-二苯乙烯.作者发现延长反应时间能够提高Z-型产物的比例.此外, 光敏剂的存在对Z-型二苯乙烯的生成具有及其关键的作用, 标准条件下, 市售的E-二苯乙烯能够以96%的收率转化为Z-二苯乙烯, 说明该体系中可能也存在能量转移过程, 需要机理研究进一步确证.这种两步一锅多米诺反应为合成1, 2-双取代Z-烯烃提供了一种高效的方法.

相比C(sp2)—H键, C(sp3)—H具有更高的键能, 其参与的化学转化在反应性和选择性方面存在更高的挑战. 2017年, Polyzos等[15]通过借助5-氯-8-氨基喹啉的强配位作用, 将可见光与钯协同催化体系拓展到了非活化C(sp3)—H键的芳基化(Scheme 10).该反应使用芳基重氮盐为芳基化试剂, 通过利用位阻效应对β-甲基C(sp3)—H键进行选择性识别, 实现了一系列含α-季碳中心的脂肪族酰胺类化合物的芳基化.

2015年, 肖文精和陆良秋等[16]通过结合可见光催化与钯催化, 实现了氧化还原中性条件下α-氨基C(sp3)—H键的烯丙基化反应(Scheme 11).该反应操作简单, 底物适用范围广, 不同的烯丙基醇衍生物, 包括烯丙基乙酸酯、碳酸酯、磷酸二乙酯, 甚至游离烯丙基醇均可以高效地转化为相应的目标产物.机理研究表明激发态光敏剂将胺类化合物氧化为氨基阳离子自由基, 经去质子化得到α-氨基烷基自由基.同时, 钯催化循环中产生的π-烯丙基钯中间体被还原态光敏剂还原为烯丙基自由基.最后, 经α-氨基烷基自由基与烯丙基自由基的自由基偶联得到相应的目标产物.

2014年, Tunge等[17]将可见光催化与钯催化相结合, 实现了自由基脱羧烯丙基化反应(Scheme 12).该反应底物适用性好, 一系列α-氨基脂肪羧酸烯丙基酯或苯乙酸烯丙基酯均可以在可见光氧化条件下实现自由基型脱羧, 进而在温和条件下实现向烯丙基化产物的转化.值得一提的是, 含氨基的游离苯乙酸类化合物在类似条件下也可实现烯丙基化反应.

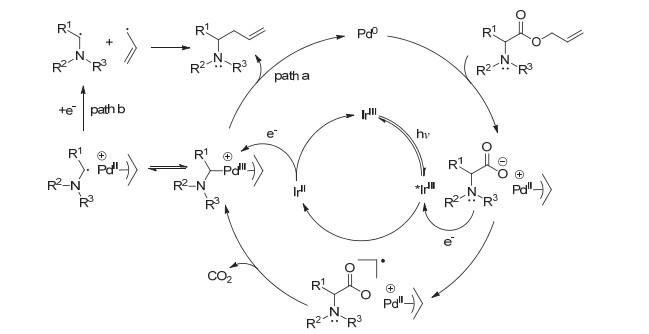
随后, 该课题组[18]对α-氨基脂肪羧酸烯丙基酯的脱羧烯丙基化反应进行了进一步研究, 并提出以下可能的反应机理(Scheme 13):首先, 零价钯与烯丙酯氧化加成生成π-烯丙基钯(II)阳离子和相应的α-氨基酸阴离子; 随后, 该α-氨基酸阴离子被激发态的光敏剂氧化, 并引发自由基脱羧, 生成相应的α-氨基烷基自由基.作者认为稳定性差的烷基自由基会被π-烯丙基钯(II)捕获生成相应的高价PdIII中间体, 随后经光诱导的单电子还原以及还原消除得到相应的烯丙基化产物(path a); 而相对稳定的烷基自由基则会通过自由基偶联的方式得到相应的烯丙基化产物(path b).
众所周知, 自由基参与的有机化学转化通常较难控制其対映选择性.最近, 俞寿云等[19]利用光与钯协同催化策略, 以4-烷基-1, 4-二氢吡啶为烷基源, 首次实现了温和条件下自由基型不对称烯丙基化反应(Scheme 14).通过在钯催化体系中引入轴手性膦配体, 顺利地实现了非对称取代的乙酸烯丙基酯的动态动力学拆分, 以较高的对映选择性、区域选择性和Z/E选择性获得不对称烷基化产物.该反应具有较好的底物适用性和官能团兼容性, 卤素、烷氧基、酯基、酰胺和杂芳环等均能在反应中兼容.作者认为反应由激发态光敏剂氧化4-烷基- 1, 4-二氢吡啶启动, 生成相应的烷基自由基; 而后, 该烷基自由基被烯丙基钯中间体捕获生成PdIII中间体, 经还原消除得到烷基化产物, 生成的PdI中间体被还原态光敏剂还原, 完成催化循环.

2020年, 俞寿云等[20]利用类似的策略实现了N-甲基苯胺的对映选择性α-烯丙基化反应(Scheme 15).不同于之前使用的活化的烷基亲核试剂, 该工作直接以易得的未活化的N-甲基苯胺为烷基亲核试剂, 并且乙酸是唯一的副产物, 体现了步骤及原子经济性.

2018年, 那日松与尚睿等[21]通过结合可见光催化与钯催化, 在温和条件下实现了二级和三级烷基脂肪羧酸与苯乙烯衍生物的脱羧Heck型反应(Scheme 16).该反应通过巧妙地结合传统金属有机化学中金属氢化合物的迁移插入与还原消除, 以及可见光催化中的能量转移和单电子转移, 高选择性地合成一系列Z-型β-烷基化苯乙烯.一系列烷基脂肪羧酸, 包括天然和非天然氨基酸均能顺利地参与到反应之中.同时, 利用过量的烯烃作为氢受体, 该反应避免了传统氧化剂的使用.

随着可见光/钯协同催化体系在不对称烯丙基化转化方面的发展, 一些研究人员也将关注点转向不对称烷基化相关的有机化学反应.最近, 俞寿云等[22]在前期工作的基础上发展了光/钯协同催化外消旋仲碳酸酯与4-烷基-1, 4-二氢吡啶的对映选择性烷基化反应(Scheme 17).该工作提供了一种由外消旋二芳基甲基碳酸酯经动态动力学不对称转化(dynamic kinetic asymmetric transformation, DYKAT)高效制备具有光学活性的二芳基烷烃的方法, 具有重要应用价值.

分子内氧化羰基化反应是构建杂环分子的有效途径. 2016年, 雷爱文等[23]结合可见光与钯协同催化, 以CO为羰基源, 通过烯酰胺的氧化羰基化反应高产率地合成了一系列1, 3-噁嗪-6-酮氮杂环衍生物(Scheme 18).该方法通过可见光催化策略, 使用氧气作为直接的氧化剂, 从而避免了传统的化学计量的高价金属盐氧化剂的使用, 为合成1, 3-噁嗪-6-酮类化合物提供了一种温和环保的方法.机理方面, 作者认为反应经由以下反应历程:在酰基的辅助作用下, 高活性的烯基C—H与二价钯作用形成相应的烯基钯中间体, 经CO的配位插入生成相应的酰基钯物种, 并在1, 4-二氮杂二环[2.2.2]辛烷(DABCO)的作用下发生异构化, 再经还原消除得到相应的目标产物和Pd0.与此同时, 生成的Pd0在氧气参与的光催化循环中被氧化为PdII参与下次循环.

2015年, 李品华与王磊等[24]以有机染料Eosin Y为光敏剂, 通过结合钯催化, 实现了N-乙酰苯胺邻位的脱羧酰基化反应(Scheme 19).该反应具有良好的底物适用性和官能团兼容性, 无论是芳香族的α-羰基羧酸, 还是脂肪族的α-羰基羧酸, 都能在温和条件下转化为相应的邻乙酰基乙酰苯胺.与传统单一过渡金属催化相比, 利用氧气作为最终氧化剂, 为该类反应提供了一条更为绿色的通道.

根据控制实验结果, 他们提出了以下反应机理:首先, α-羰基羧酸与激发态的光敏剂经还原淬灭产生苯甲酰自由基和还原态的光敏剂; 接下来, 氧分子将还原态的光敏剂氧化再生, 完成光催化循环, 同时生成氧化性更强的超氧负离子自由基(O2·-).与此同时, 酰胺导向钯催化C—H键活化产生的环钯物种捕获苯甲酰基自由基生成三价的环钯物种PdIII; 紧接着, 该环钯中间体被O2·-单电子氧化为更高价的四价钯中间体PdIV, 最后经还原消除得相应的目标产物, 并再生PdII完成钯催化循环.作者通过实验证实了上述提到的苯甲酰基自由基和超氧负离子自由基的存在, 进一步佐证了反应经由该历程的可能性.
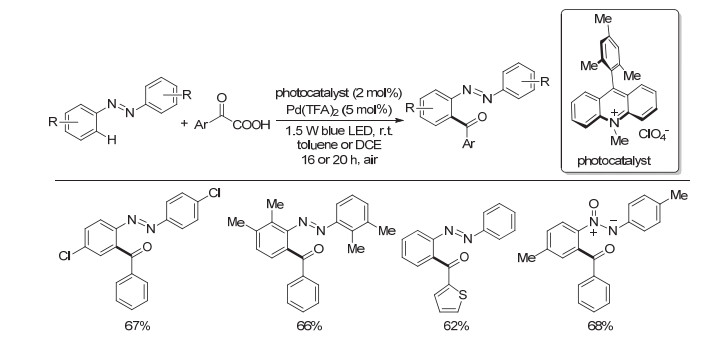
随后, 王磊等[25]将该催化体系拓展到偶氮苯及氧化偶氮苯的脱羧酰化反应之中(Scheme 20).通过结合有机光敏剂与钯催化剂, 一系列α-羰基羧酸均可顺利参与到反应之中, 实现与对称的偶氮苯和氧化偶氮苯向芳基酮的转化.该体系可以直接利用空气中的氧气为氧化剂, 与之前需氧气氛围相比, 进一步增加了反应的安全性, 还使反应变得更加绿色环保.
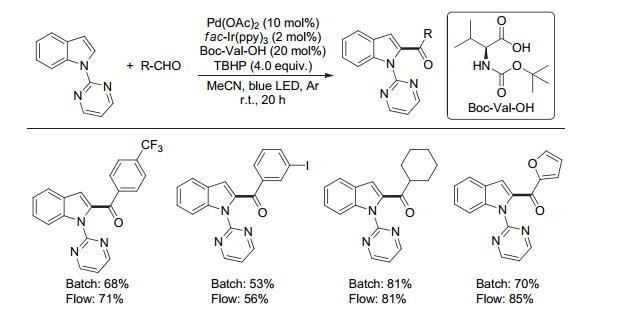
2017年, Van der Eycken等[26]以嘧啶为导向基, 通过结合钯催化导向C—H键活化与可见光催化, 以醛类化合物为酰基化试剂, 高产率地实现吲哚2位酰基化反应(Scheme 21).该反应具有较好的底物适用性, 一系列醛类化合物, 包括芳香醛、一级和二级脂肪醛都能顺利地参与到反应之中, 高产率地得到相应的目标产物.此外, 作者通过对比发现:使用连续流技术时, 反应时间可以缩短到常规方法的十分之一, 并且相应目标产物产率有一定的提升.同年, Jana等[27]使用钌光敏剂也报道了一个类似的转化.
2015年, 尚睿和傅尧等[28]利用可见光与钯协同催化体系实现了芳基卤代物与α-羰基羧酸的脱羧偶联反应(Scheme 22).该方法具有较好的底物适用性, 能够以较高的产率合成各类二芳酮、杂芳酮以及芳/杂芳酰胺类化合物.在DFT计算的基础上, 作者提出以下反应机理:首先, 激发态的光敏剂与α-羰基羧酸盐发生单电子转移生成还原态的光催化剂和相应的α-羰基羧基自由基, 该自由基随即脱二氧化碳生成稳定的酰基自由基; 酰基自由基进而被芳基卤代物与零价钯氧化加成生成的二价钯中间体捕获, 生成三价钯中间体; 该三价钯中间体被还原态的光敏剂还原为二价钯中间体, 同时再生光敏剂完成光催化循环; 最后, 二价钯中间体还原消除得相应的目标产物, 同时再生零价钯完成钯催化循环.

2018年, 尚睿和傅尧等[29]成功地将该催化体系应用于芳基卤代物与乙醛酸的脱羧甲酰化反应之中(Scheme 23).该方法以乙醛酸为酰基源, 有机染料2, 4, 5, 6-四(9-咔唑基)间苯二腈(4CzIPN)为光敏剂, 为芳香醛或杂芳香醛的制备提供了一种简单实用的方法.

作为二氧化碳利用的有效途径之一, 卤代烃的脱卤羧基化反应是合成羧酸的有效途径, 可用于各类羧酸的合成.这类反应主要依赖于过渡金属催化剂对C—X键的氧化加成以形成亲核性有机金属中间体, 同时这类反应中往往需要用到大量的金属还原剂.然而这些策略并不符合绿色化学发展的理念, 为避免化学计量金属还原剂的使用, 2017年, Iwasawa等[30]利用可见光与钯协同催化体系, 成功实现了溴代芳烃以及氯代芳烃与CO2的羧基化反应, 高产率地实现一系列芳基羧酸的合成(Scheme 24).相比以往的反应, 该体系以二异丙基乙基胺(DIPEA)为电子给体, 避免了化学计量的金属还原剂的使用, 使得反应变得更加绿色环保.作者认为反应可能经历以下历程:首先, 卤代芳烃与零价钯的氧化加成启动整个催化循环.接下来, 生成的ArPdIIXL可能会经历以下两种途径进行反应: (1) CO2插入到C—Pd键之中, 生成O—Pd键中间体(ArCOO)PdIIXL; 随后, 在可见光催化下, 该中间体经单电子还原生成一价钯中间体(ArCOO)PdIL (path I). (2) CO2先与ArPdIIXL发生配位作用, 生成ArPdIIX(CO2)L; 而后, 同样在可见光催化下, 经单电子还原生成一价钯中间体ArPdI(CO2)L, 作者认为该中间体更容易发生CO2的插入, 生成一价的O—Pd键中间体(ArCOO)PdIL (path II).最后, 该一价钯中间体经进一步的单电子还原得到相应的目标产物, 同时再生零价钯催化剂完成催化循环.

2019年, 几乎在同一时间, Iwasawa课题组[31]和Jana课题组[32]分别将该协同催化体系拓展到拟卤素的羧基化之中, 实现了芳基、烯基三氟甲磺酸酯与CO2的还原羧基化反应.
相对碳溴键与碳氯键而言, 碳氟键键能较高, 通常难于发生氧化加成, 因此脱氟羧基化是一个很大的难题.最近, 冯超等[33]通过可见光与钯协同催化, 实现了偕二氟烯烃与CO2的脱氟羧基化反应, 高产率地得到了一系列α-氟代丙烯酸类化合物(Scheme 25).该方法具有较好的底物适用范围和官能团兼容性, 甲氧基、三氟甲氧基、三氟甲基、酰胺、砜基、酯基和氰基等都能够兼容.在控制实验的基础上, 作者提出以下反应机理:偕二氟烯烃被还原态光催化剂单电子还原产生氟烯基自由基, 其被零价钯催化剂捕获生成氟烯基钯中间体; 经二氧化碳配位、碳氧双键插入得到羧基钯中间体; 最后, 羧基钯中间体经光催化剂介导的单电子还原得到羧基化产物, 同时再生钯催化剂完成催化循环.与此同时, 作者认为动力学控制为主的E式氟烯基钯中间体导致了较高比例的Z式产物.

除了碳碳键构建之外, 也可以通过可见光与钯协同催化体系实现碳杂原子键的高效构建.
咔唑类化合物是众多天然产物或药物中间体的重要组成部分, 一直以来, 化学家们对其合成具有浓厚的兴趣. 2015年, Choi等[34]成功地应用可见光与钯协同催化体系实现了C(sp2)—H键的胺化反应, 通过N-取代联苯的分子内胺化反应, 实现了咔唑类化合物的高效合成(Scheme 26).根据光化学和电化学研究结果, 作者认为环钯物种与激发态光敏剂之间的单电子转移是整个反应发生的关键; 同时, 氧分子在光催化循环以及钯催化循环中均起到重要的作用.

2018年, Singh等[35]以吡啶或苯并噻唑为导向基, 实现了2-芳基吡啶及2-芳基苯并噻唑的羟基化(Scheme 27).该反应以4CzIPN为光敏剂, Pd(OAc)2为催化剂, 在氧气气氛下成功合成一系列具有特殊发光性能的功能分子.与前人报道的需氧氧化不同, 作者认为氧分子仅仅是起到提供氧源的作用, 而没有参与到光催化循环之中:该体系以三氯溴甲烷(BrCCl3)为氧化剂与激发态光敏剂作用产生氧化态的光催化剂, 同时生成的三氯甲基自由基(·CCl3)与甲苯或环己烷经氢原子转移(HAT)过程产生相应的苄基自由基或环己基自由基, 该类自由基被氧分子捕获产生相应的过氧自由基; 最后, 由过氧自由基释放出的羟基自由基参与到PdII/PdIII/PdIV循环之中完成C—O键的构建.

与传统的交叉偶联反应相比, 两个碳氢键的交叉脱氢偶联反应具有较高的原子经济性. 2014年, Rueping等[36]将可见光与钯协同催化体系应用到了交叉脱氢偶联反应中, 通过释放氢气, 实现了N-芳基烯胺的分子内偶联, 高效地合成一系列吲哚衍生物(Scheme 28).与其他需氧氧化相似, 氧分子在该反应中对光敏剂的再生及钯催化循环均起到了至关重要的作用.

2017年, Kanai等[37]实现了四异氢喹啉及二氢吲哚的脱氢氧化, 通过结合可见光催化与钯催化, 利用氢气的释放以及芳构化的动力, 成功实现了四氢异喹啉及二氢吲哚的脱氢芳构化(Scheme 29).对于更具挑战的四氢萘类型底物, 也可以通过三元催化体系(即在可见光与钯协同催化体系中引入有机硫醇作HAT催化剂), 成功实现脱氢芳构化.作者认为杂环底物之所以能够顺利发生脱氢, 是因为激发态的光敏剂很容易与胺类化合物通过单电子氧化生成相应的氨基自由基, 进而参与到钯催化循环之中; 对于四氢萘的反应而言, 可见光与钯协同催化体系不能促使其产生相应的苄基自由基, 只有加入硫醇作为HAT催化剂, 才能促使苄基自由基的生成以完成后续转换.

综上所述, 可见光与钯协同催化在有机转化中受到了广泛关注, 近年来已经取得了重要进展.通过引入可见光催化, 可以在更加温和的条件下实现以往需要苛刻条件的钯催化有机化学转化, 使反应具有更好的底物适用范围和官能团兼容性.同时, 催化量的光敏剂在光催化循环中既可以得失电子, 也可以提供能量, 促进反应进行, 从而既提升了反应安全性、操控性, 也使得反应变得更加绿色环保.
尽管如此, 该协同催化体系依然面临着一些问题: (1)反应类型较集中, 目前较多的工作集中在sp2杂化碳的相关转化, 涉及sp杂化以及sp3杂化碳的转化较少; (2)成键类型单一, 该体系现阶段的运用集中在碳-碳键的构建, 碳-杂键的构建有待加强[38]; (3)该体系在对映选择性合成中的应用目前只有个例, 需要进一步研究; (4)该反应在天然产物和药物分子等复杂分子合成中的应用尚待开发.
可见光与钯协同催化体系在有机合成中已展现出其独特的魅力, 具有十分广阔的发展前景.随着该领域的继续蓬勃发展, 相信在不久的将来, 可见光与钯协同催化的反应类型必将更为丰富, 产品结构更为多样化, 过程更加绿色环保, 在有机合成、材料科学和医药工业等领域具有更加广泛的应用.
For select reviews: (a) Torborg, C.; Beller, M. Adv. Synth. Catal. 2009, 351, 3027.
(b) Knappke, C. E. I.; Jacobi von Wangelin, A. Chem. Soc. Rev. 2011, 40, 4948.
(c) Enthaler, S.; Company, A. Chem. Soc. Rev. 2011, 40, 4912.
(d) Kambe, N.; Iwasaki, T.; Terao, J. Chem. Soc. Rev. 2011, 40, 4937.
(e) Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Org. Chem. Front. 2015, 2, 1107.
For select reviews: (a) Liu, Q.; Dong, X.; Li, J.; Xiao, J.; Dong, Y.; Liu, H. ACS Catal. 2015, 5, 6111.
(b) Zhou, W.-J.; Zhang, Y.-H.; Cao, G.-M.; Liu, H.-D.; Yu, D.-G. Chin. J. Org. Chem. 2017, 37, 1322 (in Chinese).
(周文俊, 张逸寒, 曹光梅, 刘惠东, 余达刚, 有机化学, 2017, 37, 1322.)
For select reviews: (a) Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785.
(b) Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527.
(c) Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102.
(d) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322,
(e) Tan, F.; Xiao, W. Acta Chim. Sinica 2015, 73, 85 (in Chinese).
(谭芬, 肖文精, 化学学报, 2015, 73, 85.)
(f) Xuan, J.; Zhang, Z.-G.; Xiao, W.-J. Angew. Chem., Int. Ed. 2015, 54, 15632.
(g) Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075.
(h) Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044.
(i) Gui, Y.-Y.; Zhou, W.-J.; Ye, J.-H.; Yu, D.-G. ChemSusChem 2017, 10, 1337.
(j) Cheng, X.; Hu, X.; Lu, Z. Chin. J. Org. Chem. 2017, 37, 251 (in Chinese).
(程骁恺, 胡新根, 陆展, 有机化学, 2017, 37, 251.)
(k) Ding, K.; Xiao, W.; Wu, L. Acta Chim. Sinica 2017, 75, 5 (in Chinese).
(丁奎岭, 肖文精, 吴骊珠, 化学学报, 2017, 75, 5.)
(l) Pei, P.; Zhang, F.; Yi, H.; Lei, A. Acta Chim. Sinica 2017, 75, 15 (in Chinese).
(裴朋昆, 张凡, 易红, 雷爱文, 化学学报, 2017, 75, 15.)
(m) Wang, D.; Zhang, L.; Luo, S. Acta Chim. Sinica 2017, 75, 22 (in Chinese).
(王德红, 张龙, 罗三中, 化学学报, 2017, 75, 22.)
(n) Zhong, J.; Meng, Q.; Chen, B.; Tung, C.; Wu, L. Acta Chim. Sinica 2017, 75, 34 (in Chinese).
(钟建基, 孟庆元, 陈彬, 佟振合, 吴骊珠, 化学学报, 2017, 75, 34.)
(o) Zhang, J.; Chen, Y. Acta Chim. Sinica 2017, 75, 41 (in Chinese).
(张晶, 陈以昀, 化学学报, 2017, 75, 41.)
(p) Ye, H.; Xiao, C.; Lu, L. Chin. J. Org. Chem. 2018, 38, 1897 (in Chinese).
(叶辉, 肖聪, 陆良秋, 有机化学, 2018, 38, 1897.)
(q) Ruan, L.; Chen, C.; Zhang, X.; Sun, J. Chin. J. Org. Chem. 2018, 38, 3155 (in Chinese).
(阮利衡, 陈春欣, 张晓欣, 孙京, 有机化学, 2018, 38, 3155.)
(r) Wang, H.; Wu, P.; Zhao, X.; Zeng, J.; Wan, Q. Acta Chim. Sinica 2019, 77, 231 (in Chinese).
(王浩, 吴品儒, 赵祥, 曾静, 万谦, 化学学报, 2019, 77, 231.)
(s) Cai, B.-G.; Xuan, J.; Xiao, W.-J. Sci. Bull. 2019, 64, 337.
(t) Kong, Y.; Xu, W.; Ye, F.; Weng, J. Chin. J. Org. Chem. 2019, 39, 3065 (in Chinese).
(孔瑶蕾, 徐雯秀, 叶飞霞翁建全, 有机化学, 2019, 39, 3065.)
(u) Ren, L.; Ran, M.; He, J.; Qian, Y.; Yao, Q. Chin. J. Org. Chem. 2019, 39, 1583 (in Chinese).
(任林静, 冉茂刚, 何佳芯, 钱燕, 姚秋丽, 有机化学, 2019, 39, 1583.)
(v) Chen, J.; Li, Y.; Mei, L.; Wu, H. Chin. J. Org. Chem. 2019, 39, 3040 (in Chinese).
(陈锦杨, 李玉涵, 梅兰, 吴红谕, 有机化学, 2019, 39, 3040.)
(w) Chen, Y.; Lu, L.-Q.; Yu, D.-G.; Zhu, C.-J.; Xiao, W.-J. Sci. China: Chem. 2019, 62, 24.
(x) Hossain, A.; Bhattacharyya, A.; Reiser, O. Science 2019, 364, eaav9713.
(y) Li, S.; Xiang, S.-H.; Tan, B. Chin. J. Chem. 2020, 38, 213.
(z) Zhang, H.-H.; Chen, H.; Zhu, C.; Yu, S. Sci. China: Chem. 2020, 63, 637.
(aa) Yin, Y.; Zhao, X.; Qiao, B.; Jiang, Z. Org. Chem. Front. 2020, 7, 1283.
For select reviews: (a) Paria, S.; Reiser, O. ChemCatChem 2014, 6, 2477.
(b) Hernandez-Perez, A. C.; Collins, S. K. Acc. Chem. Res. 2016, 49, 1557.
(c) Reiser, O. Acc. Chem. Res. 2016, 49, 1990.
(d) Parasram, M.; Gevorgyan, V. Chem. Soc. Rev. 2017, 46, 6227.
(e) Zhou, W.-J.; Cao, G.-M.; Zhang, Z.-P.; Yu, D.-G. Chem. Lett. 2019, 48, 181.
(f) Chuentragool, P.; Kurandina, D.; Gevorgyan, V. Angew. Chem., Int. Ed. 2019, 58, 11586.
For select examples for visible light-excited Pd-catalysts, see: (g) Kurandina, D.; Parasram, M.; Gevorgyan, V. Angew. Chem., Int. Ed. 2017, 56, 14212.
(h) Zhou, W.-J.; Cao, G.-M.; Shen, G.; Zhu, X.-Y.; Gui, Y.-Y.; Ye, J.-H.; Sun, L.; Liao, L.-L.; Li, J.; Yu, D.-G. Angew. Chem., Int. Ed. 2017, 56, 15683.
(i) Wang, G.-Z.; Shang, R.; Cheng, W.-M.; Fu, Y. J. Am. Chem. Soc. 2017, 139, 18307.
(j) Sun, L.; Ye, J.-H.; Zhou, W.-J.; Zeng, X.; Yu, D.-G. Org. Lett. 2018, 20, 3049.
(k) Zhou, Z.-Z.; Zhao, J.-H.; Gou, X.-Y.; Chen, X.-M.; Liang, Y.-M. Org. Chem. Front. 2019, 6, 1649.
(l) Sun, S.; Zhou, C.; Yu, J.-T.; Cheng, J. Org. Lett. 2019, 21, 6579.
(m) Kancherla, R.; Muralirajan, K.; Maity, B.; Zhu, C.; Krach, P. E.; Cavallo, L.; Rueping, M. Angew. Chem., Int. Ed. 2019, 58, 3412.
(n) Huang, H.-M.; Koy, M.; Serrano, E.; Pflüger, P. M.; Schwarz, J. L.; Glorius, F. Nat. Catal. 2020, 3, 393.
(o) Torres, G. M.; Liu, L.; Arndtsen, B. A. Science 2020, 368, 318.
For select reviews: (a) Hopkinson, M. N.; Sahoo, B.; Li, J.-L.; Glorius, F. Chem.-Eur. J. 2014, 20, 3874.
(b) Gui, Y.-Y.; Sun, L.; Lu, Z.-P.; Yu, D.-G. Org. Chem. Front. 2016, 3, 522.
(c) Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116, 10035.
(d) Levin, M. D.; Kim, S.; Toste, F. D. ACS Cent. Sci. 2016, 2, 293.
(e) Goddard, J.-P.; Ollivier, C.; Fensterbank, L. Acc. Chem. Res. 2016, 49, 1924.
(f) Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116, 10035.
(g) Tóth, B. L.; Tischler, O.; Novák, Z. Tetrahedron Lett. 2016, 57, 4505.
(h) Tellis, J. C.; Kelly, C. B.; Primer, D. N.; Jouffroy, M.; Patel, N. R.; Molander, G. A. Acc. Chem. Res. 2016, 49, 1429.
(i) Hopkinson, M. N.; Tlahuext-Aca, A.; Glorius, F. Acc. Chem. Res. 2016, 49, 2261.
(j) Fabry, D. C.; Rueping, M. Acc. Chem. Res. 2016, 49, 1969.
(k) Lang, X.; Zhao, J.; Chen, X. Chem. Soc. Rev. 2016, 45, 3026.
(l) Zhou, W.-J.; Zhang, Y.-H.; Gui, Y.-Y.; Sun, L.; Yu, D.-G. Synthesis 2018, 50, 3359.
Osawa, M.; Nagai, H.; Akita, M. Dalton. Trans. 2007, 2, 827.
(a) Kalyani, D.; Mcmurtrey, K. B.; Neufeldt, S. R.; Sanford, M. S. J. Am. Chem. Soc. 2011, 133, 18566.
(b) Kalyani, D.; Deprez, N. R.; Desai, L. V.; Sanford, M. S. J. Am. Chem. Soc. 2005, 127, 7330.
Neufeldt, S. R.; Sanford, M. S. Adv. Synth. Catal. 2012, 354, 3517. doi: 10.1002/adsc.201200738
Liang, L.; Xie, M.-S.; Wang, H.-X.; Niu, H.-Y.; Qu, G.-R.; Guo, H.-M. J. Org. Chem. 2017, 82, 5966. doi: 10.1021/acs.joc.7b00659
Khan, R.; Boonseng, S.; Kemmitt, P. D.; Felix, R.; Coles, J.; Tizzard, G. J.; Williams, G.; Simmonds, O.; Harvey, J.-L.; Atack, J.; Cox, H.; Spencer, J. Adv. Synth. Catal. 2017, 359, 3261. doi: 10.1002/adsc.201700626
Jiang, J.; Zhang, W.-M.; Dai, J.-J.; Xu, J.; Xu, H.-J. J. Org. Chem. 2017, 82, 3622. doi: 10.1021/acs.joc.7b00140
Sahoo, M. K.; Midya, S. P.; Landge, G.; Balaraman, E. Green Chem. 2017, 19, 2111. doi: 10.1039/C6GC03438A
Zhao, J.; Li, H.; Li, P.; Wang, L. J. Org. Chem. 2019, 84, 9007. doi: 10.1021/acs.joc.9b00893
Zhang, H.; Huang, X. Adv. Synth. Catal. 2016, 358, 3736. doi: 10.1002/adsc.201600704
Czyz, M. L.; Lupton, D. W.; Polyzos, A. Chem.-Eur. J. 2017, 23, 14450. doi: 10.1002/chem.201704045
Xuan, J.; Zeng, T.-T.; Feng, Z.-J.; Deng, Q.-H.; Chen, J.-R.; Lu, L.-Q.; Xiao, W.-J.; Alper, H. Angew. Chem., Int. Ed. 2015, 54, 1625. doi: 10.1002/anie.201409999
Lan g, S. B.; Nele, K. M. O.; Tunge, J. A. J. Am. Chem. Soc. 2014, 136, 13606. doi: 10.1021/ja508317j
Lang, S. B.; Nele, K. M. O.; Douglas, J. T.; Tunge, J. A. Chem. Eur. J. 2015, 21, 18589. doi: 10.1002/chem.201503644
Zhang, H.-H.; Zhao, J.-J.; Yu, S. J. Am. Chem. Soc. 2018, 140, 16914. doi: 10.1021/jacs.8b10766
Zhang, H.-H.; Zhao, J.-J.; Yu, S. ACS Catal. 2020, 10, 4710. doi: 10.1021/acscatal.0c00871
Zheng, C.; Cheng, W.; Li, H.; Na, R.; Shang, R. Org. Lett. 2018, 20, 2559. doi: 10.1021/acs.orglett.8b00712
Shen, X.; Qian, L.; Yu, S. Sci. China: Chem. 2020, 63, 687. doi: 10.1007/s11426-019-9732-5
Liu, K.; Zou, M.; Lei, A. J. Org. Chem. 2016, 81, 7088. doi: 10.1021/acs.joc.6b00965
Zhou, C.; Li, P.; Zhu, X.; Wang, L. Org. Lett. 2015, 17, 6198. doi: 10.1021/acs.orglett.5b03192
Xu, N.; Li, P.; Xie, Z.; Wang, L. Chem.-Eur. J. 2016, 22, 2236. doi: 10.1002/chem.201504530
Sharma, U. K.; Gemoets, H. P. L.; Schröder, F.; Noël, T.; Van der Eycken, E. V. ACS Catal. 2017, 7, 3818. doi: 10.1021/acscatal.7b00840
Manna, M. K.; Bairy, G.; Jana, R. Org. Biomol. Chem. 2017, 15, 5899. doi: 10.1039/C7OB01418J
Cheng, W.-M.; Shang, R.; Yu, H.-Z.; Fu, Y. Chem.-Eur. J. 2015, 21, 13191. doi: 10.1002/chem.201502286
Zhao, B.; Shang, R.; Cheng, W.; Fu, Y. Org. Chem. Front. 2018, 5, 1782. doi: 10.1039/C8QO00253C
Shimomaki, K.; Murata, K.; Martin, R.; Iwasawa, N. J. Am. Chem. Soc. 2017, 139, 9467. doi: 10.1021/jacs.7b04838
Shimomaki, K.; Nakajima, T.; Caner, J.; Toriumi, N.; Iwasawa, N. Org. Lett. 2019, 21, 4486. doi: 10.1021/acs.orglett.9b01340
Bhunia, S. K.; Das, P.; Nandi, S.; Jana, R. Org. Lett. 2019, 21, 4632. doi: 10.1021/acs.orglett.9b01532
Zhu, C.; Zhang, Y.; Liu, Z.; Zhou, L.; Liu, H.; Feng, C. Chem. Sci. 2019, 10, 6721. doi: 10.1039/C9SC01336A
Choi, S.; Chatterjee, T.; Choi, W. J.; You, Y.; Cho, E. J. ACS Catal. 2015, 5, 4796. doi: 10.1021/acscatal.5b00817
Sheri, S.; Paul, A.; Bera, M.; Venkatesh, Y.; Singh, N. D. P. Org. Lett. 2018, 20, 5533. doi: 10.1021/acs.orglett.8b01973
Zoller, J.; Fabry, D. C.; Ronge, M. A.; Rueping, M. Angew. Chem., Int. Ed. 2014, 53, 13264. doi: 10.1002/anie.201405478
Kato, S.; Saga, Y.; Kojima, M.; Fuse, H.; Matsunaga, S.; Fukatsu, A.; Kondo, M.; Masaoka, S.; Kanai, M. J. Am. Chem. Soc. 2017, 139, 2204. doi: 10.1021/jacs.7b00253
Under the revision of the manuscript, an elegant dehydrative allylation was reported, see:
Masuda, Y.; Ito, M.; Mutakami, M. Org. Lett. 2020, 22, 4467.

图式 1 可见光促进的过渡金属催化的有机反应
Scheme 1 Organic reaction by visible-light-driven transition- metal catalysis

图式 2 可见光与钯协同催化的Sonogashira偶联反应
Scheme 2 Sonogashira coupling reaction via visible-light photoredox/Pd dual catalysis

图式 3 可见光与钯协同催化的导向C(sp2)—H芳基化
Scheme 3 Directed C(sp2)—H arylation via visible-light photoredox/Pd dual catalysis

图式 8 可见光与钯协同催化的C(sp2)—H芳基化环化
Scheme 8 Arylation and annulation of C(sp2)—H bonds via visible-light photoredox/Pd dual catalysis

图式 9 可见光与钯协同催化的Heck反应及烯烃异构化
Scheme 9 Heck reactions and isomerization of alkene via visible-light photoredox/Pd dual catalysis

图式 10 可见光与钯协同催化的C(sp3)—H芳基化
Scheme 10 Arylation of C(sp3)—H bonds via visible-light photoredox/Pd dual catalysis

图式 11 可见光与钯协同催化胺类化合物的烯丙基化
Scheme 11 α-Allylation of amines via visible-light photoredox/Pd dual catalysis

图式 12 可见光与钯协同催化的脱羧烯丙基化反应
Scheme 12 Decarboxylative allylation via visible-light photoredox/Pd dual catalysis

图式 13 可见光与钯协同催化的脱羧烯丙基化反应机理
Scheme 13 Mechanism of decarboxylative allylation via visible-light photoredox/Pd dual catalysis

图式 14 可见光与钯协同催化的对映选择性烯丙基化反应
Scheme 14 Enantioselective allylation via visible-light photoredox/Pd dual catalysis

图式 15 可见光与钯协同催化N-甲基苯胺的对映选择性烯丙基化反应
Scheme 15 Enantioselective allylation of N-methyl anilines via visible-light photoredox/Pd dual catalysis

图式 16 可见光与钯协同催化的Z-选择性Heck反应
Scheme 16 Z-Selective Heck reaction via photoredox/Pd dual catalysis

图式 17 可见光与钯协同催化碳酸酯的对映选择性烷基化反应
Scheme 17 Enantioselective alkylation of carbonates via visible-light photoredox/Pd dual catalysis

图式 18 可见光与钯协同催化烯酰胺的氧化羰基化
Scheme 18 Oxidative carbonylation of enamides via visible-light photoredox/Pd dual catalysis

图式 19 可见光与钯协同催化α-羰基羧酸参与的脱羧酰基化
Scheme 19 Decarboxylative acylation of α-keto acids via visible-light photoredox/Pd dual catalysis

图式 20 可见光与钯协同催化偶氮苯和氧化偶氮苯的脱羧酰基化
Scheme 20 Decarboxylative ortho-acylation of azo- and azoxy-benzenes via visible-light photoredox/Pd dual catalysis

图式 21 可见光与钯协同催化吲哚的选择性酰基化
Scheme 21 C2 acylation of indoles via visible-light photoredox/Pd dual catalysis

图式 22 可见光与钯协同催化芳基卤代物参与的脱羧偶联反应
Scheme 22 Decarboxylative couplings of α-oxocarboxylates with aryl halides via visible-light photoredox/Pd dual catalysis

图式 23 可见光与钯协同催化卤代物与乙醛酸的脱羧甲酰化反应
Scheme 23 Decarboxylative formylation of aryl halides with glyoxylic acid via visible-light photoredox/Pd dual catalysis

图式 24 可见光与钯协同催化卤代物与CO2的羧基化反应
Scheme 24 Carboxylation of aryl halides with CO2 via visible-light photoredox/Pd dual catalysis

图式 25 可见光与钯协同催化偕二氟烯烃与CO2的羧基化反应
Scheme 25 Carboxylation of gem-difluo- roalkenes with CO2 via visible-light photoredox/Pd dual catalysis

图式 26 可见光与钯协同催化咔唑类化合物的合成
Scheme 26 Synthesis of carbazoles via visible-light photoredox/Pd dual catalysis

图式 27 可见光与钯协同催化C(sp2)—H的羟基化
Scheme 27 Hydroxylation of C(sp2)—H via visible-light photoredox/Pd dual catalysis

图式 28 可见光与钯协同催化吲哚的合成
Scheme 28 Indoles synthesis via visible-light photoredox/Pd dual catalysis

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




 下载:
下载:





 下载:
下载: