表 1
乙二醇的工业制备方法及其优缺点
Table 1.
Advantages and disadvantages of industrial preparation of ethylene glycol

聚酯切片是连接石化产品和纺织、建筑以及包装等多个行业的重要中间产品.我国作为世界上最大的聚酯生产国和消费国, 聚酯工业的发展对我国国民经济的发展有重要意义.聚对苯二甲酸乙二醇酯(PET)作为最重要的聚酯品种之一, 拥有优良的机械性能, 同时具有耐磨及尺寸稳定性好等优点[1].乙二醇(EG)是PET的主要生产原料之一.近年来, 虽然我国的EG产量取得了显著的增长, 但仍不能满足市场的强烈需求, 约70%的EG依赖于进口[2].因此, 大力发展EG的生产进而缓解供需矛盾是目前广泛研究的领域之一.
目前, 乙二醇的制备方法有乙烯直接水合法、二氯乙烷水解法、碳酸乙烯酯法、环氧乙烷水合法和草酸酯法等(表 1).乙烯直接水合法等均属于传统的石油化工路线, 原料乙烯依赖于石油的裂解(表 1, Entries 1~4).草酸酯法由合成气(CO+2H2)出发经草酸酯加氢制备EG, 属于现代煤化工路线(表 1, Entry 5).由于乙烯直接水合法、二氯乙烷水解法等存在设备腐蚀严重和成本高等不足, 到20世纪70年代就已经基本被淘汰, 当前工业上应用较多的是环氧乙烷水合法和草酸酯法[3].考虑到我国“富煤贫油”的能源结构, “煤制乙二醇”工艺路线的发展应用有望协助打破我国大宗工业用乙二醇依赖于进口的严峻局面.在草酸酯法中, 草酸酯的制备技术已经成熟, 因而高效催化草酸酯加氢制EG是这一路线的研究重点和难点[1, 4].
 下载:
导出CSV
下载:
导出CSV
| Entry | 制备方法 | 原料 | 制备过程 | 优势 | 不足 |
| 1 | 乙烯直接水合法 | 乙烯 | C2H4+1/2 O2+H2O |
原料成本较低 | 设备腐蚀严重、 分离精馏成本高 |
| 2 | 二氯乙烷水解法 | 乙烯 | C2H4+Cl2 |
— | 设备腐蚀严重、 总成本高 |
| ClCH2CH2Cl+2 H2O |
|||||
| 3 | 碳酸乙烯酯法 | 乙烯 | C2H4+1/2 O2 |
反应条件温和、 能耗低 |
设备总投资大 |
| C2H4O+CO2 |
|||||
| (CH2O)2CO+H2O |
|||||
| 4 | 环氧乙烷水合法 | 乙烯 | C2H4+1/2 O2 |
乙二醇收率高 | 催化剂选择性、 稳定性有待提高 |
| C2H4O+H2O |
|||||
| 5 | 草酸酯法 | 合成气 | 2 CO+1/2 O2+2 ROH |
反应条件温和、工艺要求低 | 催化剂选择性、稳定性有待提高 |
| ROOCCOOR+4 H2
|
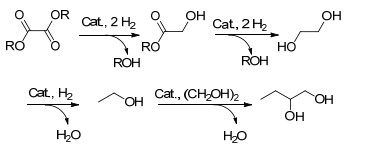
催化草酸酯加氢制EG是一连续的反应过程, 首先一个酯基加氢生成中间产物乙醇酸酯, 乙醇酸酯进一步加氢生成EG (Scheme 1).此外, 还可能发生EG过渡加氢生成乙醇以及乙醇与EG分子间脱水生成1, 2-丁二醇等副反应.由于草酸酯相邻酯基间的活化作用, 催化草酸酯部分加氢制乙醇酸酯反应难度低于催化乙醇酸酯加氢制EG[5].催化剂在这一加氢反应过程中扮演着至关重要的作用.目前, 可用于草酸酯加氢的催化剂主要有非均相负载型金属催化剂和均相金属配合物催化剂两类.相比于非均相负载型金属催化剂存在反应条件苛刻(180 ℃、20 MPa H2)、目标产物选择性低、催化加氢反应机理不明确等问题[6], 均相金属配合物催化剂可以在温和条件下高选择性催化底物加氢, 有一定的优势.同时均相催化剂明确的结构以及对反应过程的原位跟踪分析, 对于催化反应机理的研究具有重要意义.开展草酸酯均相催化加氢的研究, 一方面可以加深对这一反应过程的理解, 另一方面可以对进一步开发高效的负载型金属催化剂起指导借鉴的作用.
1980年, Grey和Pez等[5, 7]首次报道了草酸二甲酯(DMO)的均相催化加氢研究.发现钌氢负离子[(Ph3P)2-Ph2P(C6H4)RuH2]-和[(Ph3P)3(Ph2P)Ru2H4]2-可以催化DMO加氢制乙醇酸甲酯(MG); 其中[(Ph3P)3(Ph2P)-Ru2H4]2-的效果较好, 在90 ℃和0.62 MPa H2条件下反应20 h可以完成70%的DMO转化. Matteoli等[8]于1984年报道了钌羰基氢化物H4Ru4(CO)8(PBu3)4和H4Ru4- (CO)8(PPh3)4催化DMO加氢的研究.活性测试结果表明在180 ℃和13.17 MPa H2等反应条件下, H4Ru4(CO)8- (PBu3)4可以催化DMO加氢制MG (144 h, 产率51.1%), 而H4Ru4(CO)8(PPh3)4没有催化活性.这是首例关于配体可以影响钌催化剂在草酸酯加氢反应中催化性能的报道.作者研究发现催化剂在反应过程中生成新的钌金属活性物种H4Ru4(CO)9(PBu3)3和H4Ru4(CO)10(PBu3)2, 但在文中没有对膦配体的影响机制展开深入的研究和讨论. 1985年, 作者进一步将钌羰基化合物Ru(CO)2- (OAc)2(PBu3)2应用于该加氢反应研究中[9].在180 ℃、20.0 MPa H2等较为苛刻的条件下反应144 h取得了18% MG和82% EG收率的催化效果.
1997年, Elsevier等[10]发现原位催化剂体系Ru(acac)3/L1/Zn (acac:乙酰丙酮)的甲醇溶液在100 ℃、7.0 MPa H2等条件下可以有效催化DMO加氢制EG (16 h, 产率94%).甲醇和金属Zn在反应过程中均起到还原Ru(acac)3生成活性中心的作用[11].对比发现这一催化体系的催化活性有显著的提升, 且反应条件较为温和.作者也考察了不同膦配体与Ru(acac)3所构成催化剂体系的加氢性能, 结果呈现出P(C6H11)3<Ph2PC2H4PPh2<PPh3<PhP(C2H4PPh2)2≈[CH2P(Ph)C2H4PPh2]2<<MeC- (CH2PPh2)3的活性变化趋势. 2006年, Hanton等[12]进一步考察了配体L2与Ru(acac)3所构成催化剂体系的反应性能.在100 ℃、8.0 MPa H2等条件下反应136 h, DMO仅转化为MG, 无EG生成.这些结果表明配体结构对催化性能有显著影响.
|
|
综上所述, 早期用于草酸酯均相加氢的催化剂均为基于膦配体或硫配体的Ru化合物, 所需反应条件较为苛刻, 催化活性较低. 1995年, Noyori等[13]报道指出乙二胺配体可以显著促进膦-Ru(II)催化剂催化醛、酮等羰基衍生物分子加氢活性.研究发现反应体系中通过原位生成的前驱体trans-[RuCl2(phosphine)2(diamine)]形成了含有钌金属氢(RuH)的催化活性物种, 进而在RuH与配体NH的协同作用下通过与羰基发生亲核和亲电作用形成过渡态(TS)实现催化活性的提升[14].这一机理与传统的膦-Ru催化剂体系有关H2分子活化以及催化加氢反应过程均在金属中心发生的“内核加氢反应机理”截然不同[15].目前, 研究者已经成功制备出一系列具有金属-配体协同作用的Ru(II)催化剂[16]. 2007年, Saudan研究组[17]首次报道将该类型催化剂A和B应用于苯甲酸甲酯(MB)等多种酯类分子的均相加氢反应研究, 取得了极大的成功.近年来, 研究人员也将该类催化剂应用于草酸酯的均相催化加氢, 在较为温和条件下取得了优异的催化反应结果, 相比于传统的膦-Ru催化剂体系有显著的提升.本文将对典型的应用于草酸酯均相加氢且具有金属-配体协同作用的Ru(II)催化剂进行简要论述, 重点探讨催化剂结构与性质的关联以及催化加氢反应机理.
|
|
2011年以来, Takasago公司首创的螯合型Macho钌化合物(Cat. 1, 表 2)已经被成功应用于乳酸甲酯和碳酸乙烯酯等多种底物分子的加氢反应研究中[18]. Ding等[18b]研究提出RuH与配体NH的协同作用是该化合物取得优异加氢活性的关键. 2013年, Beller研究组[19]进一步考察了该类化合物催化草酸二乙酯(DEO)加氢的反应性能, 结果如表 2所示.在NaOEt/四氢呋喃(THF)溶液中于100 ℃、6.0 MPa H2等条件下反应20 h, 1可以催化DEO转化为EG (表 2, Entry 1).这一结果与Gusev等[20]报道的结构相似的Os化合物(1-CH2NHCH2- CH2PiPr2C5H4N)OsHCl(CO)不能催化草酸酯加氢的研究结果形成鲜明的对比. 1的NaBH4还原产物2在相同条件下催化活性略优于1, EG收率达96%(表 2, Entry 2).将1结构中Ph2P替换为iPr2P所得化合物3以及相应的氢化物4活性均较低, 仅可以催化DEO转化为乙醇酸乙酯(表 2, Entries 3, 4).对比化合物1(或2)和3(或4)反应结果可以看出催化剂结构对其性能有显著影响.
下载:
导出CSV
 | ||||
| Entry | Cat. | Conv./% | Yield/% of ethyl glycolate |
Yield/% of EG |
| 1 | 1 | 100 | 0 | 92 |
| 2 | 2 | 100 | 0 | 96 |
| 3 | 3 | 91 | 89 | 0 |
| 4 | 4 | 100 | 96 | 0 |
| 5 | L1/Ru(acac)3 | 0 | 0 | 0 |
| 6 | L3/Ru(acac)3 | 0 | 0 | 0 |
| 7 | L4/Ru(acac)3 | 4 | 0 | 0 |
| 8 | L5/Ru(acac)3 | 7 | 0 | 0 |
| 9b | L1/Ru(acac)3 | 100 | 0 | 96 |
| a Reaction conditions: DEO (3.7 mmol), Ru (0.0201 mmol), NaOEt (0.2 mmol), THF (20 mL), p(H2)=6.0 MPa, 100 ℃, 20 h; b MeOH as solvent. | ||||
Beller等[19]也考察了配体L1、L3~L5分别与Ru(acac)3所构成催化剂体系在相同条件下催化DEO加氢的反应性能. THF溶剂中膦配体L1(或L3、L4)与Ru(acac)3组成的催化剂体系没有催化活性(表 2, Entries 5~7);而在甲醇溶剂中, L1/Ru(acac)3催化活性与催化剂1(或2)相当(表 2, Entry 9).这一结果与Elsevier等[10]的研究结果相吻合, 表明甲醇在活化L1/Ru(acac)3生成催化活性物种这一过程中不可或缺.如前文所述, 配体L5与Ru(II)配位所构成的化合物B已经在多种酯类分子的加氢中展现出优异的催化性能[17].因此, L5/Ru(acac)3的THF溶液在反应条件下没有催化活性的原因与L1/ Ru(acac)3相似(表 2, Entry 8), 可归因于无法原位生成活性中心.考虑到反应过程中可能发生甲醇与金属中心作用生成烷氧基配合物以及分解产生CO与Ru(II)配位[21], 导致传统的膦-Ru催化剂体系中毒失活.相比之下, 基于金属-配体协同作用的化合物1(或2, 3, 4)在THF溶剂中具有较好的催化活性, 无需加入甲醇, 不易造成催化剂中毒失活, 具有独特的优越性.
围绕化合物2, Beller等[19]进一步考察了反应条件对催化性能的影响(表 3).如Entry 1所示, 1 h内2实现了DEO的完全转化, 乙醇酸乙酯和EG收率分别为21%和72%.在不使用EtONa条件下, 2也具有较好的加氢活性, 1 h后乙醇酸乙酯和EG收率分别为15%和85%(表 3, Entry 2); 4 h后EG收率高于99%(表 3, Entry 3). EtONa等碱金属醇盐在反应过程中, 可以在H2共同作用下将化合物结构中Ru-Cl置换为RuH, 生成催化活性物种[22].这一结果意味着2是1的催化活性态.如Entry 4所示, 提高底物与催化剂物质的量之比至358:1, 在60 ℃反应1 h可以取得99%的乙醇酸乙酯收率.进一步提高底物与催化剂物质的量之比、降低反应温度至室温或H2压力至3.0 MPa, 2也表现出良好的催化加氢活性(表 3, Entries 5~7).最后, 作者也考察了2催化DMO加氢的反应性能, 如Entry 8所示, 反应过程中有较多副产物生成, EG收率仅为84%.
下载:
导出CSV
| Entry | n(DEO)/n(2) | Temp./℃ | p(H2)/MPa | Time/h | Conv./% | Yield/% of ethyl glycolate | Yield/% of EG | TOFd/h-1 |
| 1b | 184 | 100 | 6.0 | 1 | 100 | 21 | 72 | 304 |
| 2 | 184 | 100 | 6.0 | 1 | 100 | 15 | 85 | 340 |
| 3 | 184 | 100 | 6.0 | 4 | 100 | 0 | >99 | 364 |
| 4 | 358 | 60 | 6.0 | 1 | 99 | 99 | 0 | 354 |
| 5 | 358 | r.t. | 6.0 | 120 | 100 | 7 | 86 | 5 |
| 6 | 358 | 60 | 3.0 | 1 | 50 | 46 | 0 | 165 |
| 7b | 6857 | 120 | 6.0 | 16 | 97 | 83 | 3 | 381 |
| 8b | 184c | 100 | 6.0 | 1 | 100 | 0 | 84 | 309 |
| a THF as solvent; b NaOEt (0.2 mmol); c DMO was used; d turnover frequency. | ||||||||
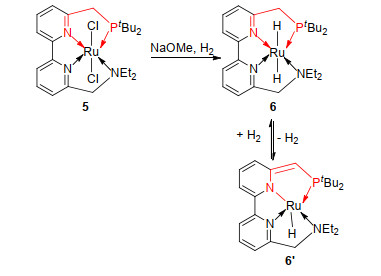
2014年, Zhou课题组[23]基于2, 2-联吡啶合成了四齿PNNN配体配位的的Ru(II)化合物5 (Scheme 2), 并将其成功应用于温和条件下多种内酯、脂肪酸酯以及芳香酸酯的加氢反应研究中.值得一提的是, 这一化合物能稳定存在于空气中, 并可以有效催化DMO加氢.在异丙醇为溶剂、5/NaOMe/DMO物质的量之为1:100:1000、5.07 MPa H2以及室温等条件下反应16 h, 体系中同时发生加氢反应和酯转移反应, 生成了99%的乙醇酸异丙醇酯.提高反应温度至100 ℃, 2 h后完全转化为EG(产率99%).作者利用核磁等表征手段成功证明反应体系中活性物种6的生成, 并进一步推测了基于配体吡啶环芳构化/去芳构化过程以及RuH与“膦臂”亚甲基氢协同作用的加氢反应机理(Scheme 2).
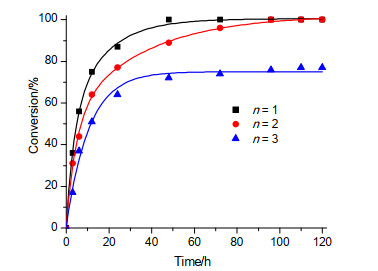
近年来, 我们课题组[24]围绕刚性o-PPh2C6H4NH2配体(L5), 在DMO均相催化加氢制MG/EG这一领域也开展了相关的研究工作.首先通过(PPh3)3RuCl2/nL5 (n=1, 2, 3)体系催化DMO加氢的原位活性测试, 探究了L5-Ru(II)化合物可能形成的结构型式.从图 1结果可以看出, n为1的反应体系在48 h内就实现了DMO的完全转化, n为2的反应体系需要约96 h; 然而, n为3的反应体系只能完成约77%的DMO转化, 48 h后基本失活.以上结果说明随着配体用量的增加, 原位反应体系催化活性逐渐降低.结合(PPh3)3RuCl2不能催化DMO加氢的实验结果, 我们推测上述原位反应体系中生成了配位有不同个数L5配体的Ru(II)化合物.
Reaction conditions: Ru (0.5 mol%), NaOMe (5 mol%), THF (10 mL), p(H2)=5.0 MPa, 100 ℃
我们将(PPh3)3RuCl2分别与1, 2, 3 equiv. L5配体反应, 尝试合成出不同结构的L5-Ru(II)化合物[24a].在甲苯溶剂中、100 ℃条件下分别反应12和48 h后, 从1和2 equiv.体系中成功分离得到单/双L5配体配位的化合物7和8; 3 equiv.体系在较长反应时间内均仅观察到8, 没有新化合物生成.幸运的是, 以(PPh3)3RuHCl作为金属前驱体与3 equiv. L5配体反应, 24 h后制备得到三配体配位的离子化合物9.活性测试结果显示7和8能有效催化DMO转化为MG(表 4, Entries 1 & 2), 9没有催化活性(表 4, Entry 3). 9不能催化DMO加氢可以推测主要是由于金属中心已经配位饱和, 无法活化H2分子产生RuH.上述结果说明随着反应的进行, (PPh3)3RuCl2/3L5原位反应体系中伴随着单→双→三L5配体配位的化合物生成, 最终完全转化为9(或与9具有相同结构特征的化合物), 致使反应体系失活.
下载:
导出CSV
| Entry | Cat. | n(NaOMe)/n(Ru) | Time/h | Conv./% | Yield/% of MG |
| 1 | 7 | 10 | 1 | 97 | 97 |
| 2 | 8 | 10 | 1 | 97 | 97 |
| 3 | 9 | 10 | 1 | 0 | 0 |
| 4 | 10 | 10 | 3 | 100 | 99 |
| 5 | 12 | 0 | 3 | 86 | 86 |
| 6 | 12 | 10 | 3 | 99 | 99 |
| 7 | 13 | 10 | 3 | 0 | 0 |
| 8 | 14 | 10 | 3 | 0 | 0 |
| 9 | 15 | 10 | 3 | 0 | 0 |
| 10 | A | 10 | 4 | 47 | 46 |
| 11 | C | 10 | 4 | 49 | 49 |
| 12 | D | 10 | 4 | 16 | 16 |
| a Reaction conditions: 0.5 mol% Ru, THF (10 mL), p(H2)=5.0 MPa, 100 ℃. | |||||
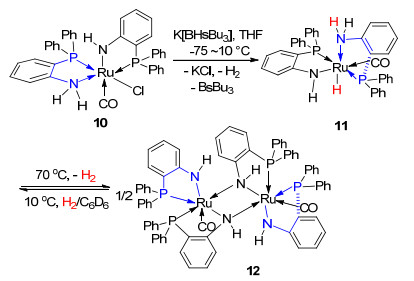
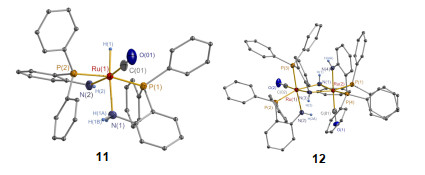
我们进一步以(PPh3)3RuHCl(CO)作为金属前驱体合成了金属中心配位有1个羰基分子的化合物10 (Scheme 3).以K[BHsBu3]作为氢化试剂, 开展了10的氢化反应并成功分离得到11.值得注意的是11稳定性较差, RuH易与NH2官能团作用脱去H2分子并二聚生成12.有趣的是, 12在H2气氛以及较低的温度下, 可以通过Ru-NH结构基元活化裂解H2分子转化为11, Ru金属价态不发生变化.这一过程有效展示了11和12相互转化的可逆性. 11和12的单晶结构(图 2)以及相关的D2实验结果有效验证了这一结论.作为对比, 我们还合成了分别与7和8具有相同分子构型、o-PPh2C6H4NMe2配体配位的(PPh3)(o-PPh2C6H4NMe2)RuCl2 (13)和(o- PPh2C6H4NMe2)2RuCl2 (14)以及金属中心配位有2个羰基分子的15.
活性测试结果表明, 10和12可以催化DMO部分加氢制MG(表 4, Entries 4, 6, );此外, 在不使用NaOMe条件下, 12也表现出一定的加氢活性(表 4, Entry 5).考虑到反应过程中, 10首先将在NaOMe和H2分子共同作用下转化成为含有RuH的活性物种[21c, 22b], 同时结合上述关于11和12可逆转化的相关认识, 我们认为11是10的活性态, 12为反应中间体.在相同反应条件下, 不含NH的配合物13和14均没有催化活性, 这表明NH在反应过程中不可或缺(表 4, Entries 7, 8). Noyori、Saudan和Ding等[17, 18b, 25]已经报道了类似的实验现象.化合物15与9相似, 在相同反应条件下没有催化活性(Entry 9, 表 4).这一结果有效验证了上文关于9不能催化DMO加氢的原因所提出的猜测.综合上述结果可以看出RuH与NH的协同作用在L5-Ru(II)催化DMO加氢过程中起着至关重要的作用.
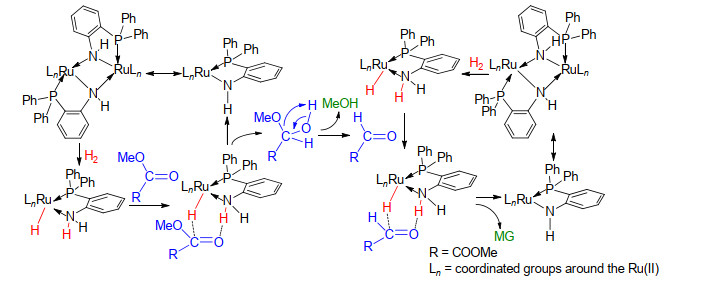
目前, Noyori[14c]、Morris[14b]以及Zhou[23]等均提出了金属-配体协同作用下Ru(II)催化剂催化苯乙酮、苯甲酸甲酯等羰基衍生物分子加氢的反应机理.催化剂通过RuH与配体NH(或亚甲基氢)协同作用分别与底物分子结构中羰基发生亲核和亲电作用, 并进一步将RuH与NH转移至羰基, 实现底物分子的加氢; 形成的催化剂中间体活化裂解H2分子回到催化活性态.基于以上研究, 为了明确草酸酯加氢的反应机理, 我们进行了原位核磁跟踪监测及同位素实验, 成功检测到RuH的形成与解离.同时我们表征了钌氢化合物11以及相应中间体12的单晶结构(图 2), 由此推测了L5-Ru(II)体系催化DMO加氢的反应机理[24a].如Scheme 4所示, 11等活性物种首先通过亲核和亲电协同作用将H-/H+等价物转移到酯基C=O上, 生成中间体半缩醛, 并进一步脱去一个MeOH分子形成MeOC(O)CHO.反应生成的催化剂中间体(如12等)活化H2分子重新形成活性态, 再经催化循环完成MeOC(O)CHO醛基加氢生成产物MG.
我们也测试了柔性膦胺配体构成的Ru(II)化合物(Ph2PCH2CH2NH2)2RuCl2 (A)、C以及D催化DMO加氢的反应活性. A(或C、D)在100 ℃、5.0 MPa H2以及4 h等反应条件下难以有效催化DMO加氢(表 4, Entries 10~12). A(和C、D)结构与8相似, 并已被成功用于催化酮、酯等多种羰基衍生物加氢制醇反应[17, 26].这些结果表明了L5-Ru(II)体系用于DMO催化加氢的优越性.
以8为例, 我们进一步考察了反应条件对L5-Ru(II)体系催化加氢性能的影响(表 5).在较低H2压力、温度或催化剂用量等反应条件下8均可以有效催化DMO部分加氢制MG (Entries 1~5).此外, 提高反应温度和延长反应时间, 8也可以催化DMO加氢制EG (Entry 6).在40 ℃、5.0 MPa H2以及1 h等反应条件下, 考察了NaOMe用量对8催化DMO加氢活性的影响.如Entries 7~11所示, 催化活性随着NaOMe用量的增加呈现火山型的变化趋势, 在n(NaOMe)/n(8)等于40时最优(MG产率81%, Entry 10).这些结果表明8与文献报道的催化剂体系相似, 反应过程中助剂碱(如NaOMe、t-BuOK等)一方面活化催化剂前驱体, 另一方面提升催化加氢活性[14c, 27].
下载:
导出CSV
| Entry | n(DMO)/n(8) | n(NaOMe)/n(8) | Temp./℃ | p(H2)/MPa | Time/h | Conv./% | Yield/% of MG | Yield/% of EG |
| 1 | 200 | 10 | 100 | 2.0 | 1 | 74 | 74 | 0 |
| 2 | 200 | 10 | 100 | 2.0 | 3 | 98 | 98 | 0 |
| 3 | 200 | 10 | 60 | 5.0 | 1 | 81 | 81 | 0 |
| 4 | 200 | 10 | RT | 5.0 | 24 | 86 | 86 | 0 |
| 5 | 2000 | 10 | 100 | 5.0 | 16 | 98 | 98 | 0 |
| 6 | 100 | 20 | 120 | 5.0 | 36 | 100 | 0 | 97 |
| 7 | 200 | 5 | 40 | 5.0 | 1 | 31 | 31 | 0 |
| 8 | 200 | 10 | 40 | 5.0 | 1 | 35 | 35 | 0 |
| 9 | 200 | 15 | 40 | 5.0 | 1 | 44 | 44 | 0 |
| 10 | 200 | 40 | 40 | 5.0 | 1 | 81 | 81 | 0 |
| 11 | 200 | 80 | 40 | 5.0 | 1 | 65 | 65 | 0 |
最后, 我们也初步考察了8催化其它酯类分子加氢的反应性能.结果表明8可以有效催化乳酸甲酯、丙酮酸甲酯和γ-戊内酯等多种酯类分子加氢制醇, 但难以催化位阻较大的芳香酸酯(如MB等)加氢, 这是一个有趣的实验现象.我们注意到A(和C、D)在催化MB加氢制苯甲醇(BA)反应中均表现出良好的催化加氢活性.这些结果意味着8和A(或C、D)在催化DMO及MB加氢反应中呈现出近乎相反的催化性能.针对这一现象, 我们进一步开展了相关的研究工作.
以8和C为例进行分析.如前文所述, 8(或C)通过RuH与NH的协同作用实现催化加氢.对比两者结构可以看出, 有无CH2链接导致它们NH2官能团局部化学环境存在一定的差异.由此可以推测, 这一差异势必影响它们在催化加氢反应过程中的协同效应, 是造成它们出现底物选择性的重要原因之一.根据这一推测, 我们提出“构筑一类兼具刚性和柔性结构特征的Ru(II)化合物, 以期该类化合物具有不同结构优势, 表现出良好底物适用性”的催化剂设计理念[24c].
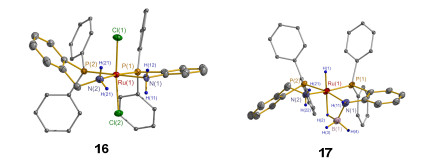
我们通过分步合成的方法成功制备得到金属中心同时配位有刚性配体L5和柔性配体o-PPh2C6H4CH2NH2 (L6)的新型Ru(II)化合物16.我们也开展了16的NaBH4还原反应, 得到相应的氢化产物17. 16和17的单晶结构如图 3所示.对比两者单晶结构可以看出, 还原前后o-PPh2C6H4CH2NH2配体中的CH2链接构象发生了翻转, 导致与之相连的N—H取向发生变化.这一过程有效地表明o-PPh2C6H4CH2NH2-Ru结构单元较为柔软的特点.我们进一步通过分步合成的方法制备了与16具有相同骨架结构、L5或L6配体NH分别被甲基化的化合物(o- PPh2C6H4CH2NH2)(o-PPh2C6H4NMe2)RuCl2 (18)和(o-P-Ph2C6H4CH2NMe2)(o-PPh2C6H4NH2)RuCl2 (19).
分别考察了化合物16~19催化DMO和MB加氢的反应性能.如表 6所示, 相比于8(或C)仅能有效催化DMO(或MB)加氢, 16在DMO和MB加氢反应中均表现出良好的催化活性(Entries 1, 2).进一步活性测试结果显示16可以有效催化多种不同结构内酯、脂肪酸酯和芳香酸酯加氢制相应的醇; 此外, 在酰胺以及碳酸酯的加氢反应中也表现出一定的催化活性.这些结果表明16兼具8和C分别在DMO和MB加氢中的优势, 具有良好的底物适用性, 有力验证了我们设计催化剂理念的正确性和有效性.
下载:
导出CSV
| Entry | Cat. | Substrate | Conv./% | Yield/% | ||
| MG or BA | EG | |||||
| 1 | 16 | DMO | 100 | 93 (MG) | 6 | |
| 2 | MB | 95 | 93 (BA) | |||
| 3 | 17 | DMO | 100 | 91 (MG) | 7 | |
| 4 | MB | 97 | 96 (BA) | |||
| 5b | 17 | DMO | 100 | 100 (MG) | 0 | |
| 6b | MB | 7 | 5 (BA) | |||
| 7 | 18 | DMO | 0 | 0 (MG) | 0 | |
| 8 | MB | 90 | 88 (BA) | |||
| 9 | 19 | DMO | 80 | 79 (MG) | 0 | |
| 10 | MB | 3 | 1 (BA) | |||
| 11 | 20 | DMO | 100 | 86 (MG) | 13 | |
| 12 | MB | 100 | 100 (BA) | |||
| 13 | 21 | DMO | 100 | 100 (MG) | 0 | |
| 14 | MB | 57 | 56 (BA) | |||
| a Reaction conditions: 0.5 mol% Ru, 5 mol% (for DMO) or 10 mol% (for MB) NaOMe, THF (10 mL), p(H2)=5.0 MPa, 100 ℃, 4 h; b No NaOMe was used. | ||||||
如表 6所示, 化合物17催化活性与16相当(Entries 3, 4).在不使用NaOMe条件下, 17也可以有效催化DMO加氢, 但在MB加氢反应中活性较低(Entries 5, 6).这一结果意味着配合物16与8等相似, 需要一定量的碱提升其催化加氢活性.与16形成鲜明对比, 18(或19)在反应条件下仅能催化MB(或DMO)加氢(Entries 7~10).这些结果表明结构基元L5-Ru和L6-Ru保留各自的性质特点, 16兼具刚性结构和柔性结构分别参与协同作用的优势.这一优势是它能有效催化DMO、MB等不同底物分子加氢的直接原因.
进一步采用柔性配体Ph2PCH2CH2NH2和Ph2P- (CH2)3NH2, 分别与L5组合制备了新型Ru(II)化合物20和21.如表 6所示, 20 (或21)在DMO和MB加氢反应中均表现出较好的催化活性(Entries 11~14).这些结果意味着新型Ru(II)化合物的设计、合成及应用具有一定的普适性.
|
|
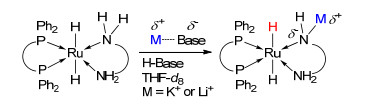
如前文所述, 具有金属-配体协同作用Ru(II)化合物在参与反应过程中多需要大量的助剂碱提升催化加氢活性[14c, 27a].然而, 过量助剂的使用会带来多方面不利的影响, 如增加反应成本和废弃物排放等.探索开发少/无助剂型Ru(II)催化剂从学术研究和工业应用的角度均具有重要的意义.目前, 研究者针对碱提升催化加氢活性的原因已经开展了多方面的研究工作.例如, Bergens课题组[28]报道指出反应过程中碱金属阳离子可以部分取代配体NH形成Nδ--Mδ+结构单元(Scheme 5).作者认为Nδ--Mδ+结构单元的生成增加了钌金属中心的电子云密度和RuH亲核性, 增强了RuH活化C=O的能力, 进而实现催化活性的提升.我们注意到有关催化加氢反应机理的研究已经明确提出Ru(II)催化剂活性与RuH亲核性间的线性关系[14c, 29].此外, Kayaki等[30]报道的化合物1结构中CO被富电子N-杂环卡宾替代所得到新化合物具有更高催化活性的实验结果进一步验证了上述观点.
基于上述研究进展, 我们提出在位阻允许的范围内将传统NH2-Ru(II)催化剂结构中伯胺官能团替代为供电性烷基取代的仲胺官能团, 希望通过N—R结构基元在一定程度上模拟原位形成Nδ--Mδ+的效果, 在提升催化加氢活性的同时降低取得最优催化活性所需的助剂用量[24d].根据这一设想, 我们制备了两类仲胺基配体配位的Ru(II)化合物.一方面, 采用单烷基取代的配体o-PPh2C6H4NHR (Me, Et, CH2Ph), 成功制备出化合物22~24.另一方面, 采用乙二胺以及N, N'-二烷基取代的乙二胺配体(CH2NHR)2 (R=H, Me, Et, iPr), 制备得到化合物25~28.值得注意的是, 二胺基配体配位的钌催化剂虽然已经成功应用于催化醛、酮加氢[14a, 31], 但用于酯类分子催化加氢还鲜有报道.
|
|
在0.05 mol% Ru、0.5 mol% NaOMe、5.0 MPa H2以及100 ℃等反应条件下考察了8、22~28催化DMO加氢的反应性能.如表 7所示, 22~24的催化活性相比于8均有了不同程度的提升, 其中以22最优(表 7, Entries 2~4).计算可得22相应的TOF值为1520 h-1(表 7, Entry 2), 远高于8(表 7, Entry 1).二胺配体构成的化合物25~28呈现出不同的催化活性.乙二胺配体配位的化合物25在反应条件下没有催化活性(表 7, Entry 5).相比之下, N, N'-二甲基乙二胺和N, N'-二乙基乙二胺配位的化合物26和27催化活性截然不同, 分别取得了36%和98%的MG收率(表 7, Entries 6, 7).此外, 在27参与催化的反应体系中还检测到少量EG生成(产率1%, 表 7, Entry 7).事实上, 在0.5 h内27就已经基本实现了DMO的完全转化, TOF值达到3920 h-1(表 7, Entry 8).据我们所知这是目前催化DMO部分加氢制MG所取得的最好实验结果.对比Entries 5~7结果可以看出, 在一定程度上催化活性随着胺配位基电子云密度的增加而增加.这些结果进一步验证了Bergens课题组[28]所提出的观点.由于异丙基位阻较大, 28催化活性较低, 仅取得6%的DMO转化率(表 7, Entry 9).
下载:
导出CSV
| Entry | Cat. | Conv./% | Yield/% | TOF/h-1 | |
| MG | EG | ||||
| 1 | 8 | 8 | 8 | 0 | 160 |
| 2 | 22 | 77 | 76 | 0 | 1520 |
| 3 | 23 | 27 | 27 | 0 | 540 |
| 4 | 24 | 24 | 24 | 0 | 480 |
| 5 | 25 | 0 | 0 | 0 | - |
| 6 | 26 | 36 | 36 | 0 | 720 |
| 7 | 27 | 100 | 98 | 1 | 2000 |
| 8b | 27 | 99 | 98 | 0 | 3920 |
| 9 | 28 | 6 | 6 | 0 | 120 |
| a Reaction conditions: 0.05 mol% Ru, 0.5 mol% NaOMe, THF (10 mL), p(H2)=5.0 MPa, 100 ℃, 1 h; b 0.5 h. | |||||
我们也进一步考察了8以及22~28催化苯甲醛(或苯乙酮)加氢制BA(或1-苯乙醇)的反应活性.有趣的是, 催化活性均呈现在DMO催化加氢反应中相似的变化趋势.综合上述结果可以看出, 在位阻效应允许的范围内通过将NH2-Ru(II)催化剂结构中伯胺官能团替代为富电性仲胺官能团来提升催化活性这一设想是可行的.
最后, 围绕27考察了反应条件对催化DMO加氢性能的影响.如表 8所示, 催化活性随着NaOMe用量的增加呈现出先增加后降低的火山型变化趋势, 并在n(NaOMe)/n(27)等于10时取得峰值(表 8, Entries 1~4).虽然这一变化趋势与8等相似, 但相比之下27取得最优催化活性所需助剂用量显著降低.这一结果意味着针对NH2-Ru(II)催化剂可以通过将伯胺官能团替代为供电性烷基取代的仲胺官能团这一方式来降低催化剂所需助剂的用量[27a-27b].在较低温度和H2压力条件下, 27也可以有效催化DMO加氢制MG(表 8, Entries 5~8).值得注意的是, 在室温条件下反应20 h可以取得95%的MG收率, 相应的TOF值为24 h-1(表 8, Entry 6).这一数值远高于2在相似反应条件下催化草酸二乙酯加氢的实验结果(Entry 5, 表 3).我们也考察了27催化DMO加氢制EG的反应性能, 结果显示催化活性相比于8也有显著的提升(表 8, Entries 9, 10 vs表 5, Entry 6).
下载:
导出CSV
| Entry | n(DMO)/n(27) | n(NaOMe)/n(27) | Temp./℃ | p(H2)/MPa | Time/h | Conv./% | Yield/% | TOF/h-1 | |
| MG | EG | ||||||||
| 1 | 2000 | 5 | 100 | 5.0 | 1 | 87 | 86 | 0 | 1720 |
| 3 | 2000 | 10 | 100 | 5.0 | 1 | 100 | 98 | 1 | 2000 |
| 3 | 2000 | 15 | 100 | 5.0 | 1 | 87 | 86 | 0 | 1720 |
| 4 | 2000 | 20 | 100 | 5.0 | 1 | 68 | 67 | 0 | 1340 |
| 5 | 500 | 10 | 100 | 1.0 | 1.5 | 100 | 99 | 0 | 330 |
| 6 | 500 | 10 | r.t. | 5.0 | 20 | 96 | 95 | 0 | 24 |
| 7 | 500 | 10 | 40 | 5.0 | 2 | 100 | 99 | 0 | 247 |
| 8 | 500 | 10 | 40 | 1.0 | 16 | 95 | 94 | 0 | 29 |
| 9 | 500 | 10 | 120 | 5.0 | 4 | 100 | 0 | 97 | 242 |
| 10 | 200 | 10 | 100 | 5.0 | 4 | 100 | 0 | 99 | 99 |
开发可高效催化草酸酯加氢制乙二醇的催化剂是“煤制乙二醇”工艺路线的研究热点和难点.在过去的近10年里, 基于金属-配体协同作用的Ru(II)催化剂用于草酸酯均相催化加氢的研究取得了一定的进展, 也发展制备出一系列结构各异的Ru(II)催化剂.研究结果表明, 配体结构对金属-配体间协同作用的发挥乃至催化剂催化加氢性能有显著影响.在尽可能降低位阻效应的前提下配体与金属中心配位所形成的结构单元具有一定刚性为佳; 胺配位基具有高的电子云密度, 与金属中心配位后能有效提升RuH亲核性.在反应过程中, RuH与配体NH(或亚甲基氢)两者均不可或缺; 配体提供NH(或亚甲基氢)与RuH产生协同效应, 并分别通过亲电和亲核作用将H+/H-等价物转移至底物分子羰基上, 实现催化加氢; 生成的催化剂中间体通过Ru-NH等结构基元活化裂解H2分子重新形成活性态.具有金属-配体协同作用Ru(II)催化剂相比于传统的膦-Ru催化剂体系具有更优异的催化加氢活性和稳定性, 同时所需反应条件较为温和.
针对现有均相配合物催化剂难以分离以及循环使用等问题, 接下来可以通过配体修饰, 以物理或化学作用的形式将催化剂锚定在金属-有机框架或有机聚合物等材料上, 探索开展金属-配体协同作用Ru(II)催化剂固载化的研究.均相催化剂固载化保留了金属配合物活性位点的结构和性质, 实现了均相催化剂反应条件温和、催化效率高和非均相催化剂使用寿命长、易于分离回收再利用等优势的有机结合; 此外, 也有利于从分子层面设计新催化剂.鉴于研究人员已经成功实现多种类型均相催化剂的固载化, 相信在不久的将来固载型金属-配体协同作用Ru(II)催化剂的合成及应用研究有望取得实质性的进展.
Chen, L.; Guo, P.; Qiao, M.; Yan, S.; Li, H.; Wei, S.; Xu, H.; Fan, K. J. Catal. 2008, 257, 172. doi: 10.1016/j.jcat.2008.04.021
尹国海, 中外能源, 2012, 17, 62.Yin, G. Sino-Global Energy 2012, 17, 62 (in Chinese).
(a) Liu, Z. Chem. Ind. Eng. Prog. 2013, 32, 1214 (in Chinese).
(刘宗语, 化工进展, 2013, 32, 1214.)
(b) Chen, W.; Sun, J.; Zhang, J.; Zhang, S.; Hua, W. Chem. Ind. Eng. Prog. 2014, 33, 1740 (in Chinese).
(成卫国, 孙剑, 张军平, 张锁江, 华炜, 化工进展, 2014, 33, 1740.)
(a) Zheng, J.; Lin, H.; Wang, Y.; Zheng, X.; Duan, X.; Yuan, Y. J. Catal. 2013, 297, 110.
(b) He, Z.; Lin, H.; He, P.; Yuan, Y. J. Catal. 2011, 277, 54.
(c) Xu, C.; Chen, G.; Zhao, Y.; Liu, P.; Duan, X.; Gu, L.; Fu, G.; Yuan, Y.; Zheng, N. Nat. Commun. 2018, 9, 3367.
Grey, R. A.; Pez, G. P.; Wallo, A. J. Am. Chem. Soc. 1981, 103, 7536. doi: 10.1021/ja00415a022
(a) Turek, T.; Trimm, D. L.; Cant, N. W. Catal. Rev.: Sci. Eng. 1994, 36, 645.
(b) Pouilloux, Y.; Autin, F.; Barrault, J. Catal. Today 2000, 63, 87.
(c) Wang, H.; Zhang, T.; Zhou, X. J. Phys.: Condens. Matter 2019, 31, 473001.
Grey, R. A.; Pez, G. P.; Wallo, A.; Corsi, J. J. Chem. Soc., Chem. Commun. 1980, 783.
Matteoli, U.; Blanchi, M.; Menchi, G.; Prediani, P.; Piacenti, F. J. Mol. Catal. 1984, 22, 353. doi: 10.1016/0304-5102(84)80075-9
(a) Matteoli, U.; Bianchi, M.; Menchi, G.; Frediani, P.; Piacenti, F. J. Mol. Catal. 1985, 29, 269.
(b) Matteoli, U.; Menchi, G.; Bianchi, M.; Piacenti, F. J. Organomet. Chem. 1986, 299, 233.
Teunissen, H. T.; J. Elsevier, C. Chem. Commun. 1997, 667.
(a) Teunissen, H. T. Chem. Commun. 1998, 1367.
(b) van Engelen, M. C.; Teunissen, H. T.; de Vries, J. G.; Elsevier, C. J. J. Mol. Catal. A: Chem. 2003, 206, 185.
Boardman, B.; Hanton, M. J.; Rensburg, H. V.; Tooze, R. P. Chem. Commun. 2006, 2289.
Ohkuma, T.; Ooka, H.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 10417. doi: 10.1021/ja00146a041
(a) Doucet, H.; Ohkuma, T.; Murata, K.; Yokozawa, T.; Kozawa, M.; Katayama, E.; England, A. F.; Ikariya, T.; Noyori, R. Angew. Chem., Int. Ed. 1998, 37, 1703.
(b) Abdur-Rashid, K.; Clapham, S. E.; Hadzovic, A.; Harvey, J. N.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2002, 124, 15104.
(c) Sandoval, C. A.; Ohkuma, T.; Muñiz, K.; Noyori, R. J. Am. Chem. Soc. 2003, 125, 13490.
(d) Ikariya, T.; Murata, K.; Noyori, R. Org. Biomol. Chem. 2006, 4, 393.
(e) Dub, P. A.; Gordon, J. C. Nat. Rev. Chem. 2018, 2, 396.
(a) Clapham, S. E.; Hadzovic, A.; Morris, R. H. Coord. Chem. Rev. 2004, 248, 2201.
(b) vom Stein, T.; Meuresch, M.; Limper, D.; Schmitz, M.; Hölscher, M.; Coetzee, J.; Cole-Hamilton, D. J.; Klankermayer, J.; Leitner, W. J. Am. Chem. Soc. 2014, 136, 13217.
(c) Liu, Y.; Yue, X.; Luo, C.; Zhang, L.; Lei, M. Energy Environ. Mater. 2019, 2, 292.
(a) Dub, P. A.; Ikariya, T. ACS Catal. 2012, 2, 1718.
(b) Zhao, B.; Han, Z.; Ding, K. Angew. Chem., Int. Ed. 2013, 52, 4744.
(c) Werkmeister, S.; Junge, K.; Beller, M. Org. Process Res. Dev. 2014, 18, 289.
(d) Pritchard, J.; Filonenko, G. A.; van Putten, R.; Hensen, E. J. M.; Pidko, E. A. Chem. Soc. Rev. 2015, 44, 3808.
(e) Zhou, Y.; Khan, R.; Fan, B.; Xu, L. Synthesis 2019, 51, 2491.
(f) Dub, P. A.; Batrice, R. J.; Gordon, J. C.; Scott, B. L.; Minko, Y.; Schmidt, J. G.; Williams, R. F. Org. Process Res. Dev. 2020, 24, 415.
Saudan, L. A.; Saudan, C. M.; Debieux, C.; Wyss, P. Angew. Chem., Int. Ed. 2007, 46, 7473. doi: 10.1002/anie.200701015
(a) Kuriyama, W.; Matsumoto, T.; Ogata, O.; Ino, Y.; Aoki, K.; Tanaka, S.; Ishida, K.; Kobayashi, T.; Sayo, N.; Saito, T. Org. Process Res. Dev. 2012, 16, 166.
(b) Han, Z.; Rong, L.; Wu, J.; Zhang, L.; Wang, Z.; Ding, K. Angew. Chem., Int. Ed. 2012, 51, 13041.
Ziebart, C.; Jackstell, R.; Beller, M. ChemCatChem 2013, 5.
Spasyuk, D.; Smith, S.; Gusev, D. G. Angew. Chem., Int. Ed. 2012, 51, 2772. doi: 10.1002/anie.201108956
(a) Van der Sluys, L. S.; Kubas, G. J.; Caulton, K. G. Organometallics 1991, 10, 1033.
(b) Chen, Y. Z.; Chan, W. C.; Lau, C. P.; Chu, H. S.; Lee, H. L.; Jia, G. Organometallics 1997, 16, 1241.
(c) Hamilton, R. J.; Bergens, S. H. J. Am. Chem. Soc. 2006, 128, 13700.
(d) Shen, Y.; Zhan, Y.; Li, S.; Ning, F.; Du, Y.; Huang, Y.; He, T.; Zhou, X. ChemSusChem 2018, 11, 864.
(e) Fang, X.; Duan, N.; Zhang, M.; Zhang, C.; Liu, R.; Zhu, H. P. Chin. J. Org. Chem. 2019, 39, 1450 (in Chinese).
(方霄龙, 段宁, 章敏, 张春燕, 刘睿, 朱红平, 有机化学, 2019, 39, 1450.)
(a) Abdur-Rashid, K.; Faatz, M.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2001, 123, 7473.
(b) Hartmann, R.; Chen, P. Angew. Chem., Int. Ed. 2001, 40, 3581.
Li, W.; Xie, J. H.; Yuan, M. L.; Zhou, Q. L. Green Chem. 2014, 16, 4081. doi: 10.1039/C4GC00835A
(a) Fang, X.; Zhang, C.; Chen, J.; Zhu, H.; Yuan, Y. RSC Adv. 2016, 6, 45512.
(b) Zhang, Y. W.; Chen, Y. L.; Fang, X. L.; Yuan, Y. Z.; Zhu, H. P. Chin. J. Org. Chem. 2017, 37, 2275 (in Chinese).
(张亦伟, 陈艺林, 方霄龙, 袁友珠, 朱红平, 有机化学, 2017, 37, 2275.)
(c) Fang, X.; Sun, M.; Zheng, J.; Li, B.; Ye, L.; Wang, X.; Cao, Z.; Zhu, H.; Yuan, Y. Sci. Rep. 2017, 7, 3961.
(d) Fang, X.; Li, B.; Zheng, J.; Wang, X.; Zhu, H.; Yuan, Y. Dalton Trans. 2019, 48, 2290.
(e) Fang, X.; Zhang, M.; Duan, N.; Wang, X.; Zhu, H. P. Chin. J. Org. Chem. 2020, 40, 226 (in Chinese).
(方霄龙, 章敏, 段宁, 汪新, 朱红平, 有机化学, 2020, 40, 226.)
Noyori, R.; Ohkuma, T. Angew. Chem., Int. Ed. 2001, 40, 40. doi: 10.1002/1521-3773(20010105)40:1<40::AID-ANIE40>3.0.CO;2-5
(a) Abdur-Rashid, K.; Guo, R.; Lough, A. J.; Morris, R. H.; Song, D. Adv. Synth. Catal. 2005, 347, 571.
(b) Jia, W.; Chen, X.; Guo, R.; Sui-Seng, C.; Amoroso, D.; Lough, A. J.; Abdur-Rashid, K. Dalton Trans. 2009, 39, 8301.
(a) Drake, J. L.; Manna, C. M.; Byers, J. A. Organometallics 2013, 32, 6891.
(b) Spasyuk, D.; Smith, S.; Gusev, D. G. Angew. Chem., Int. Ed. 2013, 52, 2538.
(c) Moore, C. M.; Bark, B.; Szymczak, N. K. ACS Catal. 2016, 6, 1981.
John, J. M.; Takebayashi, S.; Dabral, N.; Miskolzie, M.; Bergens, S. H. J. Am. Chem. Soc. 2013, 135, 8578. doi: 10.1021/ja401294q
Dub, P. A.; Henson, N. J.; Martin, R. L.; Gordon, J. C. J. Am. Chem. Soc. 2014, 136, 3505. doi: 10.1021/ja411374j
Ogata, O.; Nakayama, Y.; Nara, H.; Fujiwhara, M.; Kayaki, Y. Org. Lett. 2016, 18, 3894. doi: 10.1021/acs.orglett.6b01900
(a) Ohkuma, T.; Koizumi, M.; Muñiz, K.; Hilt, G.; Kabuto, C.; Noyori, R. J. Am. Chem. Soc. 2002, 124, 6508.
(b) Guo, R.; Chen, X.; Elpelt, C.; Song, D.; Morris, R. H. Org. Lett. 2005, 7, 1757.

图式 1 催化草酸酯加氢反应过程
Scheme 1 Reaction process for the catalytic hydrogenation of oxalates

图 1 (PPh3)3RuCl2/nL5催化DMO加氢制MG的反应结果
Figure 1 Catalyzing performance of (PPh3)3RuCl2/nL5 in hydrogenation of DMO into MG
Reaction conditions: Ru (0.5 mol%), NaOMe (5 mol%), THF (10 mL), p(H2)=5.0 MPa, 100 ℃

图式 4 推测的催化DMO加氢制MG的反应机理
Scheme 4 Proposed mechanism for the catalytic hydrogenation of DMO into MG
表 1 乙二醇的工业制备方法及其优缺点
Table 1. Advantages and disadvantages of industrial preparation of ethylene glycol
| Entry | 制备方法 | 原料 | 制备过程 | 优势 | 不足 |
| 1 | 乙烯直接水合法 | 乙烯 | C2H4+1/2 O2+H2O |
原料成本较低 | 设备腐蚀严重、 分离精馏成本高 |
| 2 | 二氯乙烷水解法 | 乙烯 | C2H4+Cl2 |
— | 设备腐蚀严重、 总成本高 |
| ClCH2CH2Cl+2 H2O |
|||||
| 3 | 碳酸乙烯酯法 | 乙烯 | C2H4+1/2 O2 |
反应条件温和、 能耗低 |
设备总投资大 |
| C2H4O+CO2 |
|||||
| (CH2O)2CO+H2O |
|||||
| 4 | 环氧乙烷水合法 | 乙烯 | C2H4+1/2 O2 |
乙二醇收率高 | 催化剂选择性、 稳定性有待提高 |
| C2H4O+H2O |
|||||
| 5 | 草酸酯法 | 合成气 | 2 CO+1/2 O2+2 ROH |
反应条件温和、工艺要求低 | 催化剂选择性、稳定性有待提高 |
| ROOCCOOR+4 H2
|
 下载: 导出CSV
下载: 导出CSV
表 2 不同催化剂体系催化DEO加氢a
Table 2. Catalytic hydrogenation of DEO with different catalyst systems
| ||||
| Entry | Cat. | Conv./% | Yield/% of ethyl glycolate |
Yield/% of EG |
| 1 | 1 | 100 | 0 | 92 |
| 2 | 2 | 100 | 0 | 96 |
| 3 | 3 | 91 | 89 | 0 |
| 4 | 4 | 100 | 96 | 0 |
| 5 | L1/Ru(acac)3 | 0 | 0 | 0 |
| 6 | L3/Ru(acac)3 | 0 | 0 | 0 |
| 7 | L4/Ru(acac)3 | 4 | 0 | 0 |
| 8 | L5/Ru(acac)3 | 7 | 0 | 0 |
| 9b | L1/Ru(acac)3 | 100 | 0 | 96 |
| a Reaction conditions: DEO (3.7 mmol), Ru (0.0201 mmol), NaOEt (0.2 mmol), THF (20 mL), p(H2)=6.0 MPa, 100 ℃, 20 h; b MeOH as solvent. | ||||
下载: 导出CSV
表 3 不同反应条件下2催化DEO加氢a
Table 3. Catalytic hydrogenation of DEO with 2 under different reaction conditions
| Entry | n(DEO)/n(2) | Temp./℃ | p(H2)/MPa | Time/h | Conv./% | Yield/% of ethyl glycolate | Yield/% of EG | TOFd/h-1 |
| 1b | 184 | 100 | 6.0 | 1 | 100 | 21 | 72 | 304 |
| 2 | 184 | 100 | 6.0 | 1 | 100 | 15 | 85 | 340 |
| 3 | 184 | 100 | 6.0 | 4 | 100 | 0 | >99 | 364 |
| 4 | 358 | 60 | 6.0 | 1 | 99 | 99 | 0 | 354 |
| 5 | 358 | r.t. | 6.0 | 120 | 100 | 7 | 86 | 5 |
| 6 | 358 | 60 | 3.0 | 1 | 50 | 46 | 0 | 165 |
| 7b | 6857 | 120 | 6.0 | 16 | 97 | 83 | 3 | 381 |
| 8b | 184c | 100 | 6.0 | 1 | 100 | 0 | 84 | 309 |
| a THF as solvent; b NaOEt (0.2 mmol); c DMO was used; d turnover frequency. | ||||||||
下载: 导出CSV
表 4 不同Ru化合物催化DMO加氢制MGa
Table 4. Catalytic hydrogenation of DMO into MG with different Ru complexes
| Entry | Cat. | n(NaOMe)/n(Ru) | Time/h | Conv./% | Yield/% of MG |
| 1 | 7 | 10 | 1 | 97 | 97 |
| 2 | 8 | 10 | 1 | 97 | 97 |
| 3 | 9 | 10 | 1 | 0 | 0 |
| 4 | 10 | 10 | 3 | 100 | 99 |
| 5 | 12 | 0 | 3 | 86 | 86 |
| 6 | 12 | 10 | 3 | 99 | 99 |
| 7 | 13 | 10 | 3 | 0 | 0 |
| 8 | 14 | 10 | 3 | 0 | 0 |
| 9 | 15 | 10 | 3 | 0 | 0 |
| 10 | A | 10 | 4 | 47 | 46 |
| 11 | C | 10 | 4 | 49 | 49 |
| 12 | D | 10 | 4 | 16 | 16 |
| a Reaction conditions: 0.5 mol% Ru, THF (10 mL), p(H2)=5.0 MPa, 100 ℃. | |||||
下载: 导出CSV
表 5 不同反应条件下8催化DMO加氢
Table 5. Catalytic hydrogenation of DMO with 8 in different reaction conditions
| Entry | n(DMO)/n(8) | n(NaOMe)/n(8) | Temp./℃ | p(H2)/MPa | Time/h | Conv./% | Yield/% of MG | Yield/% of EG |
| 1 | 200 | 10 | 100 | 2.0 | 1 | 74 | 74 | 0 |
| 2 | 200 | 10 | 100 | 2.0 | 3 | 98 | 98 | 0 |
| 3 | 200 | 10 | 60 | 5.0 | 1 | 81 | 81 | 0 |
| 4 | 200 | 10 | RT | 5.0 | 24 | 86 | 86 | 0 |
| 5 | 2000 | 10 | 100 | 5.0 | 16 | 98 | 98 | 0 |
| 6 | 100 | 20 | 120 | 5.0 | 36 | 100 | 0 | 97 |
| 7 | 200 | 5 | 40 | 5.0 | 1 | 31 | 31 | 0 |
| 8 | 200 | 10 | 40 | 5.0 | 1 | 35 | 35 | 0 |
| 9 | 200 | 15 | 40 | 5.0 | 1 | 44 | 44 | 0 |
| 10 | 200 | 40 | 40 | 5.0 | 1 | 81 | 81 | 0 |
| 11 | 200 | 80 | 40 | 5.0 | 1 | 65 | 65 | 0 |
下载: 导出CSV
表 6 催化DMO加氢制MG(和/或EG)和MB加氢制BAa
Table 6. Catalytic hydrogenation of DMO into MG (and/or EG) and that of MB into BA
| Entry | Cat. | Substrate | Conv./% | Yield/% | ||
| MG or BA | EG | |||||
| 1 | 16 | DMO | 100 | 93 (MG) | 6 | |
| 2 | MB | 95 | 93 (BA) | |||
| 3 | 17 | DMO | 100 | 91 (MG) | 7 | |
| 4 | MB | 97 | 96 (BA) | |||
| 5b | 17 | DMO | 100 | 100 (MG) | 0 | |
| 6b | MB | 7 | 5 (BA) | |||
| 7 | 18 | DMO | 0 | 0 (MG) | 0 | |
| 8 | MB | 90 | 88 (BA) | |||
| 9 | 19 | DMO | 80 | 79 (MG) | 0 | |
| 10 | MB | 3 | 1 (BA) | |||
| 11 | 20 | DMO | 100 | 86 (MG) | 13 | |
| 12 | MB | 100 | 100 (BA) | |||
| 13 | 21 | DMO | 100 | 100 (MG) | 0 | |
| 14 | MB | 57 | 56 (BA) | |||
| a Reaction conditions: 0.5 mol% Ru, 5 mol% (for DMO) or 10 mol% (for MB) NaOMe, THF (10 mL), p(H2)=5.0 MPa, 100 ℃, 4 h; b No NaOMe was used. | ||||||
下载: 导出CSV
表 7 不同Ru化合物催化DMO加氢a
Table 7. Catalytic hydrogenation of DMO with different Ru complexes
| Entry | Cat. | Conv./% | Yield/% | TOF/h-1 | |
| MG | EG | ||||
| 1 | 8 | 8 | 8 | 0 | 160 |
| 2 | 22 | 77 | 76 | 0 | 1520 |
| 3 | 23 | 27 | 27 | 0 | 540 |
| 4 | 24 | 24 | 24 | 0 | 480 |
| 5 | 25 | 0 | 0 | 0 | - |
| 6 | 26 | 36 | 36 | 0 | 720 |
| 7 | 27 | 100 | 98 | 1 | 2000 |
| 8b | 27 | 99 | 98 | 0 | 3920 |
| 9 | 28 | 6 | 6 | 0 | 120 |
| a Reaction conditions: 0.05 mol% Ru, 0.5 mol% NaOMe, THF (10 mL), p(H2)=5.0 MPa, 100 ℃, 1 h; b 0.5 h. | |||||
下载: 导出CSV
表 8 不同反应条件下27催化DMO加氢
Table 8. Catalytic hydrogenation of DMO with 27 under different reaction conditions
| Entry | n(DMO)/n(27) | n(NaOMe)/n(27) | Temp./℃ | p(H2)/MPa | Time/h | Conv./% | Yield/% | TOF/h-1 | |
| MG | EG | ||||||||
| 1 | 2000 | 5 | 100 | 5.0 | 1 | 87 | 86 | 0 | 1720 |
| 3 | 2000 | 10 | 100 | 5.0 | 1 | 100 | 98 | 1 | 2000 |
| 3 | 2000 | 15 | 100 | 5.0 | 1 | 87 | 86 | 0 | 1720 |
| 4 | 2000 | 20 | 100 | 5.0 | 1 | 68 | 67 | 0 | 1340 |
| 5 | 500 | 10 | 100 | 1.0 | 1.5 | 100 | 99 | 0 | 330 |
| 6 | 500 | 10 | r.t. | 5.0 | 20 | 96 | 95 | 0 | 24 |
| 7 | 500 | 10 | 40 | 5.0 | 2 | 100 | 99 | 0 | 247 |
| 8 | 500 | 10 | 40 | 1.0 | 16 | 95 | 94 | 0 | 29 |
| 9 | 500 | 10 | 120 | 5.0 | 4 | 100 | 0 | 97 | 242 |
| 10 | 200 | 10 | 100 | 5.0 | 4 | 100 | 0 | 99 | 99 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们









 下载:
下载: