
图 1.
热销药物中烷基取代杂环化合物的部分示例
Figure 1.
Some examples of alkyl-substituted heterocyclic compounds in top selling drugs

杂环芳烃在合成药物和天然产物等生物活性分子中广泛存在, 表现出多样的生物活性.据统计, 在2008年全球零售销售额排名前200的药物中有三分之一是杂环化合物[1], 而这些杂环化合物中又有超过四分之一是烷基取代的杂环芳烃. 图 1列举了一些带有烷基取代的杂环芳烃结构的药物分子, 化合物Ⅰ[2]和Ⅱ[3]都能限制细胞内的胆固醇合成, 用作降血脂药; 化合物Ⅲ是一种胰岛素增敏剂, 用于治疗非胰岛素依赖性糖尿病[4]; 化合物Ⅳ是一种蛋白酶抑制剂, 用于治疗艾滋病[5]; 化合物Ⅴ~Ⅶ都是用于治疗高血压的药物分子[6-8]; 化合物Ⅷ是一种磷酸二酯酶选择性抑制剂, 用于治疗阴茎勃起功能障碍[9].由此可以看出, 烷基取代的杂环芳烃在药物化学与医药工业中扮演着重要的角色.因此, 发展不同的策略去合成烷基取代的杂环芳烃是国际上研究的热点之一.
早期制备烷基化的杂环芳烃是在低温无水无氧环境中, 正丁基锂作用下杂环芳烃形成对应的碳负离子, 再对卤代烷烃亲核进攻制备而成(Scheme 1, a).传统的强碱环境下的碳负离子化学合成方法也存在一些缺点, 例如条件苛刻及官能团容忍性不佳等.近半个世纪以来, 两种新的策略被运用于杂环芳烃及其衍生物的烷基化反应中:过渡金属催化C—H键活化的策略和杂环芳烃的烷基自由基加成策略.过渡金属催化C—H键活化的策略[10]相比于传统的碳负离子策略, 不再依赖化学计量的强碱, 同时对水分不敏感, 尽管有了巨大的改进, 但仍有一定的局限性:杂环芳烃经常扮演一种天然的有效配体, 极容易让金属催化循环失效, 因此需要在杂环芳烃上引入一个导向基团, 才能保持较高的区域选择性(Scheme 1, b).对比之下, 最早的杂环芳烃的烷基自由基加成策略是经典Minisci反应[11-12], 无需反应底物的预先官能化, 具有优异的步骤经济性和原子经济性, 利用羧酸产生亲核性的碳中心自由基与缺电子的喹啉或者吡啶结合生成新的C—C键, 也是合成烷基取代杂环芳烃的最具吸引力的一种方法(Scheme 1, c).早期Minisci反应只适用于缺电子含氮的杂芳环化合物的自由基C—H官能团化, 但它提供了一种自由基对杂芳环加成的烷基化思路.而就在近10年里, 化学家们基于Minisci反应的思路取得了一些突破性的进展, 已经开发出了适用于含氧、含硫原子的杂芳环的自由基烷基化反应.同时开发了断裂C—C键[13-15]、C—X键[16-19]、C—S键[20-21]、C—N键[22]和C—B键[23-24]等众多方法来产生烷基自由基, 但最具有原子经济性的方式还是断裂C(sp3)—H键的策略, 可以减少合成操作步骤, 提高合成效率, 也是本文综述的重点.我们汇总了过去10年里基于自由基历程的C(sp3)—H键断裂策略来构建新的杂环芳烃分子间C(sp2)—C(sp3)键的交叉脱氢偶联反应, 反应底物囊括了缺电子和富电子杂环芳烃以及预官能化和无需官能化的杂芳环.并根据烷基自由基前体来源, 将该策略分为四类不同的类型(以烷基醇或醚类化合物、环状脂肪叔胺类化合物、酯类或酰胺类化合物和未活化的普通烷烃作为自由基前体), 并逐一进行论述, 对其代表性反应机理进行了整理和归纳.

烷基醚类与醇类化合物在各种自由基引发剂的作用下, 其自由基中心通常产生在与氧原子相邻的C(sp3)—H键上, 因此常被用作烷基自由基前体.同时自由基的产生也包括过渡金属催化的过氧化物热裂解、无金属参与的过氧化物热裂解以及光诱导的氧化还原三种途径.
2011年, 李朝军课题组[25]报道了一例钯催化的自由基烷基化反应.该方法采用醇自身充当溶剂, PdCl2作为催化剂, 消旋的1, 1'-联萘-2, 2'-双二苯膦(rac-BINAP)作为配体, 过氧化二异丙苯(DCP)作为自由基引发剂, 在120 ℃下实现了氮杂环化合物的自由基烷基化反应(Eq. 1).作者通过一系列的机理控制实验, 认为反应是以类似于经典Minisci反应的自由基机制进行的.与经典Minisci反应相比, 该方法由DCP引发, 反应操作更简便, 且不再需要使用化学计量酸来活化氮杂芳烃.
|
|
(1) |
2013年, 强琚莉和陈兆旭课题组[26]共同报道了一例铜催化氮杂芳烃的自由基烷基化反应.该方法使用环醚自身充当溶剂, Cu(OTf)2作为催化剂, K2S2O8作为自由基引发剂, 在氮气氛围下, 60 ℃下实现了噻唑及苯并噻唑的烷基化(Eq. 2).不同的环醚化合物均适用, 而改变杂环上的取代基团, 并没发现明显的电子效应.此外, 该方法与前人方法相比, 条件较为温和.
|
|
(2) |
2013年, 崔秀玲和吴养洁课题组[27]共同报道了一例钯催化的喹啉N-氧化物自由基烷基化反应(Eq. 3).该方法采用Pd(OAc)2作为催化剂, 四丁基溴化铵(TBAB)作为添加剂, 叔丁基过氧化氢(TBHP)作为自由基引发剂, 环醚自身充当溶剂, 在100 ℃下实现了喹啉或吡啶N-氧化物的烷基化.值得一提的是, 四氢噻吩等环状硫醚也能得到相应的烷基化产物.
|
|
(3) |
2015年, Correa课题组[28]报道了一例铁催化唑类化合物的自由基烷基化反应.该方法采用FeF2作为催化剂, TBHP作为引发剂, 醚类化合物与1, 2-二氯乙烷(DCE)作为混合溶剂(体积比为1:1), 在90 ℃下实现了各种(苯并)唑类化合物的C-2位烷基化(Eq. 4).该方法的底物普适性较好, 不同类型的环状和链状醚均能较好地进行反应.
|
|
(4) |
作者通过实验捕获了自由基中间体, 另外同位素动力学实验的结果(kH/kD=3.18)也表明C(sp3)—H键的断裂与α-氧烷基自由基的形成可能是决定速率的步骤.最后结合密度泛函理论(DFT)计算, 作者推出了可能的反应机理(Scheme 2):以苯并噻唑为例, FeF2在加热条件下促进TBHP的均裂形成羟基自由基和叔丁基自由基; FeF2催化剂通过形成一种非常稳定的FeF2(OH)复合物来帮助稳定生成的羟基自由基, 接下来叔丁基自由基可以在四氢呋喃氧原子邻位的C(sp3)—H抽取一个氢原子得到四氢呋喃自由基, 然后经过单电子转移进一步被FeF2(OH)氧化成相应的氧鎓离子, 再与苯并噻唑结合, 最后去质子化得到目标产物.

2015年, Neubert课题组[29]报道了一例哒嗪的自由基烷基化反应.该方法使用醇或者醚作为溶剂, TiCl3作为路易斯酸, TBHP作为自由基引发剂, 在0 ℃下敞口接触空气反应10 min, 实现了3, 6-二氯哒嗪的烷基化(Scheme 3).虽然3, 6-二氯哒嗪及其衍生物始终无法反应完全, 同时整体底物产率非常低, 但该方法可以容忍卤素原子或者烯烃等官能团, 可以为进一步合成四氢吡啶并哒嗪提供新的合成砌块.

2015年, 蔡春课题组[30]报道了一例镍催化选择性吲哚C-2或C-3位烷基化的反应.该方法选用NiF2作为催化剂, PPh3作为配体, 过氧化二叔丁基(DTBP)作为自由基引发剂, 1, 4-二氧六环作为溶剂, 120 ℃下能实现吲哚C-2位烷基化; 其他条件不变, 移除PPh3配体, Zn(OTf)2作为添加剂, 催化剂更换为Ni(acac)2, 氩气氛围下可以实现吲哚C-3位烷基化(Eqs. 5, 6).同时底物普适性较好, 并且能容忍包括羧基、酯基、氰基及甲酰基等众多可进一步转化的官能团.动力学同位素效应的结果(KH/KD=6.7)表明, 1, 4-二氧六环的氧原子邻位的C(sp3)—H键断裂可能是这个反应的速率决定步骤之一.作者进行了自由基诱捕实验, 成功捕获到环醚自由基中间体, 因此认为反应符合Minisci类型的自由基烷基化过程.
|
|
(5) |
|
|
(6) |
2016年, 李亚波和吴养洁课题组[31]共同报道了一种铜催化苯并呋喃的自由基烷基化反应.该方法采用环状醚类化合物作为溶剂, CuI作为催化剂, KI作为添加剂, DTBP作为自由基引发剂, 在120 ℃下实现了苯并呋喃的C-2位烷基化(Eq. 7).该方法底物普适性较好, 除了苯并呋喃, 呋喃、噻吩、苯并噻吩、苯并噻唑、苯并噁唑和吡咯均是良好的底物.
|
|
(7) |
2016年, 李桂根和陆红健课题组[32]共同报道了一例钴催化的(苯并)噁唑自由基烷基化反应.该方法以醚自身充当溶剂, 用CoCO3作为低负载(1.0 mol%)的廉价催化剂, TBHP作为自由基引发剂, 在120 ℃下获得了一些重要的生物活性氮杂芳烃的醚衍生物(Eq. 8).
|
|
(8) |
2019年, 王乃兴等[33]报道了一例稀土金属催化氮杂芳烃的自由基烷基化反应.该方法采用醚类化合物自身充当溶剂, Y(OTf)3作为催化剂, DTBP作为自由基引发剂, 在120 ℃下实现了氮杂芳烃的烷基化(Eq. 9).该反应的烷基前体较为丰富, 环醚、链状醚、环状硫醚和链状硫醚都是合适的底物.
|
|
(9) |
2019年, 王守锋和王文贵课题组[34]共同报道了一种银催化2-甲基喹啉的自由基烷基化反应.该方法利用AgNO3作为催化剂, 选择氟试剂作为温和的氧化剂, 醚与水混合物(体积比为3:1)为溶剂, 在50 ℃下实现了醚类化合物与2-甲基喹啉衍生物的交叉脱氢偶联(Eq. 10).该方法可耐受多种官能团, 操作简便, 条件温和, 是一种合成C-4位烷基取代的2-甲基喹啉衍生物的便捷途径.
|
|
(10) |
虽然过渡金属催化过氧化物热裂解的反应体系得到了快速的发展, 但是反应成本较高和重金属残留等问题依然亟待解决.相对而言, 无金属参与的过氧化物热裂解体系因具有经济、绿色和无金属残留的特点而受到有机工作者的广泛关注.
2011年, 王敏和王磊课题组[35]共同报道了一例无金属参与的氮杂芳烃C-2位自由基烷基化的反应(Eqs. 11, 12).该方法采用醇或者醚类化合物自身充当溶剂, TBHP作为自由基引发剂, 在120 ℃下实现苯并噻唑的烷基化.环状与链状的醇类均可以顺利地进行烷基化过程, 甚至乙二醇也能顺利进行反应.因为醚类的α-H的sp3结构与醇类非常相似, 因此也能够顺利地进行该转化.除了苯并噻唑, 苯并噁唑与苯并咪唑也是良好的反应底物.该方法不需要化学计量的酸来活化氮杂芳烃, 同时不再依赖金属, 简单且高效.
|
|
(11) |
|
|
(12) |
2015年, 蔡春课题组[36]报道了一例无金属参与的吲哚自由基烷基化反应.该方法采用异色满作为溶剂, DTBP作为自由基引发剂, 在120 ℃下实现吲哚的烷基化(Eq. 13).与前人报道的金属催化的方法相比, 虽然该方法不需要金属, 但如果换用四氢呋喃等活性较低的醚类作为反应物, 产率明显下降.作者进行了一系列控制实验, 提出了如下的机理(Scheme 4):以吲哚为例, 首先DTBP生成的叔丁氧基自由基在抽取醚类化合物的α-H原子后, 变成亲核性的醚自由基, 随后对吲哚进行自由基加成得到芳环自由基中间体, 最后被氧化去质子化得到目标产物.
|
|
(13) |

2015年, Singh课题组[37]报道了以K2S2O8作自由基引发剂, 丙酮与水(体积比为2:1)为溶剂, 在120 ℃下能实现环醚类化合物与氮杂芳烃的自由基烷基化(Scheme 5).另一方面, 如果选用环烷烃作为当烷基来源, 只需要添加1 equiv.的TBAB, 换成DCE与水混合物(体积比2:1)为溶剂, 就可以实现普通C(sp3)—H键的自由基烷基化.各种缺电子的氮杂芳烃如喹啉、异喹啉、吡啶、吡嗪、嘧啶均适用于该烷基化反应, 作者认为反应机理与早期报道的无金属过氧化物体系类似, 都经历了自由基进攻氮杂环和后续的氧化去质子化过程.
2016年, 吴一诺等[38]报道了一种无金属参与的吲哚与环醚、链状醚、环烷烃的交叉脱氢偶联反应.以DTBP作为自由基引发剂, 140 ℃氮气氛围下, 许多不活泼的环醚和环烷烃可以直接与吲哚衍生物结合得到烷基化产物(Eq. 14).该反应原料来源丰富, 底物官能团相容性好.值得一提的是, 吲哚C-2位有吸电子基团取代时, 所有的烷基化反应均发生在C-3位; 当吲哚C-3位有吸电子基团取代时, 则发生在C-2位.
|
|
(14) |
2016年里, Barriault课题组[39]报道了一例无金属参与的喹啉/吡啶与醚类化合物的自由基烷基化反应.作者发现, 使用K2S2O8与三氟醋酸(TFA)体系, 在乙腈与水(体积比为9:1)的混合溶剂中, 回流反应可以实现氮杂芳烃的烷基化(Eq. 15).底物普适性较好, 喹啉、异喹啉和吡啶均是合适的底物, 环状或者链状的醚类化合物都可以顺利地转化为产物.
|
|
(15) |
醚类化合物作为烷基自由基前体已有许多报道, 但均需要较大的剂量(在许多情况下用作溶剂)来实现有效的转化.这一缺点极大地限制了醚类化合物作为烷基前体的使用范围, 使得这种自由基烷基化反应只能使用简单和易于获得的醚充当底物. 2017年, 张永强和王卫等[40]克服这一困难, 报道了一种新型高效的N-羟基琥珀酰亚胺(NHS)参与的温和无金属的氮杂芳烃自由基烷基化反应(Eq. 16).该反应在水相中进行, 40 ℃下, 过硫酸铵(APS)作为自由基引发剂, NHS作为一种新型高效的电子转移媒介, 对环境较为友好, 成本低廉, 具有良好的工业生产前景.除了简单易得的醚类化合物, 其他结构复杂或者昂贵的醚类化合物, 均能适用于这一转化.此外, 结构上更多样化的氮杂环也是这个转化过程的合适底物.作者经过各种控制实验, 初步揭示了一种独特的非光氧化还原诱导的氢化原子转移(HAT)机制(Scheme 6):以异喹啉作为底物举例, 首先添加剂NHS与APS的氧化还原反应提供了以氮为中心的自由基阳离子A.紧接着中间体A与四氢呋喃发生HAT过程, 从而生成α-氧自由基B.随后, 中间体B与质子化的异喹啉进行亲核加成, 形成中间体C, 再去质子化得到中间体D, 最后被氧化, 得到所需的产物.
|
|
(16) |

2018年, 武志勇和赵铭钦课题组[41]共同报道了一例无金属存在下的氮杂环N-氧化物自由基烷基化反应.该方法使用TBHP作为自由基引发剂, 环状或者链状醚类自身充当溶剂, 120 ℃下实现了该烷基化偶联(Eq. 17).部分底物具有潜在应用价值, 比如吡嗪N-氧化物的烷基化产物是一种香料前体, 可以通过加热分解产生多种香料化合物.
|
|
(17) |
2020年, 王祖利课题组[42]发现报道了一例DTBP作为自由基引发剂的喹啉N-氧化物的自由基烷基化反应(Eq. 18).该方法能够同时兼容1, 4-二氧六环和环状硫醚等醚类化合物, 并在克-规模制备中保持高产率.
|
|
(18) |
虽然近年来利用金属催化或者无金属参与的过氧化物热裂解的反应体系取得了一定的研究成果, 但是这些报道通常需较高温度才能进行, 且具有易爆炸的风险.而光诱导的氧化还原反应通常在室温下进行, 且官能团耐受性非常好, 清洁安全, 因此近年来吸引了不少合成化学家的关注.
2015年, MacMillan课题组[43]报道了一种全新的双功能催化剂体系, 在蓝光照射下, 对甲苯磺酸作为添加剂, 实现了氮杂环芳烃与醇类化合物的自由基烷基化(Eq. 19).作者采用简单而丰富的醇类作为烷基化试剂, 但出人意料地发现产物中竟不含羟基基团.经过进一步研究发现, 该反应经历了生物化学中的自旋中心位移(SCS).虽然SCS是脱氧核糖核酸(DNA)生物合成中的关键步骤, 但在有机合成中却非常罕见.作者通过组合使用两种类型的催化剂来模仿这一自然现象.利用这种方法, 作者还对两种药用化合物Fasudil和Milrinone的衍生物实现了高效的烷基化.该烷基化方法效率高, 底物普属性好, 用途广泛, 将会为药物化学领域带来深远影响, 很有应用前景.推测反应机理如下(Scheme 7): Ir(ppy)2(dtbbpy)PF6作为光催化剂, 能够从有机催化剂硫醇化合物中夺走一个电子, 形成关键的巯基自由基中间体.这种中间产物的独特之处在于它能切断普通醇类化合物上的一个强C(sp3)—H键.被巯基自由基夺取氢原子后的剩余基团可以和杂环芳烃反应, 然后再以SCS途径形成最终产物.
|
|
(19) |

2015年, MacMillan[44]课题组对不久前发表的双功能催化剂体系进行了部分改动, 选用Na2S2O8搭配光催化剂[Ir{dF(CF3)ppy}2(dtbbpy)]PF6的催化体系, 以TFA作为添加剂, 在节能灯照射下以及丙腈与水(体积比为1:1)混合溶剂中, 实现了氮杂芳烃与醚类化合物的脱氢偶联反应, 得到对应的烷基化产物(Eq. 20).反应机理与作者之前发表的工作较为相似, 这里不再重复叙述.由于该光催化方法是在室温下实现的, 即使是高挥发性的乙醚底物也可以进行高效地偶联反应, 因此该方法在链状醚类化合物和氮杂芳烃底物方面均具有广阔的应用前景.
|
|
(20) |
2015年, DiRocco和Krska课题组[45]共同报道了以[Ir(dF-CF3-ppy)2(dtbpy)](PF6)作为光催化剂, 搭配过氧化苯甲酰(BPO), TFA作为添加剂, 室温蓝光照射下可以使甲醇选择性地生成羟甲基自由基, 并实现氮杂芳烃的羟甲基化(Eq. 21).虽然羟甲基化的整体产率并不高, 但该方法适用于多种氮杂环化合物, 如吡啶、吡嗪、喹啉、异喹啉和喹喔啉等, 同时能对一些药物的衍生物进行结构修饰.
|
|
(21) |
2015年, Shah课题组[46]开发了一种无金属参与的可见光诱导的氮杂芳烃与醚类化合物的交叉脱氢偶联(Eq. 22).该反应适用于苯并噻唑、吡啶、喹啉、喹喔啉及吡嗪等众多氮杂环, 在水相中进行, 不需要贵金属光催化剂, 只需用到K2S2O8在室温下进行节能灯照射, 操作简便, 并成功实现克-规模制备.由于无光照时, 该反应不能发生, 因此作者认为反应可能涉及到电子给体-受体(EDA)复合物以及硫酸根自由基阴离子的中间体.推测机理如下(Scheme 8):氮杂环与过硫酸盐的复合物或许就可以充当光催化剂, 在光照下变成激发态, 随后释放出硫酸根自由基阴离子, 对醚类化合物进行氢原子抽取过程, 形成烷基自由基, 随后发生Minisci类型的自由基烷基化过程.
|
|
(22) |
2017年, 李朝军课题组[47]利用甲醇作为甲基化试剂和溶剂, 二氯甲烷(DCM)作为共溶剂, TFA作为添加剂, 在室温、紫外光照射的条件下实现了五元和六元芳香杂环高效简洁的甲基化过程(Eq. 23).反应无需使用外加金属光催化剂, 避免了金属试剂带来的费时费力且高成本的后续分离步骤, 更符合“绿色化学”的理念.各种喹啉、异喹啉、吡啶底物都取得了中等到良好的产率, 嘌呤、尼古丁等活性分子也可以实现甲基化.作者通过空白对照光谱学数据表明酸的加入可以增加从甲醇到杂环芳香烃的电子转移效率, 说明了TFA对于甲基化反应的促进作用.而从轨道能量的角度来说, 杂环芳香烃发生质子化后LUMO轨道能量降低, 接受光照激发条件下, 更容易获取甲醇中的电子.氘代实验中, 作者证实了引入的甲基中三个氢原子其中两个来自甲醇中的甲基, 另一个来自甲醇中的羟基.作者提出了反应可能的机理:以2-甲基喹啉为例, 首先在光照条件下可能有两种途径生成羟甲基自由基A, 然后2-甲基喹啉质子化后, 与自由基A反应得到自由基正离子B, 进而通过平衡过程得到烯胺形式的自由基正离子C, 随后脱水得到中间体D, 再和甲醇发生反应, 随后发生电子迁移并消除质子, 最后芳构化得到甲基化产物(Scheme 9).
|
|
(23) |
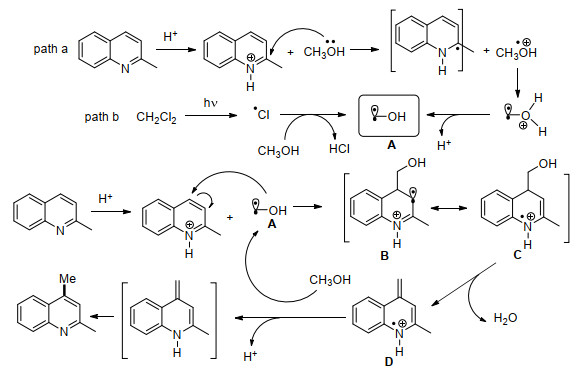
无独有偶, 同年, Scaiano和Barriault课题组[48]一起报道了与李朝军课题组[47]非常相似的光催化氮杂芳烃的烷基化反应.该反应采用醇作为烷基化试剂和溶剂, 盐酸作为添加剂, 在室温、紫外灯光照的条件下实现喹啉、吡啶等氮杂芳烃的光化学烷基化反应(Eq. 24).值得一提的是, 氘标记研究的实验结果显示了该反应如何控制CD3、CD2H和CDH2产物形成, 表明反应可能是通过烯胺中间体进行的, 这与李朝军课题组报道的结果一致, 这里不再重复叙述.
|
|
(24) |
2017年, 冯超和Loh课题组[49]共同报道了一例光催化合成对称的3, 3'-双吲哚基甲烷衍生物的反应(Eq. 25).该方法利用Ru(bpy)3(PF6)2与芳基重氮盐的单电子转移反应体系, 实现了两分子吲哚衍生物与一分子醚类或醇类化合物的偶联, 绕过了传统强氧化剂或强酸的使用, 具有广泛的官能团兼容性.
|
|
(25) |
2018年, 朱晨课题组[50]利用烷氧自由基的1, 5-氢迁移策略实现了脂肪醇与氮杂芳烃的远程自由基烷基化反应(Eq. 26).值得指出的是, 该方法无需外加金属光敏剂, 采用二乙酸碘苯(PIFA)作为唯一的自由基引发剂.在可见光照射与PIFA的作用下, 长链脂肪醇直接生成烷氧基自由基, 经过1, 5-氢迁移过程生成了烷基自由基, 后续发生类似Minisci类型的烷基化(Scheme 10).该方法操作简便, 条件温和, 底物普适性非常好, 在药物合成中有广泛的应用前景.
|
|
(26) |

2018年, Ranu课题组[51]报道了一例光照条件下钴催化8-氨基喹啉酰胺的C-4位选择性自由基烷基化反应(Eq. 27).该方法采用廉价的Co(acac)2与eosin Y有机光敏剂的体系, 在节能灯照射下合成了一系列具有重要药用价值的C-4位醚取代的喹啉酰胺衍生物.
|
|
(27) |
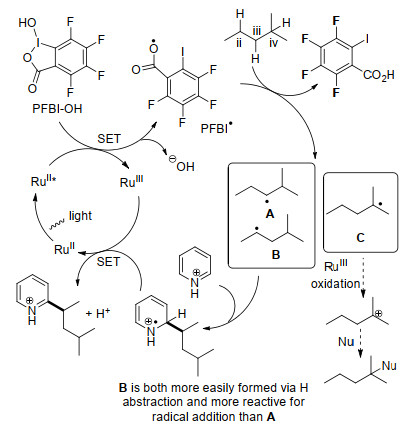
2019年, 何刚和陈弓课题组[52]共同报道了高价碘试剂(PFBI-OH)与光催化剂Ru(bpy)3Cl2在节能灯照射下30 ℃时实现了各种脂肪醇与氮杂芳烃的远程自由基烷基化反应(Eq. 28).特别指出的是, 该方法采用六氟异丙醇(HFIP)作为溶剂, 一级和二级脂肪醇的需求量显著降低(1.5~3 equiv.), 底物普适性较好, 并已被成功地应用于复杂药物分子的结构修饰.作者认为强亲电性的PFBI-OH是实现该高效转化的关键.推测的机理如下(Scheme 11):醇与PFBI-OH先进行结合, 生成高价碘中间体A, 然后被激发态的钌催化剂单电子氧化, 形成中间体B和烷氧自由基C, 自由基C经历1, 5氢迁移过程得到碳自由基D, 随后与质子化的氮杂环发生亲核加成得到中间体E, 最后再经历一次单电子转移得到目标产物.
|
|
(28) |

2019年, 雷爱文课题组[53]报道了一例选择性氟试剂作为自由基引发剂, TFA作为添加剂, 醇类作溶剂, 的光催化过程的氮杂芳烃自由基烷基化反应(Eq. 29).作者通过电子顺磁共振(EPR)研究, 认为反应的关键是蓝光照射激活选择性氟试剂的N—F键.
|
|
(29) |
2019年, 李朝军课题组[54]报道了一种新颖的在温和的可见光照射下实现无金属的氮杂芳烃自由基烷基化的策略.该方法使用二乙酰(2, 3-丁二酮)与醚类化合物作为混合溶剂(体积比为1:1), 可以无需外加光敏剂(Eq. 30).作者利用该方法还实现了奎宁、法舒地尔、尼古丁、薄荷醇和丙氨酸衍生物等复杂分子的烷基化.另外, 如果加入DTBP作为自由基引发剂, 该方法可以实现普通烷烃与氮杂芳烃的自由基烷基化(Eq. 31).该方法底物普适性好, 具有广阔的官能团容忍性.二乙酰(2, 3-丁二酮)作为一种丰富的、可持续的、可见光敏感的氢原子提取物, 具有非常大的应用前景.
|
|
(30) |
|
|
(31) |
2019年, Reisner课题组[55]利用氮化碳作为一种廉价、无毒、易于合成的碳基光催化剂, 在氧气氛围下实现了氮杂芳烃与醚、醇、酰胺的光催化自由基烷基化(Eq. 32).该方法使用的是环境友好且可自由获取的氧气作为氧化剂, 同时该光催化剂可多次循环使用, 无任何活性损失, 具有广阔的应用前景.
|
|
(32) |
2019年, 姬小趁和黄华文课题组[56]共同报道了一例LiBr促进的光催化氮杂芳烃与醚类化合物的烷基化反应(Eq. 33).该方法采用Ir[dF(CF3)ppy]2(dtbbpy)PF6和LiBr搭配的催化剂体系, 三氟甲磺酸、三氟化硼/乙醚和水作为添加剂, 为光氧化还原体系提供了新的思路.
|
|
(33) |
2019年, Hajra课题组[57]报道了一例光催化吲唑和醚类化合物的自由基烷基化反应.该反应以孟加拉红染料作为光催化剂, TBHP作为自由基引发剂, 三亚乙基二胺(DABCO)作为添加剂, 室温空气氛围中蓝光照射下实现吲哚与链状或者环状醚类的高效偶联(Eq. 34).能实现克-规模合成, 不用贵金属催化剂也是该方法的一大亮点.
|
|
(34) |
2020年, Lambert课题组[58]报道了一种光电结合催化的氮杂芳烃自由基烷基化反应(Eq. 35).该方法的反应体系非常新颖, 三氨基环丙烯鎓离子(TAC)作为唯一的氢原子转移类型的光电催化剂, 通过电和光化学能的结合, 不需要外部氧化剂, 为Minisci类型的烷基化反应提供了全新的思路.
|
|
(35) |
环状脂肪叔胺类化合物在各种自由基引发剂的作用下, 自由基中心通常产生在与氮原子相邻的C(sp3)—H键上, 作为另一种备受关注的烷基自由基前体. 2014年, Stephenson课题组[59]成功地开发了一种可见光诱导的光催化反应(Eq. 36).该方法以N-甲基吗啉与嘧啶衍生物1为起始物料, N, N-二甲基乙酰胺(DMA)与水(体积比10:1)作为混合溶剂, 较为便捷地构建了药物分子2 (LY278454, 用于治疗脊髓增生性疾病)中具有挑战性的C(sp2)—C(sp3)键, 收率中等, 结晶产物纯度高.
|
|
(36) |
酯类化合物通常被认为是活化的烷烃, 自由基中心通常生成于酯基相邻的C(sp3)—H键上, 也时常用作烷基自由基前体.
2017年, Yamaguchi和Itoh等[60]共同报道了一例不含金属的光催化噻吩与酯类化合物的烷基化反应(Eq. 37).该方法采用碘单质作为催化剂, K3PO4作为添加剂, 乙腈和水(体积比为10:1)作为混合溶剂, 在节能灯照射下构建这一新的C(sp2)—C(sp3)键.该方法的优点是反应条件温和, 操作简单, 同时催化剂廉价而丰富.
|
|
(37) |
酰胺类化合物可以看作酰基被取代的胺类化合物, 也是一类常用的烷基自由基前体.
2016年, 季海涛课题组[61]报道了一种新颖的唑类与酰胺的交叉脱氢偶联反应.该方法采用过硫酸铵作为自由基引发剂, 一水合对甲苯磺酸和苯甲醛作为添加剂, 在乙酸乙酯与酰胺的混合溶剂(体积比为1:1)中, 节能灯照射下实现了唑类化合物的自由基烷基化(Eq. 38).作者提出了独特的光催化苯甲醛促进过硫酸铵分解的反应机理(Scheme 12):光照能产生光激发态的苯甲醛(呈现双自由基状态), 促进过硫酸铵分解生成硫酸根自由基阴离子.随后, 硫酸根自由基阴离子与酰胺反应生成的中间体再对质子化的苯并噻唑C-2位进行亲核加成, 然后脱质子氧化得到所需产物.应注意的是, 作者观察发现催化量的苯甲醛也能促进反应的进行, 因此猜测双自由基激发态的苯甲醛中间体可能参与氧化步骤, 从而使苯甲醛循环再生.
|
|
(38) |

2017年, Togo课题组[62]报道了喹啉与叔酰胺在高压汞灯照射下的自由基烷基化反应(Eq. 39).该方法采用BPO作为自由基引发剂, TFA作为添加剂, 在400 W高压汞灯照射下构建新的C(sp2)—C(sp3)键.作者推测苯甲酰基自由基A是BPO在汞灯照射下均裂形成的, 随后自由基A将叔酰胺N-甲基中的一个氢原子抽离出来, 形成以碳为中心的酰胺自由基B和苯甲酸.这个亲核性的自由基B非常容易与质子化的氮杂芳烃的C-2位进行加成, 形成杂环芳基自由基阳离子C, 之后在BPO作用下氧化去质子化得到目标产物(Scheme 13).
|
|
(39) |

2018年, 汪清民课题组[63]报道了一种光催化的氮杂芳烃与叔酰胺的交叉脱氢偶联反应.该方法在前人的基础上有所改进, 添加剂三氟乙酸活化了氮杂芳烃, 氧化剂乙酸叔丁酯(t-BPA)产生自由基中间体(Eq. 40).在这种温和的条件下, 该反应具有广阔的底物范围, 并且可扩展到克-规模制备.此外, 该反应也适用于叔酰胺以外的其它烷基来源(醚类、醛类、对二甲苯类、烷烃类), 从而可以制备含不同取代基的氮杂芳烃.值得一提的是, 该方法还可以成功地应用于一些药物分子的后期功能化(N-Boc氟西汀、法舒地尔、己酮可可碱等).作者进行了一系列的机理探索实验, 得出可能的机理如下(Scheme 14):以苯并噻唑为例, [Ir(dF(CF3)ppy)2-(dtbbpy)]PF6光催化剂在蓝色LED光照下产生激发态Ir*3+, 氧化剂t-BPA与Ir*3+通过质子偶合电子转移(PCET)在酸性条件下得到一个氧化铱的物种(Ir4+)、醋酸以及叔丁氧自由基.叔酰胺在叔丁氧自由基的作用下被抽取一个氢原子形成α-氨基烷基自由基A, 随后自由基A与质子化的苯并噻唑进行亲核加成, 得到杂环自由基阳离子B, 最后被Ir4+经过单电子氧化合去质子化得到目标产物, 并生成Ir3+完成催化剂的循环再生.
|
|
(40) |

2018年, Berthelot课题组[64]也报道了非常类似的光催化的氮杂芳烃与叔酰胺的交叉脱氢偶联反应(Eq. 41).该反应的叔酰胺来源非常广泛, 各种环状的叔酰胺或者取代的吖啶类化合物以及氮杂螺环衍生物都能够很好地进行该转化.该反应体系与汪清民等[63]报道的仅在氧化剂上有所区别, 反应的机理是类似的, 这里不再重复叙述.
|
|
(41) |
2019年, 袁相爱和俞寿云课题组[65]共同报道了一例光催化的酰胺远程C(sp3)—H杂芳基化反应(Eq. 42).该方法采用3CzClIPN作为有机光敏剂, 碳酸钾作为碱, 二甲基亚砜(DMSO)作为溶剂, 90 W蓝光LED灯照射下, 实现了多种氮杂芳烃与酰胺的远程C(sp3)—H偶联.该反应具有良好的官能团耐受性和广阔的底物范围, 为复杂生物活性分子的后期功能化提供了独特的策略.作者推测的机理如下(Scheme 15):以N-苄基嘌呤为例, 光敏剂被光照后形成激发态3CzClIPN*, 与酰胺进行单电子转移后生成了3CzClIPN•+和以氮为中心的酰胺基自由基A.随后自由基A经历1, 5氢迁移过程得到了一个以碳为中心的自由基B.自由基B被N-苄基嘌呤捕获, 产生了杂芳基自由基中间体C.紧接着在碱和3CzClIPN+•的辅助下, 通过分步电子转移(ET)/质子转移(PT)过程, 得到最终产物, 同时实现光催化剂3CzClIPN的再生.
|
|
(42) |

富含C(sp3)—H键的环烷烃作为烷基来源一般用有机过氧化物作为引发剂产生烷基自由基.
2012年, 渠桂荣和郭海明课题组[66]报道了一例无金属参与的腺嘌呤核苷酸类衍生物与环烷烃的高区域选择性的C-8位烷基化反应(Eq. 43).该方法采用环烷烃作为溶剂, DTBP作为自由基引发剂.苯并噻唑、苯并噁唑和咖啡因等氮杂芳烃都是合适的底物, 此外, 环烷烃可以兼容五元环至八元环.
|
|
(43) |
2013年, Antonchick课题组[67]报道了一种以环烷烃或链状烷烃作为自由基前体, 室温下以高价碘PhI(OCOCF3)2为氧化剂, NaN3为添加剂的氮杂芳烃自由基烷基化反应(Eq. 44).通过对环状和链状烷烃的筛选, 作者发现未活化烷烃的一级碳和二级碳上C(sp3)—H键的键能相差很小, 但是在该反应体系条件下依然能将两种C(sp3)—H键选择性切断, 产生一系列的烷基自由基.另一方面, 吡啶、喹啉酮、异喹啉、喹啉类、苯并咪唑和酞嗪等氮杂芳烃在该温和条件下均能顺利地实现烷基化.作者认为PhI(OCOCF3)2可能与NaN3反应生成中间体A, 然后分解生成叠氮化物自由基B和PhIOCOCF3自由基C, 从而诱导烷基自由基D生成, 随后与质子化的氮杂芳烃E结合, 经历经典的Minisci类型的自由基加成得到杂芳基自由基F, 最后在PhIOCOCF3自由基C氧化作用以及去质子化作用下, 得到最终产物(Sheme 16).
|
|
(44) |

2015年, 于建军和王利民课题组[68]报道了一种催化量的BPO引发的N-酰胺基吡啶内鎓盐与简单环烷烃的交叉脱氢偶联反应, 生成相应的2-环烷基吡啶, 区域选择性高, 收率中等至良好, 不需要额外的还原步骤除去活化基团(Eq. 45).作者推测BPO在加热下均裂形成PhCOO•自由基, 随后普通烷烃被该自由基提取一个氢原子形成烷基自由基A, 引发链式反应, 与N-酰胺基吡啶内鎓盐形成自由基阳离子B, 随后在高温下分解释放出氢原子得到给目标产物和氮宾中间体C.而中间体C可以从环烷烃中抽象两个氢原子形成苯甲酰胺, 同时又得到烷基自由基A继续参与反应(Scheme 17).
|
|
(45) |

2016年, 易文斌课题组[69]使用DTBP促进环烷烃C(sp3)—H活化, 实现了无金属条件下具有中等至高的区域选择性的吲哚C-2、C-4、C-7位自由基烷基化的反应(Eq. 46).实验结果发现, 不同取代基的吲哚会在不同的反应位点上发生该自由基烷基化.随后作者采用密度泛函理论(DFT)研究了吲哚环上不同位置的自由基跃迁态(TS)活化能, 得出了活化能最低的反应位点就是最终烷基化的产物的结论, 因此烷基自由基进攻吲哚不同位置时, 生成的最稳定的杂芳基自由基才是主要的产物.
|
|
(46) |
2016年, 渠桂荣和郭海明课题组[70]基于之前报道工作, 再报道了一种合成多环烷基取代腺嘌呤核苷衍生物的方法.该方法同样是由DTBP引发的自由基反应完成的, 通过调节DTBP的用量和反应时间, 可以选择性合成C-6位的单环烷基或C-6、C-8位双环烷基取代腺嘌呤核苷类衍生物(Scheme 18).同时, 各种含核糖基、阿拉伯核糖基和脱氧核糖基的腺嘌呤核苷类衍生物也都能很好地进行转化.

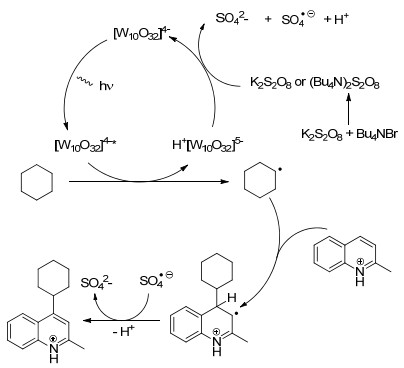
2017年, Ravelli和Ryu课题组[71]共同报道了一种以四丁基铵十聚钨酸盐(TBADT)作光催化剂, 过硫酸盐作为氧化剂, 在氙气灯照射的条件下实现了氮杂芳烃与环己烷的自由化烷基化方法(Eq. 47).该方法的烷基底物范围较广, 醚类与环烷酮也可以作为烷基来源, 同时除了喹啉, 酞嗪、喹喔啉、喹唑啉、异喹啉、苯并噻唑也是合适的偶联底物.在某些情况下, 添加TBAB作为相转移剂有助于加速这一过程, 这可能是由于K2S2O8和TBAB的置换反应形成了(Bu4N)2S2O8所致.与前人报道的反应类型的不同在于(Scheme 19):以2-甲基喹啉为例, 反应的启动需要激发态的TBADT从环烷烃中抽取一个氢原子得到环烷基自由基, 然后过硫酸盐作为氧化剂实现光催化剂的淬灭循环.随后环烷基自由基与质子化的2-甲基喹啉结合, 再被硫酸自由基阴离子氧化去质子化得到目标产物.
|
|
(47) |
2017年, 马丽芳和李子元课题组[72]报道了钴催化的噻唑/噁唑与环烷烃的交叉脱氢偶联.该反应只会发生在C-5位.该反应采用CoCl2作为催化剂, 过氧化苯甲酸叔丁酯(TBPB)或者DTBP作为自由基引发剂, 氯苯和环己烷作为混合溶剂(体积比为3:1), 120 ℃下实现这一个氮杂芳烃烷基化(Eq. 48).
|
|
(48) |
2018年, 何刚和陈弓课题组[73]基于之前报道的醚类烷基化的工作, 再次报道了一种光催化的氮杂芳烃与烷烃的高区域选择性自由基烷基化反应(Eq. 49).作者发现使用PFBI-OH与钌催化剂的体系, 对烷烃的氢原子抽取具有非常高的空间敏感性, 会优先选择空间位阻小的二级碳上的氢原子而不是三级碳的, 且适用于各种有环或者无环的烷烃.作者推测PFBI-OH上的邻位氟原子可能会影响羧基自由基中间体在抽取氢原子阶段的构象和反应方式, 从而增强其对烷烃底物的空间敏感性.随后作者通过一系列机理控制实验发现, PFBI-OH与激发态的RuⅡ*经过单电子转移, 形成PFBI•自由基, 进一步抽取烷烃上不同位置的氢原子可以形成三种烷基自由基中间体(A, B, C), 其中C自由基容易在Ru催化剂的氧化下变成碳正离子随后被亲核试剂(水)进攻得到对应的副产物.另一方面, 由于空间位阻的原因, 可能中间体B对氮杂芳烃的自由基加成过程更有利, 因此表现出较高的选择性(Scheme 20).
|
|
(49) |
2019年, 雷爱文课题组[74]将之前报道过的选择性氟试剂的光催化体系成功拓展至活化普通烷烃上, 实现了氮杂芳烃的自由基烷基化(Eq. 50).作者采用TFA作为添加剂, 烷烃与乙腈混合物(体积比为3:4)作为溶剂, 选择性氟试剂作为光催化剂, 室温下获得了喹啉或异喹啉的烷基化产物.作者推测的机理如下(Scheme 21):以喹啉为例, 首先选择性氟试剂在蓝光照射下形成自由基阳离子中间体A和氟自由基.随后夺取环烷烃中的氢原子, 形成环烷基自由基B, 之后与质子化的喹啉发生亲核加成得到杂芳基自由基中间体C, 最后经过氧化去质子化得到目标产物.
|
|
(50) |

2019年, 金健课题组[75]报道了一例与雷爱文[74]极其类似的工作, 但作者通过微调催化体系, 改用盐酸作为添加剂, 丙酮与烷烃作为混合溶剂, 极大地拓展了氮杂芳烃的底物范围, 并对一些含有杂环的药物分子进行了结构修饰(Eqs. 51, 52).
|
|
(51) |
|
|
(52) |
2019年, 吴骊珠课题组[76]报道了一例光照条件下铱催化氮杂芳烃与普通烷烃的自由基烷基化反应.该方法采用[Ir(ppy)2(dtbbpy)]PF6与K2S2O8搭配的经典体系, TFA作为活化氮杂芳烃的添加剂, 使得各种烷烃在温和条件下转化为高附加值的产品(Eq. 53).
|
|
(53) |
列举了过去10年里基于C(sp3)—H键断裂策略的自由基途径构建新的杂环芳烃分子间C(sp2)—C(sp3)键的烷基化反应.根据反应底物的不同, 涵盖了缺电子杂环芳烃(如吡啶、嘧啶和吡嗪等衍生物)、富电子杂环芳烃(如吲哚和噻吩)、预官能化的杂环芳烃(如氯代吡嗪、氨基取代的核糖核苷酸等)和无需官能化的杂环芳烃.根据烷基自由基前体的不同, 分为四大类, 其中包括以烷基醇或醚类化合物、脂肪胺类化合物、酯类或酰胺类化合物和未活化的普通烷烃.根据反应类型的不同, 主要有三大类:过渡金属参与的过氧化物热裂解反应、无金属参与的过氧化物热裂解反应以及光催化氧化还原反应.与过渡金属或者无金属参与的热反应相比, 光催化体系的反应具有绿色环保、温和高效的优点, 已成为有机合成领域新的研究方向和热点之一.并且众多光催化剂被研究者所报道, 过渡金属(如铱、钌、钨)、荧光素染料(如Rose Bengal)、有机光敏剂(如Selectfluor、单质碘、苯甲醛)、固体催化剂(如氮化碳)均是优良的断裂C(sp3)—H键的光催化剂.部分报道中还出现了双催化剂的体系以及电化学和光化学结合的反应体系, 为今后结合各种反应体系的优势提供了全新的思路.
虽然在这些方面研究者们已经取得了一些进展, 但仍存在一些问题和挑战.一方面, 基于自由基途径构建杂环芳烃分子间C(sp2)—C(sp3)键的反应, 目前集中研究的依旧是缺电子氮杂芳烃的Minisci类型反应.该类反应的区域选择性差, 如喹啉邻、对位均未被占据时, 难以调控反应位点, 经常得到邻对位双烷基化的产物.此外手性控制也存在较大难度.同时值得注意的是, 该类反应的总体效率不佳, 尤其是对于C(sp3)—H键键能较高的烷烃, 一般需要极为过量的烷基前体, 原子经济性并不理想.另一方面, 对于富电子杂环芳烃的偶联的底物范围有限, 目前仅局限于吲哚和噻吩, 而吡咯和呋喃却鲜有报道.在未来, 我们希望有更多的相关工作可以完成, 或许能在这一领域能看到一些不同体系的反应互相发挥彼此优势的新反应类型, 比如开发新的高效的多功能催化体系以减少烷基自由基前体的用量, 利用氨基杂环芳烃与手性磷酸结合来调控产物的手性, 以及拓展反应底物范围, 实现复杂杂环化合物的串联反应和复杂分子的后期功能化修饰等.
McGrath, N. A.; Brichacek, M.; Njardarson, J. T. J. Chem. Educ. 2010, 87, 1348. doi: 10.1021/ed1003806
Roth, E. M.; McKenney, J. M.; Hanotin, C.; Asset, G.; Stein, E. A. New Engl. J. Med. 2012, 367, 1891. doi: 10.1056/NEJMoa1201832
Chan, K. L.; Teo, K.; Dumesnil, J. G.; Ni, A.; Tam, J. Circulation 2010, 121, 306. doi: 10.1161/CIRCULATIONAHA.109.900027
Wysham, C.; Blevins, T.; Arakaki, R.; Colon, G.; Garcia, P.; Atisso, C.; Kuhstoss, D.; Lakshmanan, M. Diabetes Care 2014, 37, 2159. doi: 10.2337/dc13-2760
Clotet, B.; Feinberg, J.; van Lunzen, J.; Khuong-Josses, M.-A.; Antinori, A.; Dumitru, I.; Pokrovskiy, V.; Fehr, J.; Ortiz, R.; Saag, M.; Harris, J.; Brennan, C.; Fujiwara, T.; Min, S. Lancet 2014, 383, 2222. doi: 10.1016/S0140-6736(14)60084-2
White, W. B.; Weber, M. A.; Sica, D.; Bakris, G. L.; Perez, A.; Cao, C.; Kupfer, S. Hypertension 2011, 57, 413. doi: 10.1161/HYPERTENSIONAHA.110.163402
Diez, J.; Querejeta, R.; Lopez, B.; Gonzalez, A.; Larman, M.; Ubago, J. L. M. Circulation 2002, 105, 2512. doi: 10.1161/01.CIR.0000017264.66561.3D
Yusuf, S.; Teo, K.; Anderson, C.; Pogue, J.; Dyal, L.; Copland, I.; Schumacher, H.; Dagenais, G.; Sleight, P. Lancet 2008, 372, 1174. doi: 10.1016/S0140-6736(08)61242-8
Goldstein, I.; Lue, T. F.; Padma-Nathan, H.; Rosen, R. C.; Steers, W. D.; Wicker, P. A. New Engl. J. Med. 1998, 338, 1397. doi: 10.1056/NEJM199805143382001
Ackermann, L. Chem. Commun. 2010, 46, 4866. doi: 10.1039/c0cc00778a
Minisci, F.; Galli, R.; Malatesta, V.; Caronna, T. Tetrahedron 1970, 26, 4083. doi: 10.1016/S0040-4020(01)93049-2
Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinummo, M. Tetrahedron 1971, 27, 3575. doi: 10.1016/S0040-4020(01)97768-3
DiRocco, D. A.; Dykstra, K.; Krska, S.; Vachal, P.; Conway, D. V.; Tudge, M. Angew. Chem., Int. Ed. 2014, 53, 4802. doi: 10.1002/anie.201402023
Garza-Sanchez, R. A.; Tlahuext-Aca, A.; Tavakoli, G.; Glorius, F. ACS Catal. 2017, 7, 4057. doi: 10.1021/acscatal.7b01133
Gutierrez-Bonet, A.; Remeur, C.; Matsui, J. K.; Molander, G. A. J. Am. Chem. Soc. 2017, 139, 12251. doi: 10.1021/jacs.7b05899
Xiao, B.; Liu, Z.; Liu, L.; Fu, Y. J. Am. Chem. Soc. 2013, 135, 616. doi: 10.1021/ja3113752
Wu, X.; See, J. W. T.; Xu, K.; Hirao, H.; Roger, J.; Hierso, J.; Zhou, J. R. Angew. Chem., Int. Ed. 2014, 53, 13573. doi: 10.1002/anie.201408355
McCallum, T.; Barriault, L. Chem. Sci. 2016, 7, 4754. doi: 10.1039/C6SC00807K
Zhou, W.-J.; Cao, G.-M.; Shen, G.; Zhu, X.-Y.; Gui, Y.-Y.; Ye, J.-H.; Sun, L.; Liao, L.-L.; Li, J.; Yu, D.-G. Angew. Chem., Int. Ed. 2017, 56, 15683. doi: 10.1002/anie.201704513
Fujiwara, Y.; Dixon, J. A.; O'Hara, F.; Funder, E. D.; Dixon, D. D.; Rodriguez, R. A.; Baxter, R. D.; Herle, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Nature 2012, 492, 95. doi: 10.1038/nature11680
Liu, P.; Liu, W.; Li, C.-J. J. Am. Chem. Soc. 2017, 139, 14315. doi: 10.1021/jacs.7b08685
Klauck, F. J. R.; James, M. J.; Glorius, F. Angew. Chem., Int. Ed. 2017, 56, 12336. doi: 10.1002/anie.201706896
Li, G. X.; Morales-Rivera, C.; Wang, Y.; Gao, F.; He, G.; Liu, P.; Chen, G. Chem. Sci. 2016, 7, 6407. doi: 10.1039/C6SC02653B
Matsui, J. K.; Primer, D. N.; Molander, G. A. Chem. Sci. 2017, 8, 3512. doi: 10.1039/C7SC00283A
Correia, C. A.; Yang, L.; Li, C. J. Org. Lett. 2011, 13, 4581. doi: 10.1021/ol201774b
Xie, Z.; Cai, Y.; Hu, H.; Lin, C.; Jiang, J.; Chen, Z.; Wang, L.; Pan, Y. Org. Lett. 2013, 15, 4600. doi: 10.1021/ol4022113
Wu, Z.; Pi, C.; Cui, X.; Bai, J.; Wu, Y. Adv. Synth. Catal. 2013, 355, 1971. doi: 10.1002/adsc.201300111
Correa, A.; Fiserb, B.; Gόmez-Bengoa, E. Chem. Commun. 2015, 51, 13365. doi: 10.1039/C5CC05005G
Neubert, T. D.; Schmidt, Y.; Conroy, E.; Stamos, D. Org. Lett. 2015, 17, 2362. doi: 10.1021/acs.orglett.5b00861
Jin, L. K.; Wan, L.; Feng, J.; Cai, C. Org. Lett. 2015, 17, 4726. doi: 10.1021/acs.orglett.5b02217
Wang, C.; Gong, M.; Huang, M.; Li, Y.; Kim, J. K.; Wu, Y. Tetrahedron 2016, 72, 7931. doi: 10.1016/j.tet.2016.10.014
Li, Y.; Wang, M.; Fan, W.; Qian, F.; Li, G. G.; Lu, H. J. J. Org. Chem. 2016, 81, 11743. doi: 10.1021/acs.joc.6b02211
Wu, Y.-H.; Wang, N.-X.; Zhang, T.; Zhang, L.-Y.; Gao, X.-W.; Xu, B.-C.; Xing, Y.; Chi, J.-Y. Org. Lett. 2019, 21, 7450. doi: 10.1021/acs.orglett.9b02763
Wang, S.; Fan, Y.; Zhao, H.; Wang, J.; Zhang, S.; Wang, W. Synlett 2019, 30, 2096. doi: 10.1055/s-0039-1690697
He, T.; Yu, L.; Zhang, L.; Wang, L.; Wang, M. Org. Lett. 2011, 13, 5016. doi: 10.1021/ol201779n
Jin, L.; Feng, J.; Lu, G.; Cai, C. Adv. Synth. Catal. 2015, 357, 2105. doi: 10.1002/adsc.201500048
Ambala, S.; Thatikonda, T.; Sharma, S.; Munagala, G.; Yempalla, K. R.; Vishwakarma, R. A.; Singh, P. P. Org. Biomol. Chem. 2015, 13, 11341. doi: 10.1039/C5OB01268F
Yang, Q. J.; Choy, P. Y.; Wu, Y. N.; Fan, B. M.; Kwong, F. Y. Org. Biomol. Chem. 2016, 14, 2608. doi: 10.1039/C6OB00076B
McCallum, T.; Jouanno, L. A.; Cannillo, A.; Barriault, L. Synlett 2016, 27, 1282. doi: 10.1055/s-0035-1561338
Liu, S.; Liu, A.; Zhang, Y.; Wang, W. Chem. Sci. 2017, 8, 4044. doi: 10.1039/C6SC05697K
Lai, M.; Li, Y.; Wu, Z.; Zhao, M.; Ji, X.; Liu, P.; Zhang, X. Asian J. Org. Chem. 2018, 7, 1118. doi: 10.1002/ajoc.201800183
董道青, 李光辉, 陈德茂, 孙媛媛, 韩晴晴, 王祖利, 徐鑫明, 于贤勇, 有机化学, 2020, 40, 1766. doi: 10.6023/cjoc201709017Dong, D.-Q.; Li, G.-H.; Chen, D.-M.; Sun, Y.-Y.; Han, Q.-Q.; Wang, Z.-L.; Xu, X.-M.; Yu, X.-Y. Chin. J. Org. Chem. 2020, 40, 1766(in Chinese). doi: 10.6023/cjoc201709017
Jin, J.; MacMillan, D. W. C. Nature 2015, 525, 87. doi: 10.1038/nature14885
Jin, J.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2015, 54, 1565. doi: 10.1002/anie.201410432
Huff, C. A.; Cohen, R. D.; Dykstra, K. D.; Streckfuss, E.; DiRocco, D. A.; Krska, S. W. J. Org. Chem. 2016, 81, 6980. doi: 10.1021/acs.joc.6b00811
Devariab, S.; Shah, B. A. Chem. Commun. 2016, 52, 1490. doi: 10.1039/C5CC08817H
Liu, W.; Yang, X.; Zhou, Z. Z.; Li, C. J. Chem 2017, 2, 688. doi: 10.1016/j.chempr.2017.03.009
McCallum, T.; Pitre, S. P.; Morin, M.; Scaiano, J. C.; Barriault, L. Chem. Sci. 2017, 8, 7412. doi: 10.1039/C7SC03768F
Ye, L.; Cai, S.-H.; Wang, D.-X.; Wang, Y.-Q.; Lai, L.-J.; Feng, C.; Loh, T.-P. Org. Lett. 2017, 19, 6164. doi: 10.1021/acs.orglett.7b03073
Wu, X.; Zhang, H.; Tang, N.; Wu, Z.; Wang, D.; Ji, M.; Xu, Y.; Wang, M.; Zhu, C. Nat. Commun. 2018, 9, 3343. doi: 10.1038/s41467-018-05522-9
Ghosh, T.; Maity, P.; Ranu, B. C. Org. Lett. 2018, 20, 1011. doi: 10.1021/acs.orglett.7b03955
Li, G.-X.; Hu, X.; He, G.; Chen, G. Chem. Sci. 2019, 10, 688. doi: 10.1039/C8SC04134B
Niu, L.; Liu, J.; Liang, X.-A.; Wang, S.; Lei, A. Nat. Commun. 2019, 10, 467. doi: 10.1038/s41467-019-08413-9
Huang, C.-Y.; Li, J.; Liu, W.; Li, C.-J. Chem. Sci. 2019, 10, 5018. doi: 10.1039/C8SC05631E
Vijeta, A.; Reisner, E. Chem. Commun. 2019, 55, 14007. doi: 10.1039/C9CC07348E
Wang, Z.; Ji, X.; Han, T.; Deng, G.-J.; Huang, H. Adv. Synth. Catal. 2019, 361, 5643. doi: 10.1002/adsc.201901168
Singsardar, M.; Laru, S.; Mondal, S.; Hajra, A. J. Org. Chem. 2019, 84, 4543. doi: 10.1021/acs.joc.9b00318
Huang, H.; Strater, Z. M.; Lambert, T. H. J. Am. Chem. Soc. 2020, 142, 1698. doi: 10.1021/jacs.9b11472
Douglas, J. J.; Cole, K. P.; Stephenson, C. R. J. J. Org. Chem. 2014, 79, 11631. doi: 10.1021/jo502288q
Sudo, Y.; Yamaguchi, E.; Itoh, A. Org. Lett. 2017, 19, 1610. doi: 10.1021/acs.orglett.7b00428
Zhang, Y.; Teuscher, K. B.; Ji, H. Chem. Sci. 2016, 7, 2111. doi: 10.1039/C5SC03640B
Okugawa, N.; Moriyama, K.; Togo, H. I. J. Org. Chem. 2017, 82, 170. doi: 10.1021/acs.joc.6b02303
Dong, J.; Xia, Q.; Lv, X.; Yan, C.; Song, H.; Liu, Y.; Wang, Q. Org. Lett. 2018, 20, 5661. doi: 10.1021/acs.orglett.8b02389
Bosset, C.; Beucher, H.; Bretel, G.; Pasquier, E.; Queguiner, L.; Henry, C.; Vos, A.; Edwards, J. P.; Meerpoel, L.; Berthelot, D. Org. Lett. 2018, 20, 6003. doi: 10.1021/acs.orglett.8b00991
Chen, H.; Fan, W.; Yuan, X.-A.; Yu, S. Nat. Commun. 2019, 10, 4743. doi: 10.1038/s41467-019-12722-4
Xia, R.; Niu, H.-Y.; Qu, G.-R.; Guo, H.-M. Org. Lett. 2012, 14, 5546. doi: 10.1021/ol302640e
Antonchick, A. P.; Burgmann, L. Angew. Chem., Int. Ed. 2013, 52, 3267. doi: 10.1002/anie.201209584
Fang, L.; Chen, L.; Yu, J.; Wang, L. Eur. J. Org. Chem. 2015, 1910. doi: 10.1002/ejoc.201403479
Xiu, J.; Yi, W. Catal. Sci. Technol. 2016, 6, 998. doi: 10.1039/C5CY01907A
Wang, D.-C.; Xia, R.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M. Org. Biomol. Chem. 2016, 14, 4189. doi: 10.1039/C6OB00596A
Quattrini, M. C.; Fujii, S.; Yamada, K.; Fukuyama, T.; Ravelli, D.; Fagnoni, M.; Ryu, I. Chem. Commun. 2017, 53, 2335. doi: 10.1039/C6CC09725A
Wang, X.; Lei, B.; Ma, L.; Zhu, L.; Zhang, X.; Zuo, H.; Zhuang, D.; Li, Z. Chem.-Asian J. 2017, 12, 2799. doi: 10.1002/asia.201701258
Li, G.-X.; Hu, X.; He, G.; Chen, G. ACS Catal. 2018, 8, 11847. doi: 10.1021/acscatal.8b04079
Liang, X.-A.; Niu, L.; Wang, S.; Liu, J.; Lei, A. Org. Lett. 2019, 21, 2441. doi: 10.1021/acs.orglett.9b00744
Zhao, H.; Jin, J. Org. Lett. 2019, 21, 6179. doi: 10.1021/acs.orglett.9b01635
Huang, C.; Wang, J.-H.; Qiao, J.; Fan, X.-W.; Chen, B.; Tung, C.-H.; Wu, L.-Z. J. Org. Chem. 2019, 84, 12904. doi: 10.1021/acs.joc.9b01603

图 1 热销药物中烷基取代杂环化合物的部分示例
Figure 1 Some examples of alkyl-substituted heterocyclic compounds in top selling drugs

图式 1 合成烷基取代杂环化合物的策略
Scheme 1 Strategies for the synthesis of alkyl substituted heterocyclic compounds

图式 2 铁催化自由基烷基化反应机理推测
Scheme 2 Proposed mechanism for iron-catalyzed radical alkylation

图式 3 自由基介导的3, 6-二氯哒嗪的C—H功能化及四氢吡啶并哒嗪的合成
Scheme 3 Radical mediated C—H functionalization of 3, 6- dichloropyridazine and synthesis of tetrahydropyridopyridazines

图式 4 无金属参与的吲哚自由基烷基化机理推测
Scheme 4 Proposed mechanism for radical alkylation of indoles without metal

图式 6 NHS介导的氮杂环自由基烷基化机理推测
Scheme 6 Proposed mechanism for NHS-mediated radical alkylation of N-heteroarenes

图式 7 双功能催化体系的模拟SCS机理推测
Scheme 7 Proposed mechanism for difunctional catalytic system of simulated SCS

图式 9 光催化氮杂芳烃与甲醇的甲基化反应机理推测
Scheme 9 Proposed mechanism for photocatalytic methylation of N-heteroarenes with methanol

图式 10 无金属参与的光催化氢迁移机理推测
Scheme 10 Proposed mechanism for metal-free photocatalytic hydrogenatom transfer

图式 11 光催化远程C(sp3)—H杂芳基化机理推测
Scheme 11 Proposed mechanism for photocatalytic remote C(sp3)—H heteroarylation

图式 12 苯甲醛介导的氮杂芳烃与酰胺的光催化烷基化反应机理推测
Scheme 12 Proposed mechanism for a benzaldehyde-mediated photocatalytic alkylation of N-heteroarenes with amides

图式 13 光催化喹啉与叔酰胺的烷基化机理推测
Scheme 13 Proposed mechanism for photocatalytic alkylation of quinolines with tertiary amides

图式 14 光催化的氮杂芳烃与叔胺烷基化反应机理推测
Scheme 14 Proposed mechanism for photocatalytic alkylation of N-heteroarenes with tertiary amines

图式 15 选择性远程惰性C(sp3)—H键官能化机理推测
Scheme 15 Proposed mechanism for selective functionalization of remote inert C(sp3)—H bonds

图式 16 简单烷烃与氮杂芳烃选择性氧化的交叉偶联反应
Scheme 16 Proposed mechanism for selective oxidative cross-coupling of simple alkanes with N-heteroarenes

图式 17 简单烷烃与N-酰胺基吡啶内鎓盐烷基化机理推测
Scheme 17 Proposed mechanism for alkylation of simple alkanes with N-iminopyridinium ylide

图式 18 选择性合成单/双环烷基取代的嘌呤核苷
Scheme 18 Selective synthesis of monocycloalkyl or dicycloalkyl substituted purine nucleosides

图式 19 十聚钨酸盐催化氮杂芳烃烷基化机理推测
Scheme 19 Proposed mechanism for TBADT-catalyzed alkylation of N-heteroarenes

图式 20 光催化高选择性氮杂芳烃烷基化机理推测
Scheme 20 Proposed mechanism for high selective photocatalytic alkylation of N-heteroarenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:























































 下载:
下载:



 下载:
下载: