
图式 1.
光氧化还原催化的亚胺自由基的主要生成方法和反应模式
Scheme 1.
Generation and reaction modes of iminyl radicals under photoredox catalysis

含氮化合物被广泛应用于药物、农药、食品添加剂和含能材料中[1].基于含氮化合物的重要性, 开发构建碳-氮(C—N)键的方法非常重要, 而且化学家们越来越多地关注于提高C—N键的构建效率、选择性和可持续性. C—N键的构建已经取得了很多进展, 经典的C—N键的构建大都利用氮原子的亲核性.最近基于氮自由基C—N键的构建方法作为传统离子型反应的补充开始引起人们的关注[2].然而, 缺乏产生氮自由基的可靠高效清洁的方法, 极大地制约了氮自由基化学的发展.可见光氧化还原催化介导的自由基型反应, 因环境友好、条件温和以及反应易处理等优点, 受到化学家们的青睐[3].利用可见光氧化还原催化技术可以高效地产生氮自由基, 使得氮自由基化学得到很大的发展.目前氮自由基化学的研究也成为有机合成领域的一个热点, 特别是在光氧化还原催化方面[4].
亚胺自由基(iminyl radical)是经典的氮自由基主要类型之一(还包括酰胺自由基、胺基自由基及其铵正离子自由基等), 由于其分子内存在C(sp2)—N键, 属于σ型自由基, 既有亲电性也有亲核性[5].肟的衍生物由于分子内存在较弱的N—O键(BDE≈209 kJ/mol)[6], 成为产生亚胺自由基的优良前体.目前研究较多的主要有三类肟衍生物: (1) O-酰基肟衍生物, (2) O-芳基肟衍生物和(3) α-亚胺氧杂酸(α-imino-oxy acid)衍生物(Scheme 1).前两类肟的衍生物可以在可见光氧化还原催化条件下被激发态的光敏剂还原产生亚胺自由基, 第三类肟的衍生物可以被激发态的光敏剂氧化产生亚胺自由基.亚胺自由基参与的反应模式主要有以下四种: (1)对芳烃的加成反应; (2)分子内的氢原子迁移及后继反应; (3)对烯烃的加成反应; (4) Norrish-I型断裂(α-位的C—C键断裂)及后继反应(Scheme 1).本文主要总结了近几年来在光氧化还原催化的条件下, 以肟的衍生物作为前体, 亚胺自由基的产生及其参与的典型反应.
2015年, 我们课题组[7]利用可见光氧化还原催化技术, 从O-酰基的肟1高效清洁地产生亚胺自由基.该自由基随后对芳烃进行分子内加成, 发生分子内亚胺化, 制备了一系列的含氮杂芳环2, 包括吡啶、喹啉和菲啶等(Scheme 2a).该反应利用O-酰基的肟与激发态光敏剂的单电子转移来还原断裂N—O键, 从而生成亚胺自由基(Scheme 2b).由于O-酰基的引入增加了底物的氧化性和离去基团的离去性能, 所以是该策略取得成功的关键原因.为了进一步提高反应效率, 制备了新试剂O-苯甲酰基羟胺(BHA), 该试剂可以在布朗斯特酸帮助下与醛3原位生成O-酰基的肟, 再在光氧化还原催化条件下发生分子内的亚胺自由基对芳烃的加成反应.从醛出发, “一锅法”合成含氮芳杂环4[8](Scheme 2c). 2017年, 我们[9]又报道了一种以O-2, 4-二硝基苯肟醚5为原料, 在没有光催化剂存在的情况下使用可见光照射, 同样可以合成菲啶和喹啉类化合物6 (Scheme 2d).该反应是借助O-2, 4-二硝基苯肟醚和Et3N形成电子给体受体(EDA)复合物介导的电子转移实现的.

2016年, 谢红旗和程辟课题组[10]开发了一种无金属可见光氧化还原催化O-2, 4-二硝基苯氧肟醚分子7的环化反应(Scheme 2e).该策略使用的有机染料型光催化剂eosin Y相较于Ru或Ir配合物光敏剂, 价格便宜, 反应条件简单、温和, 环保. Akita与Koike等[11]报道了由N-(2-芳基苄基氨基)吡啶盐9选择性合成取代菲啶类化合物的方法(Scheme 2f).该方法中, 光激发中间体11中的吡啶基从末端芳基处获得一个电子, 生成关键的自由基中间体, 具有较高三重态能量的光催化剂, 在前体的能量传递中也起着至关重要的作用.
C(sp3)—H直接官能团化是有机化学领域极具挑战性的课题, 其经典解决方案之一是Hofmann-Löffler- Freytag反应(HLF反应), 反应的关键步骤是分子内的1, 5-氢原子迁移(HAT). HLF反应的发现已经超过一百年, 该反应还有一些明显的缺点, 比如, 反应条件非常苛刻, 经典的HLF反应通常需要在强酸性介质中, 紫外光解(或者加热)胺的氯化物产生氮自由基; HLF反应通常实现远程C(sp3)—H的卤化或者分子内的胺化, 其他类型的官能团化难以引入, 特别是远程C—C键的构建非常具有挑战性.
2015年, 我们[12a]使用可见光促进的光氧化还原催化技术, 利用N-氯磺酰胺作为酰胺自由基前体, 通过酰胺自由基的1, 5-HAT过程, 成功地在近中性的条件下实现了远程C(sp3)—H的氯化和分子内酰胺化反应.该方案首次使用光氧化还原催化技术解决远程C(sp3)—H官能团化问题, 避免了经典HLF反应的强酸性和紫外光照射等苛刻条件.在这一工作的激发下, 我们[12b]进一步通过亚胺自由基的1, 5-HAT过程实现了C(sp3)—H官能团化.使用O-酰基肟衍生物14作为亚胺自由基前体, 在光氧化还原的条件下产生亚胺自由基, 发生1, 5-HAT过程, 远程C(sp3)—H使用烯基硼酸15作为碳自由基捕获剂, 实现了远程C(sp3)—H烯基化, 将经典的HLF反应拓展到C—C键构建领域(Scheme 3).
2017年, Nevado等[13]同样使用O-酰基肟衍生物17作为亚胺自由基前体, 在可见光氧化还原催化的条件下, 利用1, 5-HAT过程实现了分子内的C(sp3)—H芳基化和亚胺化反应(Scheme 4).当使用醋酸作为添加剂时, 1, 5-HAT发生分子内芳基化, 生成苯并环己酮衍生物18.当反应加入碱(DABCO)时, 1, 5-HAT发生分子内亚胺化, 构建C—N键, 生成2H-吡咯类化合物19.

2018年, 段新华等[14]也使O-酰基肟衍生物20作为亚胺自由基前体, 在可见光氧化还原催化的条件下, 利用1, 5-HAT过程, 使用苯乙烯衍生物21作为碳自由基的捕获剂, 分别实现了远程C(sp3)—H烷基化和烯基化(Scheme 5).他们发现反应的溶剂和添加剂对反应的结果至关重要, 当反应加入水时, 烷基自由基被氧化为碳正离子后, 被水捕获, 得到远程羟基烷基化产物22 (Scheme 5a).各种醇(甲醇, 乙醇和异丙醇等)也作为亲核试剂捕获碳正离子, 获得相应的醚化产物. 2019年, 他们课题组[15]在此基础上实现了烯基化反应.用同样的苯乙烯衍生物作为碳自由基受体, 将反应溶剂改为二甲基亚砜(DMSO), 并用布朗斯特酸作为添加剂, 得到相应远程烯基化产物23 (Scheme 5b).
2017年, Leonori等[16]采用α-亚胺氧杂酸衍生物24作为亚胺自由基前体, 实现了一系列远程C(sp3)—H卤化和叠氮化反应(Scheme 6a).与O-酰基肟通过氧化淬灭激发态的光敏剂产生亚胺自由基不同, α-亚胺氧杂酸是通过还原淬灭激发态的光敏剂产生亚胺自由基.结果表明该反应有广泛的底物适应性, 并且同样适用于天然产物的后期修饰.几乎同时, Studer等[17]利用α-亚胺氧杂酸衍生物27作为亚胺自由基前体, Michael受体28作为碳自由基捕获剂, 以类似的策略实现了酮远程γ-位烷基化反应(Scheme 6b).

随后, 利用亚胺自由基1, 5-HAT的策略实现C—N、C—S以及N—S键的构建也得到发展.付华[18]与吴劼[19]课题组先后报道了简单实用的可见光促进的亚胺自由基介导的分子内C—H活化串联环化反应和分子间插SO2反应.两种反应只需要可见光照射, 不需要额外添加催化剂、氧化剂、酸和碱.以上反应是借助O-2, 4-二硝基苯肟醚与胺或DABCO·(SO2)2形成EDA复合物, 随后N—O键断裂生成亚胺自由基, 再进行后续反应实现咪唑31以及噻嗪类衍生物34的合成(Scheme 7).

亚胺自由基对烯烃的加成环化反应是构建含氮杂环类化合物的重要方法之一.早在1997年, Zard和Newcomb等[20]发现, 酮腙的衍生物35在Bu3SnH的作用下, 能够发生N—N键均裂产生亚胺自由基36, 随后亚胺自由基对C=C双键进行分子内加成, 发生5-exo- trig环化反应, 最后攫取氢原子得到产物37 (Scheme 8a).随后他们发现在镍粉作为还原剂的条件下, O-乙酰基肟38被单电子还原产生亚胺自由基, 该自由基发生环化形成的碳自由基有三种不同的反应途径: (1)攫取氢原子得到产物39, (2)被氧化成碳正离子后消除得到烯烃40, (3)被氧化成碳正离子后被醋酸捕获得到醋酸酯41 (Scheme 8b)[21].

Narasaka等[22]发现在紫外光激发下, 1, 5-二甲氧基萘(DMN)与O-芳基肟醚衍生物发生单电子转移, 形成亚胺自由基, 对C=C双键进行分子内加成后从1, 4-环己烯中攫取氢原子得到2H-吡咯类化合物43 (Scheme 9).这些早期的例子启发了可见光氧化还原催化的条件下, 亚胺自由基对烯烃加成反应的研究.
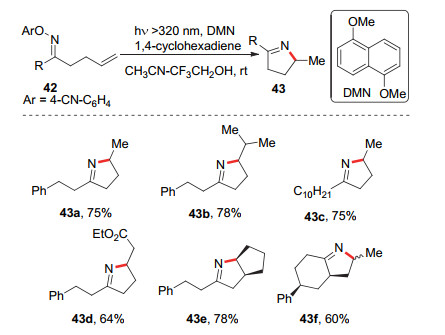
2015年, Leonori等[23]发现了可见光氧化还原催化的条件下, O-芳基肟醚衍生物44可以氧化淬灭激发态的光催化剂产生亚胺自由基, 对烯烃进行分子内加成, 最后攫取氢原子得到最终产物45.其反应途径如Scheme 10中path a所示, 反应始于肟醚44与激发态的光敏剂的单电子转移, 产生亚胺自由基, 随后发生5-exo-trig环化形成烷基自由基47, 自由基47从环己二烯中攫取一个氢原子得到产物45.同时, 他们还发现O-芳基肟醚衍生物44可以与三级胺形成EDA复合物, 该复合物可以被可见光激发, 电子转移后, O-芳基肟醚发生碎片化, 生成亚胺自由基和2, 4-二硝基苯酚负离子.该亚胺自由基同样发生5-exo-trig环化.与可见光氧化还原催化的条件的后继反应不同, 环化产生的烷基自由基48没有攫取氢原子, 而是被2, 4-二硝基苯酚负离子氧化成醇类化合物52(Scheme 10, path b).

2016年, 冯超和Loh等[24]报道一个亚胺自由基启动的O-酰基肟衍生物53与烯醇硅醚54的自由基串联反应(Scheme 11).与前面的反应类似, 亚胺自由基发生环化后得到烷基自由基, 该自由基进一步被烯醇硅醚54捕获, 最终得到高度官能团化的2H-吡咯衍生物55.

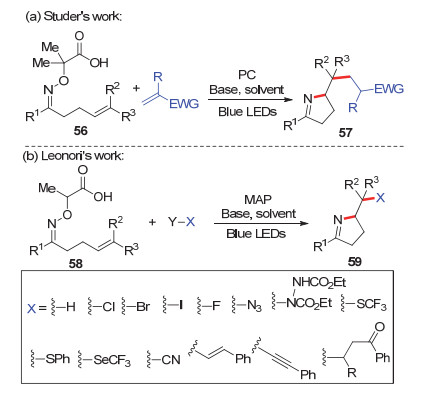
2017年, Studer和Leonori分别独立地发展了α-亚胺氧杂酸衍生物56或者58作为亚胺自由基前体及其启动的烯烃参与串联反应(Scheme 12). Studer等[25]使用缺电子的烯烃作为烷基自由基的捕获剂, 得到烷基化的2H-吡咯衍生物57 (Scheme 12a). Leonori等[26]使用类似的策略, 考察了一系列烷基自由基的捕获剂, 一系列高度官能团化的2H-吡咯衍生物59, 包括卤化、硫三氟甲基化、叠氮化、氰基化、烯基化、炔基化、胺化、硫化等, 产物的产率从中等到优良(Scheme 12b).这些反应都有一个问题没有解决, 当产物有多个手性中心时, 产物的非对映选择性很不理想.
亚胺自由基的Norrish-I型断裂(α位的C—C键断裂)是由亚胺自由基产生其他自由基(特别是碳中心自由基)的高效方法, 伴随着生成一个稳定的氰基.其中开环断裂反应为官能团化脂肪腈的合成提供了一种重要的方法[27].这一反应类型中, 研究最为充分的是环丁酮衍生物的开环官能团化反应.由于四元环的张力较大, 环丁酮衍生的亚胺或者肟的衍生物通过加热或光照发生N—X键(X=O, S, N等)的断裂形成亚胺自由基, 发生开环反应形成烷基自由基, 该自由基可以进一步反应引入不同的官能团.这一领域的先驱者Zard等[28]发现N-巯苯基环丁酮亚胺60在Bu3SnH的促进下发生N—S键均裂, 能够生成亚胺自由基61, 环张力导致亚胺自由基的α位C—C键断裂, 发生连续的开环反应, 生成烷基自由基62, 被缺电子的烯烃捕获后最终得到双环化合物63 (Scheme 13).

由于可见光氧化还原催化条件的温和与高效性, 可以兼容更多的烷基自由基的捕获剂, 实现更多类型的化学转化.最常用的烷基自由基的捕获剂为烯烃及其衍生物. 2017年, 肖文精和陈加荣等报道了一系列烯烃作为烷基自由基捕获剂的工作.他们报道可见光促进的环酮肟酯52经历亚胺自由基开环后, 烷基自由基被芳基乙烯类化合物捕获, 实现了烯基化反应[29]以及氧化烷基化反应(Scheme 14a)[30].他们尝试了多种环酮肟酯, 包括杂环、螺环、环张力较小环戊酮、环己酮以及樟脑衍生物, 能够得到中等到优良的产率.此种反应策略可以将烯烃拓展为烯基叠氮、烯基腙等, 具有优良的底物适应性.

随后, 他们报道了3-芳基环丁酮肟酯73和缺电子烯烃74的自由基加成/环化串联反应.这种温和且氧化还原中性的策略提供了一种高效实用的方法来获得各种氰烷基化的1, 2, 3, 4-四氢菲产物75, 具有良好的区域选择性和产率[31].周磊等[32]报道了可见光促进环丁酮肟酯76与烯烃77和醇的三组分反应.他们对环丁酮肟酯、烯烃以及亲核试剂进行了研究, 以中等到优良的产率得到了一系列双官能团化反应(Scheme 14b).我们课题组[12b]也研究了环酮肟酯67开环官能团化反应(Scheme 14c).用烯基硼酸68作为烷基自由基的捕获剂, 实现了开环烯基化反应.激发态的光敏剂被环丁酮肟酯氧化淬灭, 生成亚胺自由基, 开环后得到烷基自由基, 对烯基硼酸进行加成, 形成的自由基中间体被高价态光敏剂氧化成碳正离子, 脱除硼酸后得到目标产物69 (Scheme 14d).
Leonori等[16]报道了可见光促进的环戊酮肟羧酸衍生物开环氟化反应(Scheme 15a).他们采用还原淬灭的方式从环戊酮肟羧酸衍生物79获得亚胺自由基, 开环形成烷基自由基, 随后与亲电试剂氟化剂反应最终生成氟化产物80.该反应的立体选择性不能很好地控制, 产物的非对映选择性比较差. Waser等[33]使用炔基的高价碘化合物82作为烷基自由基捕获剂, 实现了开环炔基化反应(Scheme 15b).四元和五元环酮衍生的肟羧酸均能有效裂解, 在温和的条件下以良好的收率得到含有炔烃的腈类化合物83.

可见光促进的环酮肟酯经历亚胺自由基开环后烷基自由基也可以进行SO2插入反应, 得到一系列氰基砜类化合物(Scheme 16).比如, 吴劼和叶盛青等[34]报道了在光催化下, 通过亚胺自由基和二氧化硫的插入实现了环酮肟酯的多组分磺酰化反应.在可见光的照射下, O-酰基肟84被激发态的光催化剂还原产生亚胺自由基, 开环后得到烷基自由基, 该自由基随后经历SO2插入、氧化单电子转移和亲核进攻等一系列转化生成官能团丰富的β-烷氧基(或者羟基)砜87 (Scheme 16a).陆红健等[35]报道了烯醇的三氟磺酸酯或者烯丙基三氟甲基砜89作为自由基受体和SO2源, 无需外部加入SO2, 构建各类结构丰富多样的β-酮砜和烯丙基砜90 (Scheme 16b).唐课文等[36]报道了使亚甲基环丙烷92作为自由基受体, 反应经历亚胺自由基的形成、C—C键断裂、SO2插入、对亚甲基环丙烷的加成、三元环开环和环化等一系列过程, 合成了2-氰基磺化-3, 4-二氢萘93 (Scheme 16c).

环酮肟酯开环后烷基自由基也可以被含氮杂芳环捕获, 实现Minisci类型的氮杂芳环的烷基化.比如, 夏吾炯等[37]报道了可见光促进的Minisci型氮杂芳环氰烷基化.这种光化学方法为氰基取代杂芳烃的合成提供了方便, 对药物的开发具有一定的意义(Scheme 17).

肖文精和陈加荣等[38]使用有机染料作为光敏剂借助分子间的HAT过程产生亚胺自由基.该反应始于O-苄基肟醚97与激发态的光敏剂发生分子间的HAT过程, 生成的苄基自由基发生β-断裂产生亚胺自由基, C—C键裂解形成的烷基自由基, 对烯烃98进行加成, 攫取氢原子得到最后的产物99 (Scheme 18).这种通过HAT过程产生亚胺自由基方法与通过电子转移产生亚胺自由基的方法互为补充, 能够实现更多的化学转化.

肖文精和陈加荣等[39]最近报道了一系列可见光促进的铜催化的环酮肟酯参与的多组分反应, 比如环丁酮肟酯100/苯乙烯101/芳基硼酸102 (Scheme 19a)、环丁酮肟酯104/烯烃105/末端炔106 (Scheme 19b)[40]和环丁酮肟酯108/一氧化碳/苯胺109 (Scheme 19c)[41]的三组分自由基交叉偶合反应.最近, 他们课题组又开发了一种可见光驱动铜催化的环丁酮肟酯112与酚类或醇类113的C(sp3)—O交叉偶联反应[42].这一策略也适用于苄基叠氮化、硫氰化和硫醚化(Scheme 19d).在这些反应中, 无需外加贵金属钌或者铱的配合物作为光敏剂, 廉价金属铜的配合物作为吸收光子的物种, 扮演光敏剂的作用, 同时金属铜还参与化学键的断裂和形成, 是一个双功能催化剂(Scheme 19e).该策略进一步拓展了亚胺自由基参与的反应类型, 提供了构建有价值的官能团丰富的有机腈类化合物.

为了进一步拓展光催化反应的适用范围, 过渡金属与光协同催化的反应模式受到了关注.在这一反应模式中, 光催化剂可以与过渡金属发生能量或者电子转移, 过渡金属参与化学键的形成或者断裂.这一反应模式大大拓展了光催化反应的适用范围, 在亚胺自由基化学中也可以得到应用. Leonori等[43]报道了使用可见光氧化还原与镍协同催化策略可以实现环酮肟羧酸衍生物开环芳基化、烯基和烷基化反应(Scheme 20).该策略采用环丁酮肟羧酸衍生物115在可见光氧化还原催化下脱羧和丙酮生成亚胺自由基, 开环形成的烷基自由基在镍催化下实现芳基化(116)、烯基化(117)和烷基化(118).

肖文精和陈加荣等[44]实现了光氧化还原与铜协同催化环酮肟酯119的对映选择性的开环氰基化反应(Scheme 21a).该策略展示了广泛的底物范围, 可以制备各种具有光学活性的烷基二腈, 具有高收率和优良的对映体选择性.几乎同时, 王细胜等[45]报道了四元和五元环酮肟酯122的自由基介导的开环和对映选择性氰基化反应(Scheme 21b).该策略可通过铜催化或者光氧化还原与铜协同催化两种反应条件分别构建1, 5-和1, 6-二氰手性化合物.这两个工作是为数不多的亚胺自由基参与的对映选择性反应.

除了环酮肟酯的开环反应外, 亚胺基自由基还可以经历非环状底物的Norrish-I型断裂反应.杨尚东[46]与吴骊珠等[47]发现由O-酰基肟衍生物产生的亚胺自由基可以发生Norrish-I型断裂反应, 分别产生膦中心自由基碳中心自由基, 同时释放小分子腈类化合物.膦中心自由基或酰基自由基可以被苯乙烯衍生物捕获, 得到的苄位再经历氧化、Ritter/Mumm重排反应或与胺以及羧酸的亲核环化生成β-氨基膦酸酯127, 131, 132 (Schemes 22a, 22b)或β-氨基酰基化产物135, 138, 139 (Schemes 22c, 22d). Ritter/Mumm重排反应序列在氮自由基启动的烯烃加成反应中也同样被观察到[48].烷基自由基经历氧化后, 直接接受亲核试剂(醇或胺)的进攻, 生成醚类143或胺类产物144 (Scheme 22e).该方法的特点是能够在不需要氧化剂和碱的情况下构建C(sp3)—P和C(sp3)—N以及C(sp3)—O键(Scheme 22).

亚胺自由基为中间体参与的反应是构建羰基以及含氮化合物的重要方法.可见光还原氧化催化反应具有环境友好、温和的反应条件以及操作简单等优点, 其原理是通过激发态的光敏剂与底物实现单电子转移生成自由基或自由基离子中间体, 进而实现下一步的转化.本文详细总结了光氧化还原催化条件下以肟的衍生物作为前体, 亚胺自由基的产生及其参与的反应, 主要包括对芳烃的加成反应、分子内的氢迁移、对烯烃的加成反应以及Norrish-I型断裂反应等.在光氧化还原催化条件下亚胺自由基的产生方便高效, 其参与的反应类型也较为丰富.但鉴于亚胺自由基的高活性, 其参与反应的立体选择性通常较差, 对映选择性的反应极少报道.亚胺自由基参与的反应立体选择性(包括非对映选择性和对映选择性)控制是该领域亟需解决的问题, 也是未来重要的研究方向.
Lawrence, S. A. Amines: Synthesis Properties and Applications, Cambridge University, Cambridge, 2005, p. 1016.
Zard, S. Z. Chem. Soc. Rev. 2008, 37, 1603. doi: 10.1039/b613443m
(a) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322.
(b) Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116, 10035.
(c) Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075.
(d) Chen, Y.; Lu, L.-Q; Yu, D.-G; Zhu, C.-J; Xiao, W.-J. Sci. China Chem. 2019, 62, 24.
(e) Chen, F.; Chen, H.; Wu, Q.-A.; Luo, S.-P. Chin. J. Org. Chem. 2020, 40, 339 (in Chinese).
(陈锋, 陈浩, 吴庆安, 罗书平, 有机化学, 2020, 40, 339.)
(a) Davies, J.; Morcillo, S. P.; Douglas, J. J.; Leonori, D. Chem. Eur. J. 2018, 24, 12154.
(b) Kärkäs, M. D. ACS Catal. 2017, 7, 4999.
(c) An, X.-D.; Yu, S. Tetrahedron Lett. 2018, 59, 1605.
(d) Jackman, M. M.; Cai, Y.; Castle, S. L. Synthesis 2017, 49, 1785.
(e) Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044.
(f) Xiao, L; Li, J.-H; Wang, T. Acta Chim. Sinica 2019, 77, 841 (in Chinese).
(肖丽, 李嘉恒, 王挺, 化学学报, 2019, 77, 841.)
(g) Xu, X.-L.; Wan, X.; Geng, Y.; Zhang, J.-S.; Xu, H.-J. Chin. J. Org. Chem. 2011, 31, 453 (in Chinese).
(徐小岚, 万薪, 耿烨, 张家松, 许华建, 有机化学, 2011, 31, 453).
(h) Xiong, T.; Zhang, Q. Chem. Soc. Rev. 2016, 45, 3069.
(i) Song, H.; Liu, X.-Y.; Qin, Y. Acta Chim. Sinica 2017, 75, 1137 (in Chinese).
(宋颢, 刘小宇, 秦勇, 化学学报, 2017, 75, 1137.)
(j) Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.; Xiao, W.-J.; Chen, J.-R. Acc. Chem. Res. 2020, Acc. Chem. Res. 2020, 53, 1066.
Walton, J. C. Acc. Chem. Res. 2014, 47, 1406. doi: 10.1021/ar500017f
Luo, Y.-R. Handbook of Bond Dissociation Energies in Organic Compounds, CRC, Boca Raton, 2003, p. 392.
Jiang, H.; An, X. D.; Tong, K.; Zheng, T.; Zhang, Y.; Yu, S. Angew. Chem., Int. Ed. 2015, 54, 4055. doi: 10.1002/anie.201411342
An, X.-D.; Yu, S. Org. Lett. 2015, 17, 2692. doi: 10.1021/acs.orglett.5b01096
Sun, J.-J; He, Y.-Y; An, X.-D; Zhang, X; Yu, L; Yu, S. Org. Chem. Front. 2018, 5, 977. doi: 10.1039/C7QO00992E
Liu, X.-B.; Qing, Z.-X.; Zheng, X.-Y.; Zeng, J.-G.; Cheng, P.; Xie, H.-Q. Molecules 2016, 21, 1690. doi: 10.3390/molecules21121690
Matsushita, Y.; Ochi, R. Tanaka, Y.; Koike, T.; Akita, M. Org. Chem. Front. 2020, 7, 1243. doi: 10.1039/D0QO00271B
(a) Qin, Q.-X; Yu, S. Org. Lett. 2015, 17, 1894.
(b) Shen, X.; Zhao, J.-J.; Yu, S. Org. Lett. 2018, 20, 5523.
Shu, W.; Nevado, C. Angew. Chem., Int. Ed. 2017, 56, 1881. doi: 10.1002/anie.201609885
Ma, Z.-Y.; Guo, L.-N.; Gu, Y.-R.; Chen, L.; Duan, X.-H. Adv. Synth. Catal. 2018, 360, 4341. doi: 10.1002/adsc.201801198
Chen, L; Guo, L.-N.; Ma, Z.-Y.; Gu, Y.-R.; Zhang, J.; Duan, X.-H. J. Org. Chem. 2019, 84, 6475. doi: 10.1021/acs.joc.9b00525
Dauncey, E. M.; Morcillo, S. P.; Douglas, J. J.; Sheikh, N. S.; Leonori, D. Angew. Chem., Int. Ed. 2018, 57, 744. doi: 10.1002/anie.201710790
Jiang, H.; Studer, A. Angew. Chem., Int. Ed. 2018, 57, 1692. doi: 10.1002/anie.201712066
Li, J.-J.; Zhang, P.-P.; Jiang, M.; Yang, H.-J.; Zhao, Y.-F.; Fu, H. Org. Lett. 2017, 19, 1994. doi: 10.1021/acs.orglett.7b00533
Li, Y.-W.; Mao, R.-Y.; Wu, J. Org. Lett. 2017, 19, 4472. doi: 10.1021/acs.orglett.7b02010
Tadic-Biadatti, M.-H. L.; Callier-Dublanchet, A.; Horner, J. H.; Quiclet-Sire, B.; Zard, S. Z.; Newcomb, M. J. Org. Chem. 1997, 62, 559. doi: 10.1021/jo961530+
Boivin, J; Schiano, A.-M; Zard, S. Z; Zhang, H.-W. Tetrahedron Lett. 1999, 40, 4531. doi: 10.1016/S0040-4039(99)00720-0
Mikami, T.; Narasaka, K. Chem. Lett. 1999, 24, 338.
Davies, J.; Booth, S. G.; Essafi, S.; Dryfe, Robert A. W.; Leonori, D. Angew. Chem., Int. Ed. 2015, 54, 14017. doi: 10.1002/anie.201507641
Cai, S.-H.; Xie, J.-H; Song, S.-J.; Ye, L.; Feng, C.; Loh, T. P. ACS Catal. 2016, 6, 5571. doi: 10.1021/acscatal.6b01230
Jiang, H.; Studer, A. Angew. Chem., Int. Ed. 2017, 56, 12273. doi: 10.1002/anie.201706270
Davies, J.; Sheikh, N. S.; Leonori, D. Angew. Chem., Int. Ed. 2017, 56, 13361. doi: 10.1002/anie.201708497
(a) Yin, W.; Wang, X. New J. Chem. 2019, 43, 3254.
(b) Xiao, T.-B.; Huang, H.-T.; Anand, D.; Zhou, L. Synthesis 2020, Synthesis 2020, 52, 1585.
Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron 1994, 50, 1757. doi: 10.1016/S0040-4020(01)80850-4
Yu, X.-Y.; Chen, J.-R.; Wang, P.-Z.; Yang, M.-N.; Liang, D.; Xiao, W.-J. Angew. Chem., Int. Ed. 2018, 57, 738. doi: 10.1002/anie.201710618
He, B.-Q.; Yu, X.-Y.; Wang, P.-Z.; Chen, J.-R.; Xiao, W.-J. Chem. Commun. 2018, 54, 12262. doi: 10.1039/C8CC07072E
Wang, P.-Z.; Yu, X.-Y.; Li, C.-Y.; He, B.-Q.; Chen, J.-R.; Xiao, W.-J. Chem. Commun. 2018, 54, 9925. doi: 10.1039/C8CC06145A
Li, L.; Chen, H.; Mei, M.; Zhou, L. Chem. Commun. 2017, 53, 11544. doi: 10.1039/C7CC07347J
Vaillant, F. L.; Garreau, M.; Nicolai, S.; Grynova, G.; Corminboeuf, C.; Waster, J. Chem. Sci. 2018, 9, 5883. doi: 10.1039/C8SC01818A
Zhang, J.; Li, X.-F.; Xie, W.-L.; Ye, S.-Q.; Wu, J. Org. Lett. 2019, 21, 4950. doi: 10.1021/acs.orglett.9b01323
Zheng, M.; Li, G.-L.; Lu, H.-J. Org. Lett. 2019, 21, 1216. doi: 10.1021/acs.orglett.9b00201
Liu, Y.; Wang, Q.-L.; Chen, Z.; Li, H.; Xiong, B.-Q.; Zhang, P.-L.; Tang, K.-W. Chem. Commun. 2020, 56, 3011. doi: 10.1039/C9CC10057A
Jian, Y.; Chen, M.; Yang, Chao; Xia, W.-J. Eur. J. Org. Chem. 2020, 1439.
Wang, P.-Z.; He, B.-Q.; Cheng, Y.; Chen, J.-R.; Xiao, W.-J. Org. Lett. 2019, 21, 6924. doi: 10.1021/acs.orglett.9b02535
Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.; Chen, J.-R.; Xiao, W.-J. Angew. Chem., Int. Ed. 2018, 57, 15505. doi: 10.1002/anie.201809820
Chen, J.; He, B-Q.; Wang, P.-Z.; Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.-R.; Xiao, W.-J. Org. Lett. 2019, 21, 4359. doi: 10.1021/acs.orglett.9b01529
Lu, B.; Cheng, Y.; Chen, L.-Y.; Chen, J. R.; Xiao, W.-J. ACS Catal. 2019, 9, 8159. doi: 10.1021/acscatal.9b02830
Yu, X.-Y.; Chen, J.; Chen, H.-W.; Xiao, W.-J.; Chen, J.-R. Org. Lett. 2020, 22, 2333. doi: 10.1021/acs.orglett.0c00532
Dauncey, E. M.; Dighe, S. U.; Douglas, J. J.; Leonori, D. Chem. Sci. 2019, 10, 7728. doi: 10.1039/C9SC02616A
Chen, J.; Wang, P.-Z.; Lu, B.; Liang, D.; Yu, X.-Y.; Xiao, W.-J.; Chen, J. R. Org. Lett. 2019, 21, 9763. doi: 10.1021/acs.orglett.9b03970
Wang, T.; Wang; Y.-N.; Wang, R.; Zhang, B.-C.; Yang, C.; Li, Y.-L.; Wang, X.-S. Nat. Commun. 2019, 10, 5373. doi: 10.1038/s41467-019-13369-x
(a) Li, Y.-H.; Wang, C.-H.; Gao, S.-Q.; Qi, F.-M.; Yang, S.-D. Chem. Commun. 2019, 55, 11888.
(b) Li, C.; Qi, Z.-C.; Yang, Q.; Qiang, X.-Y.; Yang, A.-D. Chin. J. Chem. 2018, 36, 1052.
(a) Cheng, Y.-Y.; Lei, T.; Su, L.-L.; Fan, X.-W.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Org. Lett. 2019, 21, 8789.
(b) Fan, X.-W.; Lei, T.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Org. Lett. 2019, 21, 4153.
(c) Fan, X.-W.; Lei, T.; Liu, Z.; Yang, X.-L.; Cheng, Y.-Y.; Liang, G.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Eur. J. Org. Chem. 2020, 10, 1551.
Qin, Q.; Han, Y.-Y; Jiao, Y.-Y; He, Y.; Yu, S. Org. Lett. 2017, 19, 2909. doi: 10.1021/acs.orglett.7b01145

图式 1 光氧化还原催化的亚胺自由基的主要生成方法和反应模式
Scheme 1 Generation and reaction modes of iminyl radicals under photoredox catalysis

图式 2 可见光促进的亚胺自由基对芳基的加成反应
Scheme 2 Visible light-promoted addition of iminyl radicals to arenes

图式 6 α-亚胺氧杂酸衍生物的远程C(sp3)—H官能团化
Scheme 6 Remote C(sp3)—H functionalization from α-imino- oxy acids

图式 7 EDA复合物介导的远程C(sp3)—H官能团化反应
Scheme 7 EDA complex-mediated remote C(sp3)—H functionalization

图式 8 Bu3SnH介导的亚胺自由基对烯烃的分子内加成反应
Scheme 8 Bu3SnH-mediated intramolecular addition of iminyl radicals to alkenes

图式 9 紫外光诱导的亚胺自由基对烯烃的分子内加成反应
Scheme 9 UV-induced intramolecular addition of iminyl radicals to alkenes

图式 10 可见光促进的亚胺自由基对烯烃的分子内加成反应环化氢化以及羟化反应
Scheme 10 Visible light-promoted intramolecular addition of iminyl radicals to alkenes

图式 11 可见光促进的亚胺自由基引发的串联反应
Scheme 11 Visible light-promoted and iminyl radical-initiated domino reactions

图式 13 Bu3SnH介导的N-巯苯基环丁酮亚胺开环反应
Scheme 13 Bu3SnH-mediated ring-opening of N-mercapto- phenyl cyclobutanones

图式 14 可见光促进的环丁酮肟酯与烯烃的官能团化反应
Scheme 14 Visible light-promoted reactions of cyclobutanone oxime esters with alkenes

图式 15 环酮肟羧酸衍生物介导的远程氟化和炔基化反应
Scheme 15 Remote fluorination and alkynation of cycloketone oxime carboxylic acid derivatives

图式 16 环酮肟酯的开环和SO2插入反应
Scheme 16 Ring-opening and SO2 insertion of cycloketone oxime ester

图式 17 环酮肟酯的开环和Minisci型反应
Scheme 17 Ring-opening and Minisci-type reaction of cycloketone oxime ester

图式 18 O-苄基环丁酮肟醚的开环烷基化反应
Scheme 18 Ring-opening and alkylation of O-benzyl cyclobutanone oxime ether

图式 19 可见光促进的铜催化环丁酮肟酯参与的反应
Scheme 19 Visible light-mediated and copper-catalyzed reactions of cyclobutanone oxime esters

图式 20 可见光促进的镍催化的芳基化、烯基以及烷基化反应
Scheme 20 Visible light promoted Nickel catalyzed arylation, alkenyl and alkylation

图式 21 光氧化还原和铜协同催化不对称远程氰化反应
Scheme 21 Dual photoredox and copper catalyzed asymmetric remote cyanation reaction

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:





 下载:
下载: