
图式 1.
乙烯基氮杂环丙烷结构特点和反应模式
Scheme 1.
Structural properties of vinylaziridines and reaction modes

含氮化合物, 尤其是含氮杂环是一类重要的有机分子, 与人类社会密切相关.这类骨架广泛存在于许多具有生物活性的天然产物、农药和药物分子中[1-2].例如, 美国食品药品监督局(FDA)批准上市的小分子药物中约60%含有氮杂环[3], 如六元环的哌啶、吡啶、哌嗪, 五元环的β-内酰胺、吡咯烷等.因此, 含氮分子的构建和含氮杂环的合成一直是有机化学合成中的重要研究领域, 吸引着众多研究者的兴趣.围绕这一主题, 许多高效、高选择性、底物广谱的用于构建碳氮键和氮杂环的反应和催化方法相继见诸报道[4-9].另外, 化学家们还不断发展各种合适的底物和试剂来实现含氮分子的合成.其中, 乙烯基氮杂环丙烷因其结构特殊性, 同时含有烯烃和具有较强张力的氮杂环丙烷官能团, 使得其表现出丰富的反应活性和模式.作为一类重要的合成砌块, 被广泛用于各种含氮分子, 尤其是含氮杂环的合成, 如氮杂环丁烷、吡咯烷、哌啶和氮杂环庚烷等杂环化合物[10-11].近年来, 这类试剂持续吸引着众多研究者的兴趣, 在有机合成化学领域取得了快速发展[12-13].本文对近五年来乙烯基氮杂环丙烷参与的亲核性开环反应和环化反应进行了综述, 并对该领域的发展方向进行了展望.其中, 亲核性开环部分的代表性研究工作根据不同的亲核试剂来分类, 有关环化反应的研究进展主要根据不同的催化体系来分类.期望进一步加深人们对乙烯基氮杂环丙烷作为含氮前体在构建含氮化合物中所起重要作用的认识, 为开发新型的合成方法学和催化方法提供帮助.
乙烯基氮杂环丙烷具有较强的环张力, 因此其氮杂环丙烷部分容易与各种碳亲核试剂和杂原子亲核试剂发生亲核性开环反应(SN2).另外, 在合适条件下, 烯烃部分也可以发生反应(SN2').这些反应往往能够立体专一性地生成各种重要的官能化的胺产物.我们将根据亲核试剂的不同种类分别介绍该领域的相关代表性研究工作.
芳基取代的乙胺衍生物是一类重要的生物活性分子.芳基亲核试剂对乙烯基氮杂环丙烷的亲核性开环反应, 即Friedel-Crafts烷基化反应, 是合成多取代芳基乙胺的重要方法之一.由于乙烯基氮杂环丙烷有两个亲电性反应位点, 在反应中往往同时得到SN2和SN2'反应产物.如何控制反应的区域选择性是发展此类反应的难点[14]. 2017年, 张俊良和冯见君等[15]报道了铑催化的光学活性乙烯基氮杂环丙烷1与羟基芳烃2的Friedel- Crafts烷基化反应(Scheme 2).该反应具有优异的化学和区域选择性, 只得到碳作为亲核试剂的烷基化反应产物.各种光学活性的底物都能够很好地参与反应, 并且产物保持了优异的光学纯度.该反应为官能化的2-芳基乙胺的合成提供了一种有效、简单且原子经济的途径.

作者对该反应提出了可能的机理(Scheme 3).铑催化剂首先通过乙烯基氮丙烷的烯烃和氮原子配位得到铑配合物1-A.随后经构型保留的氧化加成得到烯基- (σ+π)-铑物种1-B, 如果1-B异构成1-B', 则会导致消旋化, 在部分例子中确实可以观察到产物ee值的下降.随后, 萘酚2a通过络合物1-B的酰胺阴离子作用发生去质子化, 以及与1-B发生烯丙位去芳香化生成中间体1-C.在这个过程中, 底物的手性中心发生翻转.最后, 中间体1-C发生芳香化给出产物, 同时实现铑催化剂再生.

同年, 为对映选择性合成β-乙烯基色胺结构单元, 张俊良和冯见君课题组[16]还将这个催化体系进一步应用于乙烯基氮杂环丙烷4和吲哚5的立体专一性烯丙基烷基化反应(Scheme 4).值得注意的是, 和Trost课题组[17]报道的钯催化体系得到吲哚N-烷基化产物不同, 该反应能够以优秀的区域选择性在吲哚3-位发生反应.并且反应底物的手性能够很好地转移到产物中.作者认为该反应也是通过烯基-(σ+π)-铑中间体进行的.都是先通过乙烯基氮丙烷烯烃和N原子与金属铑配位, 然后通过氧化加成产生烯基-(σ+π)-铑中间体, 吲哚从背面进行亲核进攻, 最后进行芳香化得到目标产物.部分例子中产物的ee值有所降低也是因为烯基(σ+π)铑中间体发生了部分消旋化.该反应也能适用于吡咯杂环体系作为亲核试剂的反应.

2015年, Krische课题组[18]发展了手性铱催化的乙烯基氮杂环丙烷与醇和醛的极性反转的开环不对称烯丙基化反应(Scheme 5).该手性铱催化剂可由[Ir(cod)-Cl]2、烯丙基醋酸酯、4-氰基-3-硝基-苯甲酸和(R)-MeO- BIPHEP制备、且可柱层析分离.该催化体系对各种烷基醇和芳香醇都表现出良好的兼容性, 能够以中等到较高的产率和非对映选择性、以及优异的立体选择性得到相应的(胺甲基化)烯丙基化产物.对于醛底物, 通过添加异丙醇(300 mol%)作为氢源, 反应同样有着比较好的收率和立体选择性.两类反应的产物都以反式构型为主.值得一提的是, 该方法学同样适用于1, 3-二醇底物, 为构建结构更为复杂的手性二醇提供了重要方法.在机理方面, 作者认为反应首先通过醇与铱催化剂反应生成关键铱-氢中间体8-C, 同时醇脱氢氧化原位生成醛9a.在碱性条件下, 该中间体对乙烯基氮杂环丙烷进行氧化加成开环形成亲核性的烯丙基铱络合物8-E和8-F.随后, 烯丙基铱络合物对先前原位形成的醛进行立体选选择性的烯丙基化反应生成最终的产物.

2018年, 石枫课题组[19]报道了一例铱催化支链选择性的吲哚13与乙烯基氮杂环丙烷12的开环烷基化反应(Scheme 6).该反应在吲哚3-位发生烯丙基烷基化反应, 得到较好的产率.基于乙烯基环丙烷的类似反应过程, 该反应的关键步骤也有可能是先通过铱催化剂活化乙烯基氮杂环丙烷形成铱配位的两性离子中间体, 随后经过质子化形成亲电性的π-烯丙基铱络合物再与吲哚发生亲核加成得到最终产物.

2014年, Hyland课题组[20]利用邻菲罗啉作为二齿配体发展了钯催化的芳基和杂芳基硼酸15对乙烯基氮杂环丙烷12的SN2'加成反应, 得到烯烃为Z-构型为主的烯丙基磺酰胺产物(Scheme 7).硼酸芳基的电性对反应有较大影响, 对于芳环上含有缺电子基团的硼酸底物, 反应能给出优异的产率; 而对于含有富电子基团的硼酸, 该反应的产率为中等.对于反应机理, 作者认为邻菲罗啉配体、Pd(OAc)2、以及AgSbF6先形成阳离子钯络合物, 再与硼酸发生转移金属化形成芳基钯物种.随后, 乙烯基氮杂环丙烷的氮保护基与钯有配位作用促使其与芳基钯的插入反应. 最后, 由于钯与Ts基团之间的空间位阻作用, 使得中间体12-C通过反式β-氮消除得到烯烃为Z式的开环产物的路径更为有利.

2017年, Booker-Milburn课题组[21]发现利用光化学克级规模合成的三环乙烯基氮杂环丙烷17在钯(0)催化下容易和各种碳亲核试剂以及苯酚发生亲核性的SN2'的开环反应(Scheme 8).该反应以中等的收率和非对应选择性得到相应的多取代双环产物.有意思的是, 作者利用含有π-体系的底物, 如异腈酸酯、缺电子烯烃、醛以及亚胺, 发展了钯催化的[3+2]环化反应来构建结构更为复杂的稠三环化合物.

2018年, Clavier课题组[22]利用乙烯基氮杂环丙烷 21作为烯丙基前体, 发展了钴催化的芳环和杂芳环的C—H键烯丙基化反应(Scheme 9).该反应以中等到较高的产率生成相应的烯丙基化产物22.值得注意的是, 除了取代的砜基, 当乙烯基氮杂环丙烷的N原子上含有其他吸电子基团时, 反应也能顺利地进行.作者认为该反应首先通过原位形成的钴催化活性物种, 然后在底物导向基作用下活化其邻位C—H键形成钴碳环中间体.随后发生乙烯基氮杂环丙烷中的烯烃插入该络合物以及顺式β-N消除/开环/质子化得到最终产物.该反应对产物中的烯烃构型没有选择性(E:Z=0.4:1~1:1), 也说明该反应在烯烃插入这一步没有选择性.

近年来, 无钯、镍以及其他过渡金属催化的乙烯基氮杂环丙烷的亲核性开环二硼烷基化反应也受到了化学工作者的广泛关注[23]. 2019年, Fernández和Gava等[24]用偕二硼烷23与LiTMP试剂原位形成的α-二硼烷基碳负离子作为亲核试剂, 发展了这类硼烷基锂盐对乙烯基氮杂环丙烷的亲核性开环反应(Scheme 10).有趣的是, 这个反应的区域选择性取决于底物乙烯基氮杂环丙烷的取代基模式.例如, 对于本身较小空间位阻的1-甲苯磺酰基-2-乙烯基氮杂环丙烷而言, 反应在位阻最小的位置优先进行SN2开环反应生成24 (Scheme 8a).对于2-甲基-1-甲苯磺酰基-2-乙烯基氮杂环丙烷, 反应专一性地在烯烃末端发生SN2'反应, 从而以中等到优异的产率生成直链产物26 (Scheme 8b).当使用环状乙烯基氮杂环丙烷时, 则在烯丙位发生SN2开环反应, 立体选择性地生成反式构型的高烯丙基二硼产物28 (Scheme 8c).在大多数例子中, 产物的双硼片断不受影响.

2018年, Hong课题组[25]报道了含磷亲核试剂对乙烯基氮杂环丙烷的区域选择性可调的亲核性开环反应(Scheme 11).通过反应条件的改变可以很好地控制反应的区域选择性.作者发现在氮气氛围、铜催化条件下, 由二苯基氧膦产生的自由基与乙烯基氮杂环丙烷发生SN2'反应, 生成δ-氨基取代的烷基氧膦产物31 (Scheme 11a).另一方面, 当在空气氛围银催化条件下, 二苯基氧膦会被氧气进一步氧化为磷酸, 再与乙烯基氮杂环丙烷发生SN2反应, 形成膦酸酯氧取代的胺产物33 (Scheme 11b).利用酯基取代的乙烯基氮杂环丙烷底物, 可以合成一系列磷取代的氨基酸衍生物.

作者利用2, 2, 6, 6-四甲基哌啶-氮-氧化物(TEMPO)进行了自由基抑制实验和控制实验, 证明了该反应可能是经历自由基过程.随后, 作者提出了该反应可能的机理(Scheme 12).催化剂银或铜与乙烯基氮杂环丙烷络合形成配合物12-A, 在氮气氛围下, 膦酰基自由基30-A通过SN2'的方式区域选择性地加成到该配合物的烯烃末端碳上, 从而形成线性产物31.反之, 在氧气条件下, 膦酰基自由基被Ag催化氧化生成磷酸根阴离子, 随后与乙烯基氮杂环丙烷以SN2方式发生开环反应, 从而形成磷酸化的支链型产物33.

由于乙烯基氮杂环丙烷同时含有亲核性和亲电性位点, 因此容易作为三元合成子, 与一些二元、三元以及四元合成子发生环加成反应, 形成五元、六元和七元等含氮杂环.常见的不饱和体系如α, β-不饱和烯酮、异氰酸酯、碳酸酯、烯烃以及炔烃等.乙烯基氮杂环丙烷的环化反应通常需要在过渡金属, 如钯、铑、铱和镍等催化下发生反应.在有些例子中, 路易斯酸或者碱也作为催化剂或活化试剂促使其环加成反应.
乙烯基氮杂环丙烷与零价钯催化剂作用容易发生开环反应形成具有高反应活性的π-烯丙基钯络合物[26-27].自从2002年Yamamoto[28]和Aggarwal课题组等[29-30]先后报道了钯催化的乙烯基氮杂环丙烷与活化烯烃的[3+2]环加成反应以来, 这类反应引起了许多研究者的兴趣.
2015年, 侯雪龙和丁昌华等[31]利用(Rphos, Ra)- SIOCPhox作为手性配体, 发展了钯催化的乙烯基氮杂环丙烷34与α, β-不饱和酮35的不对称[3+2]环加成反应(Scheme 13).虽然α, β-不饱和酮中烯烃只有一个羰基活化基团, 该反应仍然表现出优异的非对映选择性和对映选择性.另外, 当乙烯基氮杂环丙烷的烯烃-2位有取代基时(如甲基), 反应的非对映选择性可以得到极大地提高.因此, 这个反应为3, 4-二取代吡咯烷36的不对称合成提供了一个重要的方法.

2016年, 在研究乙烯基环丙烷的[3+2]反应过程中, 姚和权和林爱俊等[32]发现, 他们发展的钯/膦-硫脲体系同样能够有效地催化乙烯基氮杂环丙烷12和对亚甲基苯醌37的[3+2]环化反应(Scheme 14).该反应能够以92%的产率和76:24的dr值得到相应的螺环化合物38.在此反应中膦-硫脲作为双功能配体, 硫脲部分通过氢键作用活化对亚甲基苯醌, 膦部分和金属钯配位, 从而使得该反应具有比较高的非对映选择性.虽然作者只报道了一个例子, 该工作为发展相关催化不对称反应奠定了基础.

利用钯催化乙烯基氮杂环丙烷形成具有偶极子性质的烯丙基钯络合物的特点, 2016年, 陆良秋等[33]报道了钯催化的乙烯基氮杂环丙烷39与亚甲基吲哚酮40的不对称[3+2]环化反应(Scheme 15).在该反应中, 钯/手性亚膦酰胺催化剂首先与乙烯基氮杂环丙烷39形成钯螯合的N1-1, 3-偶极子中间体39-A, 随后与亚甲基吲哚酮的缺电子烯烃部分发生环加成反应, 以较高的产率、非对映选择性和对映选择性得到一系列手性的3, 3'-吡咯啉螺氧化吲哚产物41.在底物的普适性方面, 虽然乙烯基氮杂环丙烷局限于氮上取代基的改变, 但是, 亚甲基吲哚酮在烯烃部分和芳环上可以引入不同的官能团.值得注意的是, 该反应放大量到克级规模, 同样能够拿到优异的结果, 说明该反应方法学具有潜在的合成应用价值.通过添加剂的控制实验研究, 作者认为在烯丙基钯络合物中氮阴离子提供辅助配位作用, 使得该络合物在发生反应时有更好的立体诱导作用.

2017年, Hyland和Ryan等[34]利用3-硝基吲哚作为缺电子烯烃底物, 发展了钯/菲罗啉催化的其与乙烯基氮杂环丙烷12的[3+2]环加成反应, 以优异的产率和反式选择性得到多取代的吡咯并吲哚啉产物43 (Scheme 16).该反应对吲哚底物同样表现出宽广的底物适用范围.但是, 3-位上强吸电子性的硝基对反应是必不可少的.值得注意的是, 当吲哚4-位有取代基(如甲基、酯基)时, 反应得到顺式选择性的产物44.
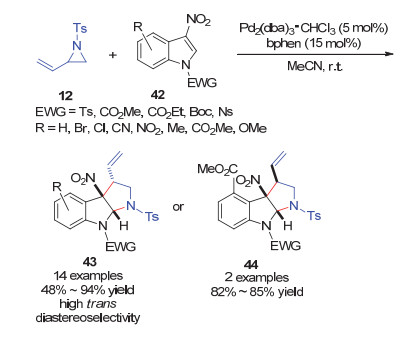
对于该反应可能的机理(Scheme 17), 作者认为钯(0)/菲罗啉催化剂首先与乙烯基氮杂环丙烷发生氧化加成生成1, 3-两性离子偶极子12-A.随后, 该偶极子12-A对缺电子吲哚42的C2处进行可逆的亲核进攻, 形成硝基烯醇盐中间体, 最后与π-烯丙基钯部分发生环化反应生成产物.这步过程中有两个可能的过渡态TS-I和TS-II.在过渡态TS-I中, π-烯丙基阳离子部分与富含电子的吲哚骨架之间的相互作用使得其更稳定, 从而形成反式主产物43a.反之, 当在吲哚的C4-位存在取代基时, 由于空间位阻效应, 这种有利的静电相互作用被破坏, 从而形成顺式产物44a.该工作为发展3-硝基吲哚催化不对称的反应提供了基础.

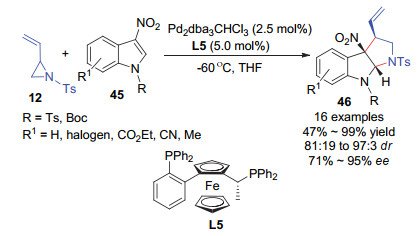
例如, 2018年, 侯雪龙和丁昌华等[35]利用商业可得的Walphos作为手性双膦配体发展了钯催化的乙烯基氮杂环丙烷12与3-硝基吲哚45的不对称[3+2]环加成反应(Scheme 18).该反应以较高收率以及高非对映和对映选择性得到多取代的顺式吡咯并吲哚啉产物46.该反应可以放大量至克级规模, 并且反应产物易于进一步转化为氨基吡咯并吲哚啉和其他吡咯并吲哚啉衍生物.
同年, 王兴旺课题组[36]发现了钯/双噁唑啉催化体系也能有效地催化乙烯基氮杂环丙烷与3-硝基吲哚47的立体选择性[3+2]环加成反应(Scheme 19).该反应可以顺利地以高收率、高立体选择性获得一系列含有三个连续手性中心的反式吡咯啉并吲哚啉产物48.该催化体系也能够成功应用于乙烯基环丙烷底物.

除了缺电子烯烃, 亚胺底物也能参与乙烯基氮杂环丙烷的环化反应.例如, 2018年, de Figueiredo课题组[37]报道了钯催化的乙烯基氮杂环丙烷49与环状N-砜基亚胺50的非对映选择性[3+2]环加成反应(Scheme 20). 该反应具有宽广的底物适用范围, 能以优异的产率和非对映选择性得到一系列含季碳的环状咪唑啉产物.有趣的是, 对于空间位阻较小的乙烯基氮杂环丙烷底物, 作者发现加入LiCl作为添加剂并在-30 ℃的条件下可以明显地提升反应的非对映选择性.

对于该反应可能的机理, 作者认为也是通过钯(0)与乙烯基氮杂环丙烷原位形成的成两性离子π-烯丙基钯中间体49-A (Scheme 21).随后, 该中间体49-A的阴离子部分与环状亚胺50生成49-B, 这一步骤是分子间C—N键的形成.接着, 中间体49-B再经一次分子内的C—N键形成反应, 从而得到产物51.作者认为添加剂LiCl有利于中间体49-B与49-C之间发生快速的非对映异构体转化; 高非对映选择性地形成第二个N—C键主要取决于第一步的C—N键形成.有意思的是, 当使用手性配体(R)-Taniaphos时, 反应获得一定的立体选择性控制(19% ee), 说明该反应有发展催化不对称版本的潜力.此后不久, 该课题组发现当使用含有联萘骨架的手性亚膦酰酰胺作为配体时, 该反应的立体选择性可以得到明显地提高[38].

乙烯基氮杂环丙烷一般容易发生[3+2]环加成反应, 而其作为五原子片断参与的[5+2]环加成反应则很少见诸报道. 2015年, 张俊良课题组[39]报道了首例铑(I)催化的易制备的光学纯乙烯基氮杂环丙烷52与非活化炔烃的分子内形式的杂[5+2]环加成反应(Scheme 22).尽管该反应存在潜在的[3+2]以及乙烯基氮杂环丙烷自身的重排等竞争性反应, 作者发现商品化的催化剂[Rh(NBD)2]BF4表现出最好的催化效果. 反应对于内炔烃和末端炔烃、以及炔烃和氮杂环丙烷的取代模式改变都具有很好的兼容性.在该反应中乙烯基氮杂环丙烷作为杂五原子片断参与反应.手性底物的光学纯度能够完全地转移到产物中, 以优异的非对映选择性和对应选择性得到手性稠双环吖庚因产物.由于底物两种光学纯的对映体都容易由文献方法制备, 因此, 对映体的[5+2]环化产物都能够以优异的光学纯度得到.该催化体系同样适用于克级规模的反应.重要的是, 产物可以通过简单转化来构建更多的手性中心.通过控制实验, 作者发现含Z-构型烯烃的底物反应速率明显比含E-构型烯烃的底物要慢, 但是同样有很高的对映选择性.

在机理方面, 作者认为首先烯烃部分的Re-面和Si-面都能和铑配位通过1, 6-烯炔的氧化环金属化非对映选择性地形成金属环戊烯中间体52-B和52-F.随后发生协同的氮杂环丙烷开环和形成含有Z-式烯烃的中间体52-D和52-H则需要经历中间体52-C和52-G.而在52-C的构象更容易发生这一过程.最后通过还原消除生成2, 5-二氢稠双环吖庚因产物.反应之所以有很好的手性保持, 作者认为有可能是氮杂环丙烷不可逆开环导致.总体而言, 该方法学具有反应条件温和、手性底物易得以及普适性广等优点, 为发展乙烯基氮杂环丙烷的新环化模式奠定基础.
基于该底物手性传递策略, 随后, 张俊良课题组[40]还成功发展了一价铑催化的手性乙烯基氮杂环丙烷与烯烃的分子内杂-[5+2]环加成反应(Scheme 23).利用该反应构建了一系列富含sp3-碳和3个连续手性中心的稠双环吖庚因产物55.在这个反应中, 作者同样发现乙烯基氮杂环丙烷中的烯烃的E/Z构型影响环化反应产物的顺反选择性.据此, 通过调控底物的构型作者合成了8个理论异构体中的6个立体异构体.

基于这些分子内的反应, 张俊良课题组[41]继续发展了铑(I)催化乙烯基氮杂环丙烷57和炔烃58分子间可调控的环加成反应, 即[3+2]和[5+2]环加成(Scheme 24).基于相同的反应底物, 作者发现使用不同的铑催化剂可以实现反应路径的调控.当使用[Rh(NBD)2]- BF4时候, 反应发生[3+2]环化, 并且底物的光学纯度能够完全得到保持, 得到一系列重要的手性二氢吡咯烷产物58 (Scheme 24a).当使用[Rh(η6-C10H8)(COD)]SbF6作为催化剂时, 反应发生[5+2]环化反应, 得到吖庚因产物59 (Scheme 24b).有趣的是, 作者发现其他常用的金属盐(如Pd2(dba)3, AgSbF6, FeCl3和Sc(OTf)3等)并不能催化这类反应.另外, 底物氮上的强吸电子性砜基有重要作用.具体反应机理方面, 作者认为铑催化剂通过构型保持的方式和底物乙烯基氮杂环丙烷发生氧化加成, 形成可以相互转换的烯基-(σ+π)-铑中间体56-B和56-C.随后, 中间体56-B和56-C分别和炔烃通过区域选择性的插入反应形成可以相互转换的烯基-(σ+π)-铑中间体56-D和56-E.最后, 中间体56-D和56-E分别通过不可逆的还原消除生成相应的[3+2]和[5+2]环化产物.随后, 黄跟平课题组[42]对该反应进行了理论计算研究, 支持了作者提出的机理.

尽管有分子内版本的零星报道[43], 乙烯基氮杂环丙烷与非活化烯烃的分子间反应仍是一个挑战. 2016年, 张俊良和冯见君课题组[44]报道了铑(I)催化的乙烯基氮杂环丙烷和累积二烯的[3+2]环化反应(Scheme 25).作者发现催化剂[Rh(NBD)2]BF4同样显示出最好的催化效果.有意思的是, 当使用N-累积二烯胺底物时, 邻近氮原子的C=C双键发生环化反应, 并且光学纯的乙烯基氮杂环丙烷的手性得到很好的保留, 以优异的产率和立体选择性得到相应的3-亚甲基吡咯烷产物62 (Scheme 25a).反之, 当使用芳基或烷基取代的普通累积二烯时, 远端的C=C双键发生[3+2]环化反应, 同样以优异的立体选择性得到2-亚甲基吡咯烷产物64 (Scheme 25b).产物中引入的双键可以进一步衍生化, 得到官能团更丰富的吡咯烷.

2018年, 张俊良和冯见君课题组[45]又成功实现了铑(I)催化的光学纯乙烯基氮杂环丙烷65和烯醇硅醚66的[3+2]环加成反应(Scheme 26).有意思的是, 对于含有大位阻烷基团的底物80且使用[Rh(NBD)2]BF4作为催化剂时, 反应以中等到较好的产率、以及优异的立体选择性生成官能化吡咯烷81 (Scheme 26a).反之, 对于含有较小体积三甲基硅基的底物, 使用催化体系[Rh-(NBD)Cl]2/AgPF6则使得氮杂环丙烷部分发生亲核性开环反应, 得到官能化的γ-氨基酮产物(Scheme 26b).底物的光学纯度同样能够在产物中得到保留.

在机理方面(Scheme 27), 作者认为乙烯基氮杂环丙烷65与铑催化剂通过氧化加成生成构型保留的烯基- (σ+π)-铑物种65-B.然后底物烯基烯硅醚从背面向中间体65-B发生亲核进攻生成中间体65-D.在此过程中, 发生绝对构型净反转并再生铑催化剂.最后, 对65-D发生分子内环化得到[3+2]环加成产物. [3+2]环加成产物水解开环或者中间体65-D直接水解生成直链产物68.在反应中, 如果中间体65-B发生异构化为65-C, 则会导致产物的ee值下降.

张俊良和冯见君课题组发现乙烯基氮杂环丙烷不仅可以作为五原子片断参与[5+2]环加成反应, 同时也可以作为含氮三原子片断参与[4+3]环化反应.例如, 2017年, 该团队[46]报道了首例铑(I)催化的光学纯乙烯基氮杂环丙烷69与硅基二烯基醚70的[4+3]环加成反应(Scheme 28).在反应中, 氮杂环丙烷部分作为氮杂三原子片断参与反应.该反应体系有着宽广的底物适用范围, 底物的手性在环化产物中也得到了很好地保留.和以往的[5+2]环化反应相比, 该反应为合成吖庚因化合物71提供了一个重要的补充方法.机理上而言, 该反应也是通过铑催化剂活化乙烯基氮杂环丙烷经历氧化加成生成烯基-(σ+π)-铑物种, 随后与硅基二烯基醚发生亲核性进攻和环化过程生成产物.

基于该工作, 张俊良和冯见君课题组[47]利用C, N-环偶氮甲碱亚胺73作为偶极子以及[Rh(NBD)Cl]2作为催化剂, 发展了其与光学纯乙烯基氮杂环丙烷72的[3+3]环化反应(Scheme 29).该反应同样表出良好的底物普适性和官能团兼容性, 以优异的立体选择性得到相应的1, 2, 4-六氢三嗪稠环化合物74.

张俊良和冯见君等[48]还发现催化体系[Rh(NBD)-Cl]2/AgSbBF5能够有效地催化光学纯乙烯基氮杂环丙烷75和肟醚76的[3+2]环加成反应(Scheme 30).在该反应中, 醛肟和酮肟都能作为合适的底物参与反应, 以中等到较好的产率和优异立体选择性得到相应重要的手性咪唑烷类化合物77.

2019年, Liu课题组[49]发现当乙烯基氮杂环丙烷含有共轭羰基时, 其与末端炔发生铑(I)催化的[3+2]环化反应得到二氢吡咯烷(Scheme 31).该类产物通过一锅连续的碱处理和酸处理可以进一步发生双键迁移从而得到相应的吡咯化合物.

当选择合适的底物来发展乙烯基氮杂环丙烷C-2位的开环和后续环化可以为γ-内酰胺合成提供重要方法, 但是广谱的方法尚未见文献报道[50]. 2017年, 张俊良和冯见君课题组[51]报道了第一例铱催化的乙烯基氮杂环丙烷82与β-酮羰基化合物83的多米诺开环/环化反应(Scheme 32).在相同反应条件下, 对于α-位取代的1, 3-二羰基底物, 反应以中等到优异的非对映选择性生成带有两个相邻的sp3-碳中心的2-亚甲基吡咯烷产物84.反之, 当使用α-位未取代的1, 3-二羰基底物时, 反应以较好的收率得到多取代的2, 3-二氢吡咯产物85.机理方面, 作者认为也是通过铱催化剂与乙烯基氮环丙烷先通过环丙烷的氧化加成形成(η3-烯丙基)铱(III)活性物种82-B.随后, β-酮羰基化合物对该中间体的进行亲核进攻得到中间体82-C.基于1, 3-二羰基底物的α-位取代模式, 中间体82-D分别发生环内或者环外脱水生成相应的产物.

2018年, 石枫课题组[52]利用对位-亚甲基苯醌87作为四原子偶极子发展了铱催化的与乙烯基氮丙烷86的[4+3]环化反应(Scheme 33).反应表现出宽广的底物适用范围, 以中等到较好的收率和优秀的非对映选择性得到七元苯并氧杂吖庚因产物88.当使用手性钯催化剂时, 作者还发展了该反应的催化不对称版本, 获得了较高的非对映和对映选择性.

除了过渡金属外, 路易斯酸也常用于活化或催化乙烯基氮杂环丙烷的环加成反应. 2014年, Chiu课题组[53]报道了硝基乙烷作溶剂、三氟乙酸(1.2~5.0 equiv.)介导的乙烯基氮杂环丙烷硅醚89和呋喃以及环戊二烯的[4+3]环加成反应(Scheme 34).值得一提的是, 该反应需要在-90 ℃下进行, 升温会使得产率明显降低.在最佳条件下, 反应能以优异的产率和中等的非对映选择性得到七元环酮的endo-和exo-的混合物.底物中含有大位阻取代基时, 产物的非对映选择性能得到明显地提高.当使用光学纯的乙烯基氮杂环丙烷硅醚作为底物时, 虽然反应的非对映选择性一般, 但是两种非对映体都能以优异的对映选择性得到.

2017年, 在研究氮杂环丙烷的环化反应过程中, Ghorai课题组[54]发现以水为溶剂以及使用LiClO4/ Bu4NBF4作双催化剂体系时候, 光学纯的乙烯基氮杂环丙烷12能够与硝酮92发生多米诺SN2-开环/环化反应(Scheme 35).该反应能以较高的产率和优异的立体选择性得到相应的1, 2, 4-氧杂二嗪烷产物93.

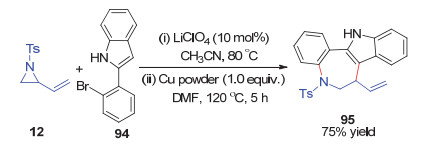
随后, Ghorai课题组[55]还报道了一例两步一锅的乙烯基氮杂环丙烷12与2-(2-溴苯基)-1H-吲哚94的开环/偶联环化(Scheme 36).在LiClO4的催化活化下, 该反应首先发生吲哚的C-3位对乙烯基氮杂环的SN2开环反应, 随后在铜介导作用下发生分子内C—N键偶联环化, 得到形式[4+3]环化产物, 吲哚并七元氮杂环95.
基于乙烯基氮杂丙烷SN2开环/偶联环化策略, Ghorai课题组[56]还发展了乙烯基氮杂环丙烷12与2-溴酚96的一锅开环/偶联环化反应得到形式[3+3]环化反应产物97 (Scheme 37).该反应为合成乙烯基取代3, 4-二氢-2H-苯并[b]-[1, 4]噁嗪提供了一个实用的方法.

2016年, 肖文精和陆良秋等[57]利用t-BuOK和BEt3试剂原位活化螯合吲哚98的N—H键, 该策略有效阻止了氮原子的亲核性, 成功发展了3-取代吲哚98与乙烯基氮杂环丙烷12的[3+2]环加成反应(Scheme 38).该反应以中等到较好的收率和中等的非对映选择性得到了一系列乙烯基取代的吡咯吲哚啉产物113.与文献中常用的路易斯酸活化乙烯基氮杂环丙烷的策略不同, 该方法使得可以直接使用氮原子上无需保护基的吲哚作为底物.

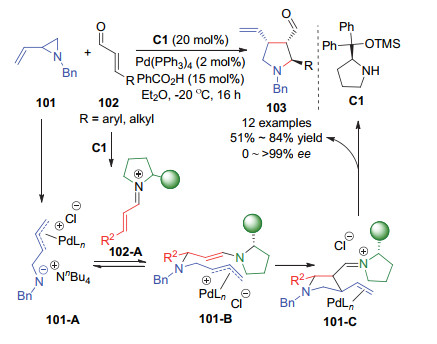
金属催化和有机催化协同催化模式为底物活化和反应选择性控制提供了一个重要的策略, 利用该方法往往能获得单一催化模式难以实现的催化效果[58]. 2017年, Jørgensen课题组[59]报道了金属催化和有机胺催化协同催化的乙烯基氮杂环丙烷101与α, β-不饱和醛102的不对称[3+2]加成反应(Scheme 39).该反应能以较好的产率、中等的非对映选择性、以及优异的对映选择性得到含有三个手性中心的吡咯烷产物103.在反应机理方面, 作者认为钯(0)催化剂与乙烯基氮杂环丙烷发生开环的氧化加成形成两性的π-烯丙基-钯(II)中间体101-A, 添加剂nBu4NCl的作用被认为是通过阻碍氮负离子部分与钯的配位来提升该中间体的亲核性; 另一方面, 手性胺催化剂与α, β-不饱和醛原位生成α, β-不饱和亚胺离子中间体102-A.随后, 中间体101-A与中间体102-A发生可逆的分子间氮杂-Michael加成和不可逆的分子内环化以及水解释放处手性胺催化剂和最终产物.
2019年, 陈应春和杜玮等[60]报道了首例非手性三级胺和手性铱络合物协同催化的靛红衍生的MBH碳酸酯104和乙烯基氮杂环丙烷105的[3+3]环加成反应(Scheme 40).该反应能以中等的产率、优异的立体选择性得到一系列多取代的氧化吲哚螺四氢哌啶的产物.关于反应理, 作者认为经过协同催化的模式, 即非手性的路易斯碱4-哌啶基吡啶活化颠红衍生的Morita-Baylis- Hillman碳酸酯从而形成两性离子, 烯丙基叶立德中间体104-A.另一方面, 手性的铱催化剂活化乙烯基氮杂环丙烷开环形成三原子的π-烯丙基铱络合物两性偶极子.随后, 这两个活性物种发生形式[3+3]环化反应.

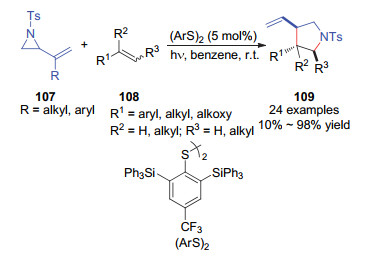
杂原子自由基通常被用于碳-杂原子键的构建以及碳-氢键的活化[61-62].其中, 硫自由基是一类重要的自由基中间体. 1988年, Oshima[63]和Feldman[64]课题组曾发现催化产生的硫自由基能够活化的乙烯基环丙烷, 并发展了乙烯基环丙烷与烯烃的[3+2]环化反应.在此工作基础上, 2016年, Maruoka课题组[65]设计了一类大位阻苯硫酚自由基前体-双硫醚(Scheme 41).随后, 利用光引发产生苯硫酚自由基, 作者发展了乙烯基氮杂环丙烷107和烯烃108的自由基[3+2]环化反应.该反应能以中等到优异的产率和中等的非对应选择性得到多取代的吡咯烷109.合理设计的具有较大空间位阻的硫自由基是该反应成功的关键, 该结构特点既避免苯硫酚自由基降解, 又避免了其对烯烃加成的竞争性副反应.作者还把这个方法学成功应用到富勒烯的结构修饰中.这类新型的硫自由基催化剂也进一步丰富了硫自由基在有机合成中的应用.
在机理方面(Scheme 42), 催化活性的硫基由由相应的芳基二硫化物光解产生.产生的硫自由基首先加成到对乙烯基氮杂环丙烷的烯烃末端, 从而引发氮杂环丙烷开环生成中间体氮自由基116.随后, 该氮自由基116与烯烃117发生分子间加成以及分子内环化得到[3+2]环化产物.同时, 硫自由基再生并进入下一个催化循环.在此反应中, 存在两种可能的竞争性途径:一种途径是关键中间体胺氨基自由基116发生攫氢形成副产物120; 另一种是两分子硫自由基加到烯烃119中生成生成烯烃双硫化副产物.

2020年, 基于前期在可见光驱动氮自由基化学方面的工作基础[66], 陈加荣等[67]报道了可见光驱动氮自由基催化的乙烯基氮杂环丙烷110与烯烃111的[3+2]环化反应(Scheme 43).在该策略中, 氮自由基通过可见光驱动的脱质子光致电子转移过程活化前体腙的氮-氢键来产生氮自由基.催化剂前体中sp3杂环的氮原子使得其空间位阻和电性很容易通过取代基来调控.另外, 氮自由基和还原态的光催化剂之间存在反电子转移过程也有利于调控氮自由基的浓度.该催化体系具有广谱的底物适用范围, 线型、支链以及环状烷基的富电子的单取代烯烃都能很好地参与反应.同时, 当乙烯基氮杂环丙烷的烯烃部分含有各种取代基时, 反应也能顺利地进行.通过该环化反应, 以良好的收率和中等到较好的非对映选择性生成多取代的吡咯烷112.与硫自由基催化相比, 该反应避免使用有气味的硫醇催化剂前体.

基于一系列的机理研究和控制实验, 作者认为该反应可能是通过光催化和氮自由基协同催化机理进行的.首先, 在K2CO3存在下, 由腙原位生成的氮阴离子NCR1-A被光激发的*IrIII光催化剂单电子氧化得到氮自由基NCR1-B和还原态的光催化剂IrII.接下来, 氮自由基NCR1-B的乙烯基氮杂环丙烷123发生自由基加成/开环反应与生成磺酰胺氮自由基中间体110a-A.随后, 自由基中间体110a-A与烯烃111a发生分子间加成和分子内环化反应得到碳最终产物112a(路径b), 并重新生成氮自由基NCR1-B.原则上, 原位催化形成的氮自由基NCR1-B也可以与和富电子的烯烃底物发生可逆加成反应.然而, 由于环丙烷的开环C—N键断裂, 会使得其与乙烯基氮杂环丙烷的加成速率更快.氮自由基NCR1-B也可以与还原态的光催化剂IrII之间发生反向电子转移被还原为氮阴离子NCR1-A, 以及重新产生基态的光催化剂IrIII, 从而完成光催化循环(路径a).
2019年, 王启卫和李俊龙等[68]发现由缺电子二烯烃和两亲氮试剂通过形式氮杂-[2+1]环化反应制备得到的乙烯基氮杂环丙烷113在加热条件下容易发生重排环化反应(Scheme 41).该反应对烯烃部分和氮杂环丙烷部分都有较好的官能团兼容性, 以优异的产率得到一系列多取代的二氢吡咯烷产物114.机理方面, 作者认为在加热条件下, 乙烯基氮杂环丙烷先发生碳-碳键断裂生成亚胺叶立德中间体113-A.随后, 烯丙基阴离子部分发生异构化以及对亚胺的分子内亲核加成, 从而得到最终产物114.值得一提的是, 作者还发展了克级、一锅的氮杂环丙烷化/重排串联反应.

综上所述, 经过化学家们的不懈努力, 近五年来乙烯基氮杂环丙烷在有机合成中的应用取得了进一步的发展.在含氮分子合成, 尤其是含氮杂环合成方面继续表现出巨大的潜力, 各种新型的[3+2]、[3+3]、[4+3]、[5+2]等环化反应相继被报道.利用合适的手性催化体系实现了相应的立体选择性控制.最近, 随着自由基催化和光氧化还原催化等策略的引入, 富电子烯烃也能很好地与乙烯基氮杂环丙烷发生环化反应, 进一步拓展了底物的适用范围, 也为新反应的发展和创新提供了新的方向.但是在该研究领域还存在着一些挑战性的问题[69-70].继续寻找合适的、廉价绿色的催化体系来进一步拓宽乙烯基氮杂环丙烷的底物范围和取代模式、实现更大环系的构建、以及反应立体选择性控制等将成为新的研究热点[71-72].例如, 最近Studer和Yudin课题组[73]以及Sun和Williams课题组[74]报道了乙烯基氮杂环丙烷还可以与高活性的苯炔或者硅氧基炔发生[5+2]环化反应.同时, 进一步拓展这些反应在生物活性天然产物和药物分子合成中的应用、以及相关机理的研究也将是重要的努力方向.
Lawrence, S. A. Amines: Synthesis Properties and Applications, Cambridge University Press, Cambridge, 2008, p. 371.
Das, P.; Delost, M. D.; Qureshi, M. H.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2019, 62, 4265. doi: 10.1021/acs.jmedchem.8b01610
Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257. doi: 10.1021/jm501100b
Naito, T. Chem. Pharm. Bull. 2008, 56, 1367. doi: 10.1248/cpb.56.1367
Bariwal, J.; Van der Eycken, E. Chem. Soc. Rev. 2013, 42, 9283. doi: 10.1039/c3cs60228a
Kim, H.; Chang, S. ACS Catal. 2016, 6, 2341. doi: 10.1021/acscatal.6b00293
Trowbridge, A.; Walton, S. M.; Gaunt, M. J. Chem. Rev. 2020, 120, 2613. doi: 10.1021/acs.chemrev.9b00462
Yuan, J.-W.; Liu, C.; Lei, A.-W. Chem. Commun. 2015, 51, 1394. doi: 10.1039/C4CC08116A
Ye, Z.; Zhang, F. Chin. J. Chem. 2019, 37, 513. doi: 10.1002/cjoc.201900049
Heo, Y. M.; Paek, S. M. Molecules 2013, 18, 9650. doi: 10.3390/molecules18089650
Ohno, H. Chem. Rev. 2014, 114, 7784. doi: 10.1021/cr400543u
Ilardi, E. A.; Njardarson, J. T. J. Org. Chem. 2013, 78, 9533. doi: 10.1021/jo401776s
Feng, J.-J.; Zhang, J. ACS Catal. 2016, 6, 6651. doi: 10.1021/acscatal.6b02072
Ling, J.; Lam, S. K.; Lo, B.; Lam, S.; Wong, W.-T.; Sun, J.; Chen, G.; Chiu, P. Org. Chem. Front. 2016, 3, 457. doi: 10.1039/C5QO00333D
Lin, T.-Y.; Wu, H.-H.; Feng, J.-J.; Zhang, J. Org. Lett. 2017, 19, 2897. doi: 10.1021/acs.orglett.7b01136
Lin, T.-Y.; Wu, H.-H.; Feng, J.-J.; Zhang, J. ACS Catal. 2017, 7, 4047. doi: 10.1021/acscatal.7b00870
Trost, B. M.; Osipov, M.; Dong, G.-B. J. Am. Chem. Soc. 2010, 132, 15800. doi: 10.1021/ja1071509
Wang, G.; Franke, J.; Ngo, C. Q.; Krische, M. J. J. Am. Chem. Soc. 2015, 137, 7915. doi: 10.1021/jacs.5b04404
Mei, G.-J.; Yu, L.; Zhu, Z.-Q.; Sun, M.; Shi, F. Synthesis 2018, 51, 1655.
Yin, J. X.; Mekelburg, T.; Hyland, C. Org. Biomol. Chem. 2014, 12, 9113. doi: 10.1039/C4OB01786B
Blackham, E. E.; Knowles, J. P.; Burgess, J.; Booker-Milburn, K. I. Chem. Sci. 2016, 7, 2302. doi: 10.1039/C5SC04062K
Kong, L.; Biletskyi, B.; Nuel, D.; Clavier, H. Org. Chem. Front. 2018, 5, 1600. doi: 10.1039/C8QO00173A
Sanz, X.; Lee, G. M.; Pubill-Ulldemolins, C.; Bonet, A.; Gulyás, H.; Westcott, S. A.; Bo, C.; Fernández, E. Org. Biomol. Chem. 2013, 11, 7004. doi: 10.1039/c3ob41328d
Salvado, O.; Gava, R.; Fernandez, E. Org. Lett. 2019, 21, 9247. doi: 10.1021/acs.orglett.9b03672
Kang, D.; Kim, T.; Lee, H.; Hong, S. Org. Lett. 2018, 20, 7571. doi: 10.1021/acs.orglett.8b03309
Fugami, K.; Morizawa, Y.; Ishima, K.; Nozaki, H. Tetrahedron Lett. 1985, 26, 857. doi: 10.1016/S0040-4039(00)61948-2
Spears, G. W.; Nakanishi, K.; Ohfune, Y. Synlett 1991, 91.
Aoyagi, K.; Nakamura, H.; Yamamoto, Y. J. Org. Chem. 2002, 67, 5977. doi: 10.1021/jo025747h
Lowe, M. A.; Ostovar, M.; Ferrini, S.; Chen, C. C.; Lawrence, P. G.; Fontana, F.; Calabrese, A. A.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2011, 50, 6370. doi: 10.1002/anie.201101389
Arena, G.; Chen, C. C.; Leonori, D.; Aggarwal, V. K. Org. Lett. 2013, 15, 4250. doi: 10.1021/ol4020333
Xu, C.-F.; Zheng, B.-H.; Suo, J.-J.; Ding, C.-H.; Hou, X.-L. Angew. Chem., Int. Ed. 2015, 54, 1604. doi: 10.1002/anie.201409467
Yuan, Z.; Wei, W.; Lin, A.; Yao, H. Org. Lett. 2016, 18, 3370. doi: 10.1021/acs.orglett.6b01512
Li, T.-R.; Cheng, B.-Y.; Fan, S.-Q.; Wang, Y.-N.; Lu, L.-Q.; Xiao, W.-J. Chem. Eur. J. 2016, 22, 6243. doi: 10.1002/chem.201600735
Rivinoja, D. J.; G, Y. S.; Gardiner, M. G.; Ryan, J. H.; Hyland, C. J. T. ACS Catal. 2017, 7, 1053. doi: 10.1021/acscatal.6b03248
Suo, J.-J.; Liu, W.; Du, J.; Ding, C.-H.; Hou, X.-L. Chem. Asian J. 2018, 13, 959. doi: 10.1002/asia.201800133
Zhang, J.-Q.; Tong, F.; Sun, B.-B.; Fan, W.-T.; Chen, J.-B.; Hu, D.; Wang, X.-W. J. Org. Chem. 2018, 83, 2882. doi: 10.1021/acs.joc.8b00046
Spielmann, K.; van der Lee, A.; de Figueiredo, R. M.; Campagne, J.-M. Org. Lett. 2018, 20, 1444. doi: 10.1021/acs.orglett.8b00228
Spielmann, K.; Tosi, E.; Lebrun, A.; Niel, G.; van der Lee, A.; de Figueiredo, R. M.; Campagne, J.-M. Tetrahedron 2018, 74, 6497. doi: 10.1016/j.tet.2018.09.040
Feng, J.-J.; Lin, T.-Y.; Wu, H.-H.; Zhang, J. J. Am. Chem. Soc. 2015, 137, 3787. doi: 10.1021/jacs.5b01305
Feng, J.-J.; Lin, T.-Y.; Wu, H.-H.; Zhang, J. L. Angew. Chem., Int. Ed. 2015, 54, 15854. doi: 10.1002/anie.201509185
Feng, J.-J.; Lin, T.-Y.; Zhu, C.-Z.; Wang, H.; Wu, H.-H.; Zhang, J. L. J. Am. Chem. Soc. 2016, 138, 2178. doi: 10.1021/jacs.6b00386
Zhang, X.; Zou, H.; Huang, G. ChemCatChem 2016, 8, 2549. doi: 10.1002/cctc.201600349
Hirner, J. J.; Roth, K. E.; Shi, Y.; Blum, S. A. Organometallics 2012, 31, 6843. doi: 10.1021/om300671j
Lin, T.-Y.; Zhu, C.-Z.; Zhang, P.; Wang, Y.; Wu, H.-H.; Feng, J.-J.; Zhang, J. Angew. Chem., Int. Ed. 2016, 55, 10844. doi: 10.1002/anie.201605530
Zhu, C.-Z.; Feng, J.-J.; Zhang, J. Chem. Commun. 2018, 54, 2401. doi: 10.1039/C8CC00279G
Zhu, C.-Z.; Feng, J.-J.; Zhang, J. L. Angew. Chem., Int. Ed. 2017, 56, 1351. doi: 10.1002/anie.201609608
Zhu, C.-Z.; Feng, J.-J.; Zhang, J. Chem. Commun. 2017, 53, 4688. doi: 10.1039/C7CC02078C
Lin, T.-Y.; Wu, H.-H.; Feng, J.-J.; Zhang, J. Org. Lett. 2018, 20, 3587. doi: 10.1021/acs.orglett.8b01378
Wan, S.-H.; Liu, S.-T. Tetrahedron 2019, 75, 1166. doi: 10.1016/j.tet.2019.01.022
Ghorai, M. K.; Tiwari, D. P. J. Org. Chem. 2010, 75, 6173. doi: 10.1021/jo101004x
Lin, T.-Y.; Wu, H.-H.; Feng, J.-J.; Zhang, J. Org. Lett. 2017, 19, 6526. doi: 10.1021/acs.orglett.7b03232
Jiang, F.; Yuan, F.-R.; Jin, L.-W.; Mei, G.-J.; Shi, F. ACS Catal. 2018, 8, 10234. doi: 10.1021/acscatal.8b03410
Lam, S. K.; Lam, S.; Wong, W.-T.; Chiu, P. Chem. Commun. 2014, 50, 1738. doi: 10.1039/c3cc48266a
Wani, I. A.; Sayyad, M.; Ghorai, M. K. Chem. Commun. 2017, 53, 4386. doi: 10.1039/C7CC01033H
Pradhan, S.; Shahi, C. K.; Bhattacharyya, A.; Chauhan, N.; Ghorai, M. K. Org. Lett. 2017, 19, 3438. doi: 10.1021/acs.orglett.7b01397
Mal, A.; Wani, I. A.; Goswami, G.; Ghorai, M. K. J. Org. Chem. 2018, 83, 7907. doi: 10.1021/acs.joc.8b00788
Wang, Y.-N.; Li, T.-R.; Zhang, M.-M.; Cheng, B.-Y.; Lu, L.-Q.; Xiao, W.-J. J. Org. Chem. 2016, 81, 10491. doi: 10.1021/acs.joc.6b00991
Du, Z.-T.; Shao, Z.-H. Chem. Soc. Rev. 2013, 42, 1337. doi: 10.1039/C2CS35258C
Naesborg, L.; Tur, F.; Meazza, M.; Blom, J.; Halskov, K. S.; Jørgensen, K. A. Chem. Eur. J. 2017, 23, 268. doi: 10.1002/chem.201604995
Chen, Z.-C.; Chen, Z.; Yang, Z.-H.; Guo, L.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2019, 58, 15021. doi: 10.1002/anie.201907797
Denes, F.; Pichowicz, M.; Povie, G.; Renaud, P. Chem. Rev. 2014, 114, 2587. doi: 10.1021/cr400441m
Taniguchi, T. Synthesis 2017, 49, 3511. doi: 10.1055/s-0036-1588481
Miura, K.; Fugami, K.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1988, 29, 5135. doi: 10.1016/S0040-4039(00)80701-7
Feldman, K. S.; Romanelli, A. L.; Ruckle, R. E.; Miller, R. F. J. Am. Chem. Soc. 1988, 110, 3300. doi: 10.1021/ja00218a050
Hashimoto, T.; Takino, K.; Hato, K.; Maruoka, K. Angew. Chem., Int. Ed. 2016, 55, 8081. doi: 10.1002/anie.201602723
Hu, X.-Q.; Chen, J.-R.; Wei, Q.; Liu, F.-L.; Deng, Q.-H.; Beauchemin, A. M.; Xiao, W.-J. Angew. Chem. Int. Ed. 2014, 53, 12163. doi: 10.1002/anie.201406491
Zhao, Q.-Q.; Zhou, X.-S.; Xu, S.-H.; Wu, Y.-L.; Xiao, W.-J.; Chen, J.-R. Org. Lett. 2020, 22, 2470. doi: 10.1021/acs.orglett.0c00712
Zhang, X.; Huang, Q.-F.; Zou, W.-L.; Li, Q.-Z.; Feng, X.; Jia, Z.-Q.; Liu, Y.; Li, J.-L.; Wang, Q.-W. Org. Chem. Front. 2019, 6, 3321. doi: 10.1039/C9QO00509A
王清宇, 常宏宏, 魏文珑, 刘强, 高文超, 李彦威, 李兴, 有机化学, 2016, 36, 939. doi: 10.6023/cjoc201511039Wang, Q.; Chang, H.; Wei, W.; Liu, Q.; Gao, W.; Li, Y.; Li, X. Chin. J. Org. Chem. 2016, 36, 939 (in Chinese). doi: 10.6023/cjoc201511039
Chai, Z. Synthesis 2020, 52, 1738. doi: 10.1055/s-0039-1690857
Ling, J.; Lam, S.; Low, K. H.; Chiu, P. Angew. Chem. Int. Ed. 2017, 56, 8879. doi: 10.1002/anie.201704155
Moragas, T.; Liffey, R. M.; Regentova, D.; Ward, J. P.; Dutton, J.; Lewis, W.; Churcher, I.; Walton, L.; Souto, J. A.; Stockman, R. A. Angew. Chem., Int. Ed. 2016, 55, 10047. doi: 10.1002/anie.201604188
Kaldas, S. J.; Kran, E.; Muck-Lichtenfeld, C.; Yudin, A. K.; Studer, A. Chem. Eur. J. 2020, 26, 1501. doi: 10.1002/chem.201904727
Wu, A.; Feng, Q.; Sung, H. H. Y.; Williams, I. D.; Sun, J. Angew. Chem., Int. Ed. 2019, 58, 6776. doi: 10.1002/anie.201902866

图式 1 乙烯基氮杂环丙烷结构特点和反应模式
Scheme 1 Structural properties of vinylaziridines and reaction modes

图式 2 乙烯基氮杂环丙烷与羟基取代芳烃的亲核性开环反应
Scheme 2 Ring-opening reaction of vinylaziridines with hydroxyarenes

图式 3 乙烯基氮杂环丙烷与羟基取代芳烃的开环反应机理
Scheme 3 Reaction mechanism of ring-opening reaction of vinylaziridines with hydroxyarenes

图式 4 乙烯基氮杂环丙烷与取代吲哚的开环烷基化反应
Scheme 4 Ring-opening reaction of vinylaziridines with indoles

图式 5 铱催化的乙烯基氮杂环丙烷与醇和醛的不对称偶联反应
Scheme 5 Ir-catalyzed asymmetric coupling of vinylaziridines with alcohols and aldehydes

图式 6 铱催化的乙烯基氮杂环丙烷与吲哚的开环烯丙基化反应
Scheme 6 Ir-catalyzed ring-opening C3-allylation reaction of indole with vinylaziridine

图式 7 钯催化的芳基硼酸对乙烯基氮杂环丙烷的加成反应
Scheme 7 Pd(II)-catalyzed addition of arylboronic acid to vinylaziridines

图式 8 钯催化的三环乙烯基氮杂环丙烷的亲核性开环反应
Scheme 8 Pd(0)-catalyzed nucleophilic ring-opening of tricyclic vinylaziridines

图式 9 钴催化的乙烯基氮杂环丙烷参与的芳环和杂芳环的碳氢烯丙基化反应
Scheme 9 Co-catalyzed C—H allylation of (hetero)aromatic compounds with vinylaziridines

图式 10 二硼烷基锂盐对乙烯基氮杂环丙烷的区域选择性开环反应
Scheme 10 Nucleophilic ring-opening of vinylaziridines by diborylalkyllithium salts

图式 11 催化的区域选择性可调的磷亲核试剂对乙烯基氮杂环丙烷的亲核性开环反应
Scheme 11 Catalytic regiodivergent ring-opening reaction of vinylaziridines with phosphorus nucleophiles

图式 12 催化的乙烯基氮杂环丙烷与磷亲核试剂的反应机理
Scheme 12 Plausible mechanism for the catalytic reaction of vinylaziridines with phosphorus nucleophiles

图式 13 钯催化的乙烯基氮杂环丙烷与α, β-不饱和酮的不对称[3+2]环加成反应
Scheme 13 Pd-catalyzed asymmetric [3+2] cycloaddition reaction of vinylaziridines with α, β-unsaturated ketones

图式 14 钯催化剂/膦-硫脲催化的乙烯基氮杂环丙烷与对亚甲基苯醌的[3+2]环化反应
Scheme 14 Pd/phosphine-thiourea cooperatively catalyzed [3+2] cycloaddition of vinylaziridines with para-quinone methides

图式 15 钯催化的乙烯基氮杂环丙烷与亚甲基吲哚酮催化不对称[3+2]环加成反应
Scheme 15 Pd-catalyzed asymmetric [3+2] cycloaddition reaction of vinylaziridines and methylene indolinones

图式 16 钯催化的乙烯基氮杂环丙烷与3-硝基吲哚的[3+2]环加成反应
Scheme 16 Pd-catalyzed diastereoselective [3+2] cycloaddition of N-tosyl vinylaziridines with 3-nitroindoles

图式 17 钯催化的乙烯基氮杂环丙烷与3-硝基吲哚的[3+2]环加成反应机理
Scheme 17 Proposed mechanism for Pd-catalyzed diastereoselective [3+2] cycloaddition of N-tosyl vinylaziridines with 3-nitroindoles

图式 18 钯催化的乙烯基氮杂环丙烷与3-硝基吲哚的不对称[3+2]环加成反应
Scheme 18 Pd-catalyzed asymmetric [3+2] cycloaddition of N-tosyl vinylaziridines with 3-nitroindoles

图式 19 钯/双噁唑啉催化的乙烯基氮杂环丙烷与3-硝基吲哚的不对称[3+2]环加成反应
Scheme 19 Pd/chiral box complex-catalyzed asymmetric [3+2] cycloaddition of N-tosyl vinylaziridines with 3-nitroindoles

图式 20 钯催化的乙烯基氮杂环丙烷与环状N-砜基亚胺的非对映选择性[3+2]环加成反应
Scheme 20 Pd(0)-catalyzed diastereoselective [3+2] cycloaddition of vinylaziridine and cyclic imines

图式 21 钯催化乙烯基氮杂环丙烷与环状亚胺的[3+2]环加成反应机理
Scheme 21 Proposed mechanism for Pd-catalyzed [3+2] cycloaddition of vinylaziridines and cyclic N-sulfonyl imines

图式 22 铑催化的乙烯基氮杂环丙烷和炔烃的分子内立体选择性杂[5+2]环加成反应
Scheme 22 Rh(I)-catalyzed intramolecular stereoselective hetero-[5+2] cycloaddition reaction of vinylaziridines and alkynes

图式 23 铑催化的乙烯基氮杂环丙烷和烯烃的分子内立体选择性杂[5+2]环加成反应
Scheme 23 Rh(I)-catalyzed intramolecular stereoselective hetero-[5+2] cycloaddition reaction of vinylaziridines and alkenes

图式 24 铑催化的乙烯基氮杂环丙烷和炔烃的分子间可调的[3+2]和[5+2]环加成反应
Scheme 24 Rh(I)-catalyzed intermolecular [3+2] and [5+2] cycloaddition reaction of vinylaziridines and alkynes

图式 25 铑催化乙烯基氮杂环丙烷和联烯的区域性分子间[3+2]环化反应
Scheme 25 Rh-catalyzed regiodivergent intermolecular [3+2] cycloadditions of vinylaziridines and allenes

图式 26 铑催化的乙烯基氮杂环丙烷和烯醇硅醚的[3+2]环加成反应和亲和性开环反应
Scheme 26 Rh-catalyzed [3+2] cycloaddition and ring-open- ing reactions of vinylaziridines and silyl enol ethers

图式 27 乙烯基氮杂环丙烷和烯醇硅醚的反应机理
Scheme 27 Proposed mechanism for the reactions of vinylaziridines and silyl enol ethers

图式 28 铑(I)催化的乙烯基氮杂环丙烷与硅基二烯基醚的[4+3]环加成反应
Scheme 28 Rh(I)-catalyzed [4+3] cycloaddition reaction of vinylaziridines with silyl dienol ethers

图式 29 铑催化的乙烯基氮杂环丙烷与C, N-环偶氮甲碱亚胺的分子间[3+3]环加成反应
Scheme 29 Rh(I)-catalyzed intermolecular [3+3] cycloaddition reaction of vinylaziridines with C, N-cyclic azomethine imines

图式 30 铑(I)催化的乙烯基氮杂环丙烷和肟醚的[3+2]环加成反应
Scheme 30 Rh(I)-catalyzed [3+2] cycloaddition of vinylaziridines and oxime ethers

图式 31 铑(I)催化的乙烯基氮杂环丙烷和炔烃的[3+2]环加成/双键迁移反应合成吡咯
Scheme 31 Rh(I)-catalyzed [3+2] cycloaddition of vinylaziridines and alkynes and C=C bond migration

图式 32 铱催化的β-酮羰基化合物与乙烯基氮杂丙烷的多米诺环开环/环化反应
Scheme 32 Ir-catalyzed domino-ring-opening cyclization of vinylaziridines and β-ketocarbonyls

图式 33 铱催化的乙烯基氮杂环丙烷和对位-亚甲基苯醌的[4+3]环加成反应
Scheme 33 Iridium-catalyzed [4+3] cyclization of vinylaziridines and para-quinone methides

图式 34 布朗斯特酸介导的乙烯基氮杂环丙烷硅醚和呋喃以及环戊二烯的[4+3]环加成反应
Scheme 34 Brønsted acid-promoted [4+3] cycloaddition reactions of aziridinyl enolsilanes with furans and cyclopentadienes

图式 35 乙烯基氮杂环丙烷与硝酮的多米诺开环/环化反应
Scheme 35 Domino ring-opening/cyclization of vinylaziridines with nitrones

图式 36 乙烯基氮杂环丙烷与2-(2-溴苯基)-1H-吲哚的开环/偶联环化反应
Scheme 36 One-pot ring-opening cyclization of vinylaziridine with 2-(2-bromophenyl)-1H-indole

图式 37 乙烯基氮杂环丙烷与2-溴酚的环化反应
Scheme 37 Cyclization of vinylazapropane with 2-bromo- phenol

图式 38 3-取代吲哚与乙烯基氮杂环丙烷的[3+2]环加成反应
Scheme 38 [3+2] cycloaddition reaction of 3-substituted indoles with vinylaziridines

图式 39 钯和胺协同催化的乙烯基氮杂环丙烷与α, β-不饱和醛的不对称[3+2]环化反应
Scheme 39 Asymmetric [3+2] cycloaddition of vinylaziridines and α, β-unsaturated aldehydes by synergistic palladium and aminocatalysis

图式 40 三级胺和手性铱络合物协同催化的乙烯基氮杂环丙烷与MBH碳酸的不对称[3+3]环化反应
Scheme 40 Synergistic tertiary amine and chiral iridium complex-catalyzed asymmetric [3+3] cycloaddition of vinylaziridines and MBH carbonates

图式 41 硫自由基催化的乙烯基氮杂环丙烷与烯烃的自由基[3+2]环加成反应
Scheme 41 Thiyl radical-catalyzed radical [3+2] cycloaddition of vinylaziridines with alkenes

图式 42 硫自由基催化的乙烯基氮杂环丙烷与烯烃的[3+2]环加成机理
Scheme 42 Mechanism for the thiyl radical-catalyzed radical [3+2] cycloaddition of vinylaziridines with alkenes

图式 43 可见光驱动氮自由基催化的乙烯基氮杂环丙烷与烯烃的[3+2]环化反应
Scheme 43 Visible light-driven nitrogen radical-catalyzed [3+2] cyclization of N-tosyl vinylaziridines with alkenes

图式 40 氮自由基催化的乙烯基氮杂环丙烷与烯烃的[3+2]环化反应机理
Scheme 40 Mechanism for nitrogen radical-catalyzed [3+2] cyclization of N-tosyl vinylaziridines with alkenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

 下载:
下载: