Received Date:
25 March 2020 Revised Date:
30 April 2020 Available Online:
25 November 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21925109, 21772170), the Outstanding Young Talents of Zhejiang Province (No. ZJWR0108), the Fundamental Research Funds for the Central Universities (No. 2018XZZX001-02) and the Natural Science Foundation of Zhejiang Province (No. LR17B020001)
Abstract:
In the past decades, transition metal-catalyzed C—H activation has experienced tremendous growth and revolutionized the field of organic synthesis. Several elegant strategies have been developed to promote reactivity and control precise site-selectivity. Among which, transient directing group strategy has been recognized to be an efficient and powerful approach for selective C—H functionalization. In contrast to traditional directing groups with covalent linkage, transient directing group strategy circumvents the covalent installation and removal of directing groups, which significantly improve the synthetic efficiency and broaden the range of synthetic applications. The recent advances in imine-based transition directing groups are summarized, providing an overview of recent achievements in this cutting-edge research field over the past few years. For clarity, it is classified into two sections according to the type of substrate and the type of activated hydrocarbon bond. Emphasis is placed on the fully discussion of various transient directing groups and their applications. Finally, the limitations of previous works and perspectives on this cutting-edge area are also described.
Dedicated to the 40th anniversary of Chinese Journal of Organic Chemistry.
[1]
(a) Godula, K.; Sames, D. Science2006, 312, 67. (b) Bergman, R. G. Nature2007, 446, 391. (c) Ellman, J. A.; Ackermann, L.; Shi, B. F. J. Org. Chem.2019, 84, 12701.
[2]
For recent reviews on C-H activation, see: (a) Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed.2009, 48, 5094. (b) Lyons, T. W.; Sanford, M. S. Chem. Rev.2010, 110, 1147. (c) Pan, F.; Shi, Z. Acta Chim. Sinica2012, 70, 1679(in Chinese). (潘菲, 施章杰, 化学学报, 2012, 70, 1679.) (d) Yuan, Y.; Song, S.; Jiao, N. Acta Chim. Sinica2015, 73, 1231(in Chinese). (袁逸之, 宋颂, 焦宁, 化学学报, 2015, 73, 1231.) (e) Liu, B.; Hu, F.; Shi, B.-F. ACS Catal.2015, 5, 1863. (f) Huang, Z.; Lim, H. N.; Mo, F.; Young, M. C.; Dong, G. Chem. Soc. Rev.2015, 44, 7764. (g) Daugulis, O.; Roane, J.; Tran, L. D. Acc. Chem. Res.2015, 48, 1053. (h) He, G.; Wang, B.; Nack, W. A.; Chen, G. Acc. Chem. Res.2016, 49, 635. (i) Rao, W.-H.; Shi, B.-F. Org. Chem. Front.2016, 3, 1028. (j) Yang, Y.; Lan, J.; You, J. Chem. Rev.2017, 117, 8787. (k) He, J.; Wasa, M.; Chan, K. S. L.; Shao, Q.; Yu, J.-Q. Chem. Rev.2017, 117, 8754. (l) Huang, J.; Gu, Q.; You, S.-L. Chin. J. Org. Chem.2018, 38, 51(in Chinese). (黄家翩, 顾庆, 游书力, 有机化学, 2018, 38, 51.) (m) Ren, Q.; Nie, B.; Zhang, Y.; Zhang, J. Chin. J. Org. Chem.2018, 38, 2465(in Chinese). (任青云, 聂飚, 张英俊, 张霁, 有机化学, 2018, 38, 2465.) (n) Zhao, K.; Yang, L.; Liu, J.; Xia, C. R. Chin. J. Org. Chem.2018, 38, 2833(in Chinese). (赵康, 杨磊, 刘建华, 夏春谷, 有机化学, 2018, 38, 2833.) (o) Xu, L.; Xu, H.; Lin, H.; Dai, H. Chin. J. Org. Chem.2018, 38, 1940(in Chinese). (徐琳琳, 徐辉, 林海霞, 戴辉雄, 有机化学, 2018, 38, 1940.) (p) Zhan, B.-B.; Shi, B.-F. Chin. J. Org. Chem.2019, 39, 3602(in Chinese). (占贝贝, 史炳锋, 有机化学, 2019, 39, 3602.) (q) Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. Chem. Rev.2019, 119, 2192. (r) Han, Y.-Q.; Zhou, T. Chin. J. Chem.2020, 38, 527. (s) Liu, Y.-H.; Xia, Y.-N.; Shi, B.-F. Chin. J. Chem.2020, 38, 635.
[3]
(a) Zhang, M.; Zhang, Y.; Jie, X.; Zhao, H.; Li, G.; Su, W. Org. Chem. Front.2014, 1, 843. (b) Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M. F.; Wencel-Delord, J.; Besset, T.; Maes, B. U. W.; Schnürch, M. Chem. Soc. Rev.2018, 47, 6603. (c) Zhang, Q.; Shi, B.-F. Chin. J. Chem.2019, 37, 647. (d) Rej, S.; Ano, Y.; Chatani, N. Chem. Rev.2020, 120, 1788.
For selected examples of phosphites as transient directing groups, see: (a) Bedford, R. B.; Coles, S. J.; Hursthouse, M. B.; Limmert, M. E. Angew. Chem., Int. Ed.2003, 42, 112. (b) Bedford, R. B.; Limmert, M. E. J. Org. Chem.2003, 68, 8669. (c) Bedford, R. B.; Betham, M.; Caffyn, A. J.; Charmant, J. P.; Lewis-Alleyne, L. C.; Long, P. D.; Polo-Ceron, D.; Prashar, S. Chem. Commun.2008, 990. (d) Bedford, R. B.; Haddow, M. F.; Webster, R. L.; Mitchell, C. J. Org. Biomol. Chem.2009, 7, 3119. (e) Carrion, M. C.; Cole-Hamilton, D. J. Chem. Commun.2006, 4527. (f) Lewis, J. C.; Wu, J.; Bergman, R. G.; Ellman, J. A. Organometallics2005, 24, 5737. (g) Oi, S.; Watanabe, S.-I.; Fukita, S.; Inoue, Y. Tetrahedron Lett.2003, 44, 8665. (h) Yang, J. F.; Wang, R. H.; Wang, Y. X.; Yao, W. W.; Liu, Q. S.; Ye, M. Angew. Chem., Int. Ed.2016, 55, 14116. (i) Liu, Q.-S.; Wang, D.-Y.; Yang, J.-F.; Ma, Z.-Y.; Ye, M. Tetrahedron2017, 73, 3591.
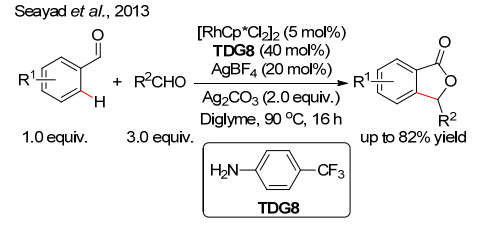
Liu, X. H.; Park, H.; Hu, J. H.; Hu, Y.; Zhang, Q. L.; Wang, B. L.; Sun, B.; Yeung, K. S.; Zhang, F. L.; Yu, J. Q. J. Am. Chem. Soc.2017, 139, 888. doi: 10.1021/jacs.6b11188
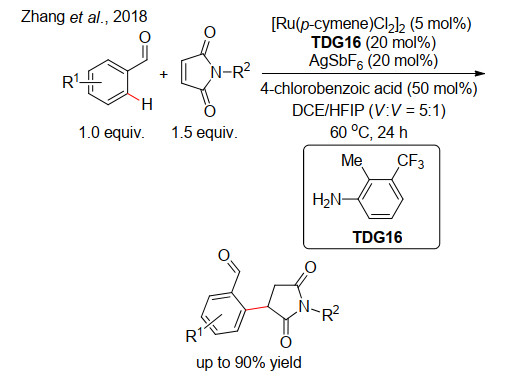
(a) Liao, G.; Yao, Q. J.; Zhang, Z. Z.; Wu, Y. J.; Huang, D. Y.; Shi, B. F. Angew. Chem., Int. Ed.2018, 57, 3661. (b) Liao, G.; Li, B.; Chen, H. M.; Yao, Q. J.; Xia, Y. N.; Luo, J.; Shi, B. F. Angew. Chem., Int. Ed.2018, 57, 17151.
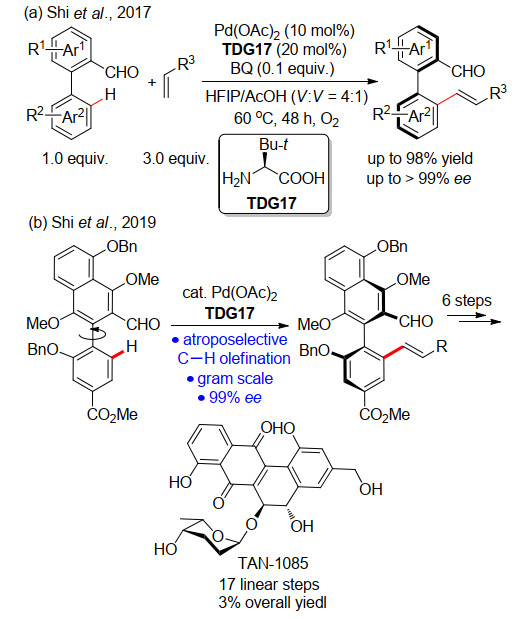
[30]
Liao, G.; Chen, H. M.; Xia, Y. N.; Li, B.; Yao, Q. J.; Shi, B. F. Angew. Chem., Int. Ed.2019, 58, 11464. doi: 10.1002/anie.201906700
Chen, Y.-Q.; Wang, Z.; Wu, Y.; Wisniewski, S. R.; Qiao, J. X.; Ewing, W. R.; Eastgate, M. D.; Yu, J.-Q. J. Am. Chem. Soc.2018, 140, 17884. doi: 10.1021/jacs.8b07109
[61]
(a) Vicente, J.; Saura-Llamas, I.; Palin, M. G.; Jones, P. G.; Ramírez de Arellano, M. C. Organometallics1997, 16, 826. (b) Largeron, M. Eur. J. Org. Chem.2013, 2013, 5225. (c) Corey, E. J.; Achiwa, K. J. Am. Chem. Soc.1969, 91, 1429. (d) Hartwig, J. F.; Richards, S.; Barañano, D.; Paul, F. J. Am. Chem. Soc.1996, 118, 3626. (e) Wolfe, J. P.; Wagaw, S.; Buchwald, S. L. J. Am. Chem. Soc.1996, 118, 7215. (f) Ryland, B. L.; Stahl, S. S. Angew. Chem., Int. Ed.2014, 53, 8824.


 下载:
下载:















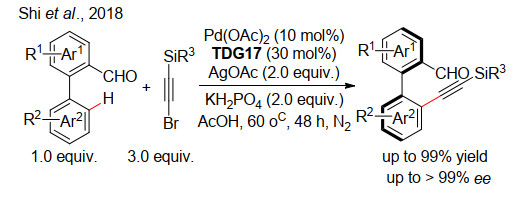





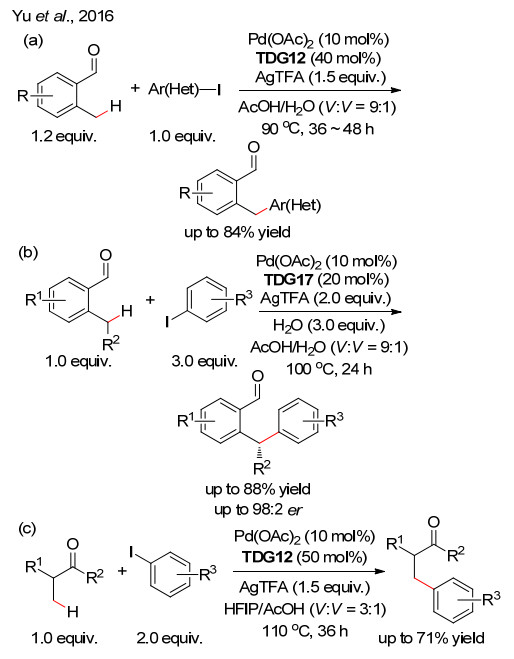






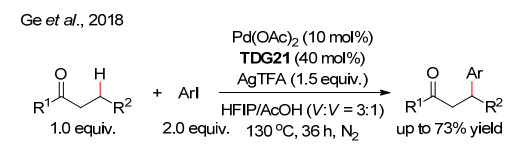

















 下载:
下载:



















































 下载:
下载:
