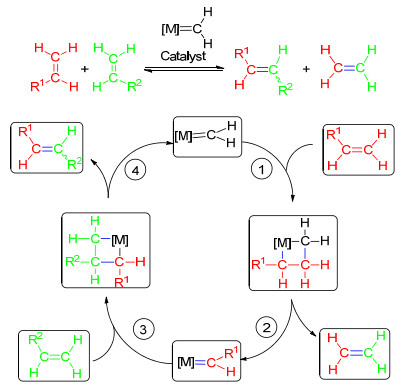
图式 1.
烯烃复分解反应机理
Scheme 1.
Mechanism of the metathesis of olefins

烯烃复分解反应(olefin metathesis)是指两种不饱和碳-碳重键(叁键或双键)之间碳骨架的重排反应, 是在金属烯烃络合物(金属卡宾配合物)催化下碳碳重键断裂后又重新组合的过程(Scheme 1).与传统的合成烯烃的工艺相比, 这种制备烯烃的方法具有操作简便、成本低廉、副反应少、后处理容易等优点.由于烯烃复分解反应条件温和, 产率较高, 绝大多数有机基团在反应中无需保护, 此类反应成为一种备受青睐的合成手段.近年来, 随着各种性能优良的催化剂不断出现, 烯烃复分解反应在复杂有机分子、生物活性化合物以及天然产物的不对称合成中的应用越来越广泛.目前研究较多的烯烃复分解反应主要有三大类型:关环复分解反应(RCM)、开环交叉复分解反应(ROCM)和交叉复分解反应(CM).
自从烯烃复分解反应被发现以来, 关于其反应机理人们进行了大量研究并提出了许多假说.目前普遍接受的催化机理是Chauvin等[1]于1971年提出的金属卡宾催化烯烃复分解反应的机理(Scheme 1).
在烯烃复分解反应过程中, 金属卡宾是催化反应的活性中心, 而钌杂环丁烷则是重要的中间体, 经过一系列[2+2]环加成反应和逆环加成反应的转化, 最终得到目标产物.催化剂中的金属卡宾首先与反应物中的一个烯烃分子结合, 形成由一个金属原子和三个碳原子构成的四元环型化合物.然后, 四元环型化合物中的两个单键断裂, 生成了一分子乙烯和新的金属卡宾, 该金属卡宾与另外的烯烃分子继续反应, 再次形成一个新的含金属四元环化合物.最后, 四元环的两个单键再次断裂, 生成了新的烯烃和金属卡宾, 而该金属卡宾又进入下一催化循环过程.这就是Chauvin机理.
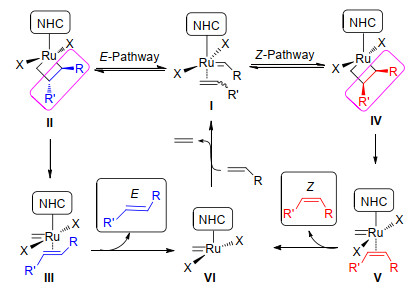
在早期研究中, 人们对于烯烃复分解反应产物的立体化学问题关注并不多, 反应的立体化学控制完全依赖于烯烃自身反应活性和反应过程的热力学差异.近年来, 随着烯烃复分解反应催化剂的发展和对烯烃复分解反应立体化学的深入研究, 人们发现在烯烃复分解反应中产物的立体化学可以通过改变催化剂的结构来改变反应的动力学, 从而控制产物的立体选择性.产物的立体选择性主要是指生成烯烃分子中双键的Z/E构型选择性和生成双键相邻的手性碳原子的R/S构型选择性.
烯烃复分解反应产物立体构型的动力学选择性主要依赖于反应中间体钌杂环丁烷的空间效应和电子效应(Scheme 2), 它们决定了产物的构型.如果钌杂环丁烷形成反式排列(Ⅱ), 环化反应结束后将产生E-烯烃.反之, 如果钌杂环丁烷与烯烃形成顺式排列(Ⅳ), 则环化反应结束后将得到Z-异构体.因此, 为了开发Z-或E-选择性催化剂, 需要将烯烃与钌卡宾的配位限制在某一侧; 同时在[2+2]环加成后产生的钌配位环需要以某种方式形成全部顺式或反式排列, 这样反应后才能选择性得到Z-烯烃或E-烯烃.
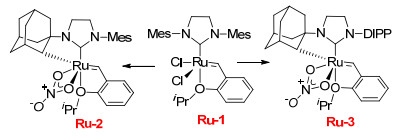
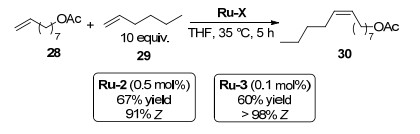
2012年, Grubbs课题组[2]为了改进现有催化剂的稳定性、活性和选择性, 对催化剂Ru-1进行了结构改造.为了增大空间位阻, 将用硝酸根代替钌原子上的氯原子, N-金刚烷基取代N-杂环卡宾上的N-Mes基团, 得到催化剂Ru-2, 用体积更大的N-2, 6-二异丙基苯基(DIPP)代替另一个N-Mes基团得到催化剂Ru-3 (Scheme 3).
2013年, Grubbs课题组[3-4]考察了上述两个改造后的催化剂在利用烯烃RCM制备大环类化合物过程中的立体选择性.结果表明, 对于多数大环RCM, Ru-2表现出中等偏上的立体选择性(65%~86% Z), 而Ru-3则表现出比较突出的Z-构型产物选择性(>95% Z)(表 1).
 下载:
导出CSV
下载:
导出CSV

|
2017年, Grubbs课题组[5]将催化剂Ru-1 N-杂环卡宾上的N-Mes基团全部换为DIPP, 发展了含有邻二硫酚配体的催化剂Ru-4.通过考察其在大环烯烃化合物合成中的立体选择性发现, Ru-4可以高选择性地得到Z-构型环烯烃产物.而且, 即使是在较低温度下, 此类反应也可在几分钟内完成.与之前反应体系相比, 催化剂表现出显著的反应活性和高效的立体选择性.这种方法适用于合成十二元环到十七元环化合物, 反应的收率在68%~79%之间, Z-构型所占比例介于95%~99%之间(表 2).
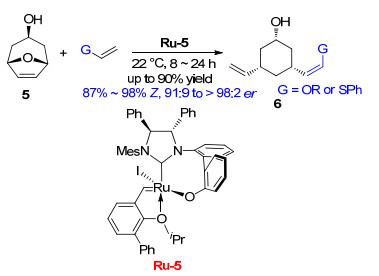
2012年, Hoveyda课题组[6]报道了用催化剂Ru-5催化双环氧杂环烯烃与烯醇醚或苯基乙烯基硫醚发生的ROCM (Scheme 4).研究结果表明, 该催化剂表现出很好的Z-构型选择性, Z构型烯烃比例最高可达98%;同时该反应的对映选择性也非常突出, 对映体比例大于98:2.
同年, Hoveyda等[7]将钌催化的烯烃ROCM应用在降冰片烯二醇与末端烯烃的反应中.结果发现(表 3), 中表现出较好的Z-选择性, Z-烯烃产物比例高达98%, 催化剂Ru-6在各种取代的末端烯烃参与的ROCM反应活性中等偏上, 收率58%~97%.
下载:
导出CSV
 |
||||
| Entry | R | Ru-6/mol% | Yield/% | Z/% |
| 1 | Ph | 1 | 92 | 97 |
| 2 | m-FC6H4 | 1 | 93 | 96 |
| 3 | (CH2)2OTBS | 5 | 68 | >98 |
| 4 | (CH2)2C(O)NHPh | 5 | 65 | >98 |
| 5 | (CH2)7Me | 5 | 58 | >98 |
| 6 |  |
5 | 93 | 93 |
| 7 |  |
5 | 97 | >98 |
| 8 |  |
2 | 84 | 91 |
| 9 |  |
5 | 80 | >98 |
| 10 | OnBu | 2 | 95 | >98 |
| 11 | SEt | 5 | 80 | 92 |
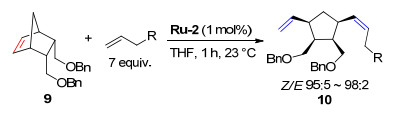
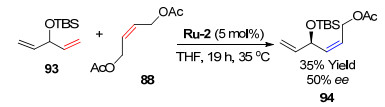
2013年, Grubbs课题组[8]研究了降冰片烯衍生物与末端烯烃发生的ROCM (Scheme 5).催化剂Ru-2在反应中表现出很高的Z-构型选择性, Z/E比高达98:2, 但是收率仅达到中等水平.
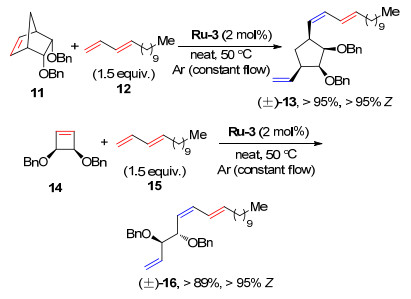
2016年, Grubbs课题组[9]报道了环烯烃与3E-1, 3-共轭二烯烃在钌催化剂作用下发生ROCM的研究.结果表明, 两者在催化剂Ru-3的作用下可以高立体选择性地得到开环共轭二烯烃(>95% Z), 收率可达95% (Scheme 6).
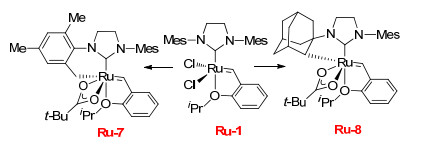
2011年, Grubbs课题组[10-11]将催化剂Ru-1结构中氯原子用体积较大的新戊羧基配体代替(Scheme 7), 制备了催化剂Ru-7.在其催化下的烯烃复分解反应中得到了当时最好的选择性(41%, Z).随后, 为了进一步增加催化剂自身的立体效应, 又设计了N-金刚烷基取代的N-杂环卡宾Ru-8, 这个催化剂具有更大的空间位阻.结果发现利用Ru-8催化烯丙基苯和顺式-1, 4-二乙酰氧基-2-丁烯的复分解反应, 获得高达88%的Z-构型烯烃产物, 而在催化末端烯烃发生的自身CM中竟获得>95%的Z-产物(表 4).
下载:
导出CSV
 |
||||
| Entry | R | Ru-8/mol% | Yield/% | Z/% |
| 1 | CH2Ph | 0.10 | >95 | >95 |
| 2 | CH2Ph | 0.01 | 74 | >95 |
| 3 | (CH2)3OH | 0.10 | >95 | >95 |
| 4 | (CH2)8CO2Me | 0.10 | 77 | 95 |
2013年, 该课题组[4, 12]将催化剂Ru-2和Ru-3应用到烯丙基苯和顺式1, 4-二乙酰氧基-2-丁烯复分解反应中, 结果发现改造后催化剂反应活性和立体选择性都大大提高.催化剂Ru-2在0.5 mol%的用量下可以获得91%的Z-选择性和高达67%的收率; 当使用Ru-3为催化剂时, 即使将催化剂的用量减少到0.1 mol%, 也可以得到不低于95%的Z-选择性烯烃产物(表 5)
下载:
导出CSV
 |
||||||
| Entry | R | R1 | R2 | Cat. (mol%) | Yield/% | Z/% |
| 1 | CH2Ph | CH2OAc | CH2OAc | Ru-2 (1.0) | 58 | 91 |
| 2 | (CH2)Me | H | H | Ru-2 (0.5) | 70 | 91 |
| 3 | (CH2)3Me | (CH2)7OAc | H | Ru-2 (0.5) | 67 | 91 |
| 4 | (CH2)Me | H | H | Ru-3 (0.5) | 71 | >95 |
| 5 | (CH2)3Me | (CH2)7OAc | H | Ru-3 (0.1) | 60 | >95 |
同年, Grubbs课题组[13]还考察了催化剂Ru-3对末端烯烃和非共轭的二烯烃发生的CM催化效果.结果发现, Ru-3表现出很好的Z-构型选择性和化学区域选择性(表 6), 最终以较高的收率得到了1, 4-二烯复分解产物.
下载:
导出CSV
 |
|||
| Entry | R | Yield/% | Z/% |
| 1 | CH2Ph | 63 | > 95 |
| 2 | (CH2)8CHO | 70 | > 95 |
| 3 | (CH2)7CO2Me | 82 | > 95 |
| 4 | CH2NHPh | 68 | > 95 |
| 5 | CH2OCO2Me | 79 | > 95 |
2014年, Grubbs小组[14]又研究了催化剂Ru-3对各种乙烯基缩醛与十二烷烯发生的CM催化活性和选择性.结果表明, 在不同的底物反应时, Ru-3都表现出很好的反应活性和立体选择性(表 7).其中, 非环二乙基缩醛Z-构型产物的选择性多数大于95%, 对于二聚缩醛产物27g, 产物的主要异构体为Z, Z-构型, 所占比例大于89%, 而且其他异构体只有Z, E-构型(11%).
2015年, Grubbs课题组[15]将催化剂Ru-2和Ru-3的应用拓展到1-己烯与8-壬烯醋酸酯的CM中, 考察其在反应中的立体选择性(Scheme 8).结果发现, 当使用0.5 mol%的催化剂Ru-2时, 反应得到67%的收率和91%的Z构型选择性; 当将Ru-2中的N-Mes换为体积较大的N-DIPP基团变为Ru-3时, 即使仅用0.1 mo%催化剂用量, 也可以得到大于98%的Z-构型产物, Z-选择性大大提高.
同年, Hoveyda课题组[16]考察了二硫代的催化剂Ru-9在顺-2-丁烯-1, 4-二醇与端烯发生的CM中的立体选择性.他们发现, 所考查的端烯大多具有较高的Z-构型选择性, 当底物为空间位阻较大的末端烯烃时, 发生CM需要增加催化剂用量.同时还发现醛类、羧酸类末端烯烃以及烯丙基醚类底物在催化剂Ru-9的作用下也可以发生此类反应(表 8).
下载:
导出CSV
 |
||||
| Entry | R | Time/h | Yield/% | Z/% |
| 1 | (CH2)9Me | 4 | 72 | 96 |
| 2 | (CH2)2OTBS | 4 | 65 | 93 |
| 3 | (CH2)2OPNP | 4 | 74 | 96 |
| 4 | (CH2)2OnBu | 12 | 57 | 91 |
| 5 | (CH2)2CO2Bn | 4 | 80 | 98 |
| 6 | (CH2)4Phth | 4 | 64 | 98 |
| 7 | (CH2)8CHO | 4 | 80 | 94 |
| 8 | (CH2)3CO2H | 4 | 70 | 96 |
| 9 | Cy | 4 | 59 | 98 |
| 10 |  |
4 | 73 | 98 |
| 11 |  |
8 | 66 | 95 |
| 12 |  |
8 | 63 | 92 |
| 13 |  |
8 | 56 | 96 |
| 14 |  |
8 | 54 | 87 |
2016年, Grubbs课题组[9]报道了末端烯烃与3E-1, 3-共轭二烯烃在钌催化剂作用下发生CM的立体选择性(Scheme 9).结果表明, 两种烯烃在催化剂Ru-3的作用下可以高选择性地得到Z-构型的共轭二烯烃(>95% Z).
2010年, Blechert课题组[17]研究了催化剂Ru-10在降冰片烯衍生物与苯乙烯发生ROCM的选择性.研究发现, 在催化剂作用下可以得到较好对映选择性的目标二烯烃产物, 而且主要是E-异构体(表 9).
下载:
导出CSV
 |
||||
| Entry | Substrate | E:Z | Coversion/% | ee/% |
| 1 |  |
24:1 13:1 |
>98 >98 |
82 90 |
| 2 |  |
21:1 23:1 |
>98 >98 |
82 90 |
| 3 |  |
19:1 | 61 | 86 |
| 4 |  |
>30:1 | >98 | 76 |
| 5 |  |
21:1 | >98 | 70 |
2013年, Grubbs课题组[3, 18]发现, 当用催化剂Ru-2催化Z/E构型混合大环烯烃发生乙烯解反应时, 其中的Z构型烯烃具有较高的反应活性, 而E构型烯烃反应活性很低.结果导致反应产物中, 除了发生开环反应得到产物外, E-构型大环烯烃的比例大幅提高, 从而得到了E构型含量高的烯烃产物(表 10).相同的现象也发生在链状烯烃的复分解反应中(表 11).
下载:
导出CSV
 |
|||
| Entry | Substrate | Initial E/% | Final E/% |
| 1 | 19 | 55 | > 95 |
| 2 | 20 | 80 | > 95 |
| 3 | 21 | 80 | > 95 |
下载:
导出CSV
 |
||||
| Entry | R | n | Initial E/% | Final E/% |
| 1 | Me | 3 | 52 | 90 |
| 2 | OAc | 7 | 78 | > 95 |
| 3 | OH | 4 | 68 | 90 |
| 4 | CO2Me | 6 | 80 | > 95 |
| 5 | NHPh | 3 | 80 | > 95 |
| 6 | C(O)Me | 2 | 72 | > 95 |
2016年, Grubbs课题组[19]在研究1-癸烯与反式-4-辛烯的CM中, 发现邻二硫酚离子配位的钌催化剂在反应中表现出很好的E-选择性(表 12).当使用Ru-4时, 得到中等E-构型选择性; 而当使用钌NHC芳基邻位取代基为体积较小甲基的Ru-9催化时, E-构型烯烃比例超过99%, 收率也有所提高; 而当钌NHC芳基邻位取代基为甲基或原子半径更小的氟时(Ru-10和Ru-11), 反应保持了高度的E-构型选择性, 同时反应收率也大大提高.
下载:
导出CSV
 |
||||
| Entry | Ru-X | Conv./% | Yield/% of 46 | E/Z |
| 1 | Ru-4 | 92 | 4 | 88/12 |
| 2 | Ru-9 | 37 | 7 | > 99/1 |
| 3 | Ru-10 | 53 | 29 | > 99/1 |
| 4 | Ru-11 | 55 | 31 | > 99/1 |
近年来, 立体化学保持的烯烃复分解反应成为非常活跃的研究领域, 人们发现基于邻二硫酚离子配位的新型钌催化剂可以使烯烃经过复分解反应得到立体保持的烯烃产物[20].也就是说, 使用Z-构型(E-构型)的烯烃为起始原料, 经过某种催化剂催化的烯烃复分解反应后可以得到构型仍然是Z-构型(E-构型)烯烃产物.
2013年, Hoveyda课题组[21]最早报道了二硫代钌卡宾催化剂Ru-6, 并利用其催化烯烃复分解反应.研究结果认为, 该催化剂为Z-构型选择性催化剂. 2016年, Grubbs课题组[19]在研究该催化剂催化CM时发现, 催化剂Ru-6确实可以促进Z-和E-烯烃的烯烃自身复分解, 但是所得产物保持了原料烯烃的立体化学. Grubbs及其同事重点研究了催化剂Ru-4和Ru-9在烯烃复分解反应中的立体选择性.结果显示催化剂Ru-4和Ru-9也有很好的立体化学保持的特性(表 13).
下载:
导出CSV
 |
||||
| Entry | Ru (mol%) | 47 | 48/% (Z/E) | 49/% (Z/E) |
| 1 | Ru-4 (0.01) | >99% Z | 24 (>99/1) | 24 (>99/1) |
| 2 | Ru-9 (0.50) | >99% Z | 10 (>99/1) | 10 (>99/1) |
| 3 | Ru-9 (7.50) | >97% E | 24 (4/96) | 24 (4/96) |
2017年Grubbs课题组[22]研究了催化剂Ru-12和Ru-13在烯烃自身复分解反应中的立体选择性.结果发现, 这两个催化剂均实现了立体保持的催化效果(表 14).
下载:
导出CSV
 |
||||
| Entry | 50 | Ru (mol%) | 51/% (E/Z) | 52/% (E/Z) |
| 1 | E-50 | Ru-12 (1.00) | 25 (>99/1) | 25 (>99/1) |
| 2 | E-50 | Ru-13 (1.00) | 6 (>99/1) | 6 (>99/1) |
| 3 | Z-50 | Ru-12 (0.10) | 25 (<1/99) | 25 (<1/99) |
| 4 | Z-50 | Ru-13 (0.05) | 25 (<1/99) | 25 (<1/99) |
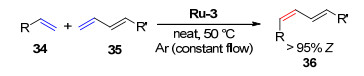
同年, Grubbs课题组[23]研究了催化剂Ru-12~Ru-14在顺式1, 4-二乙酰氧基-2-丁烯与顺式-4-辛烯发生的CM中的立体选择性(Scheme 10).结果发现, 各催化剂均具有很高的反应活性, 反应开始仅20 min, 转化率即可80%以上, 所得烯烃产物保持反应物的Z-构型, 所占比例均>99%.
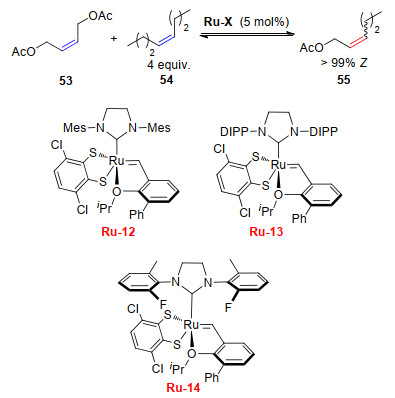
同时, 该研究还发现在反式1, 4-二乙酰氧基-2-丁烯与反式-4-辛烯发生的CM中, 催化剂Ru-14也表现出很高的反应活性, 反应25 min完成80%的转化, 同时得到>99%的E-构型保持的烯烃产物(Scheme 11).
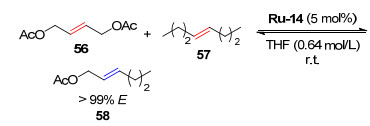
2017年, Hoveyda小组[24-25]报道了一种采用顺式-2-丁烯作为封端试剂, 在催化剂Ru-9的作用下顺式或反式烯烃与末端烯烃发生交叉复分解反应, 得到构型保持产物的案例, 并利用此性质合成了前列腺素(Scheme 12).
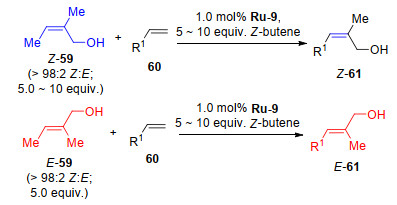
2018年, Grubbs课题组[26]利用二硫代钌催化剂(Ru-4、Ru-6)通过烯烃复分解反应实现了立体保持(选择性>99%)的E-构型大环烯烃化合物的合成, 从12元环到18元环均可实现(表 15).
下载:
导出CSV

|
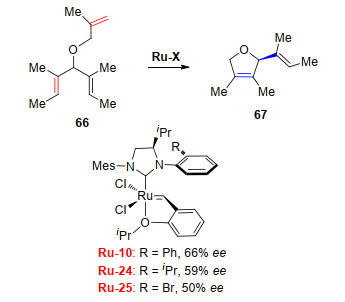
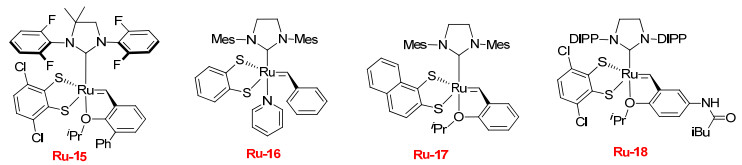
Grubbs课题组对催化剂Ru-10、Ru-14和Ru-15选择性的研究表明, NHC-配体N-芳基的邻位取代基越小, 烯烃复分解反应产物中E-构型烯烃的立体保持效率越高[22]. Hoveyda课题组[27-29], Grubbs课题组[30]和Marc Mauduit课题组[31]分别研究了催化剂Ru-16、Ru-17、Ru-18在复分解反应中的立体保持特性, 结果发现这三个催化剂效果都较差(图 1).
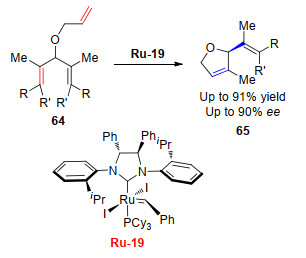
烯烃复分解反应中, 催化剂对产物烯烃结构中新产生手性碳原子构型的立体选择性的研究起源于Grubbs课题组[32], 他们以手性NHC钌络合物Ru-19为催化剂, 催化一个原本存在着对称因素的三烯化合物发生RCM, 在催化剂的作用下, 化合物64立体选择地向一侧关环, 最终反应以高对映选择性(90% ee)得到不对称RCM产物(Scheme 13).此后, 研究者在此基础上, 陆续开发了众多钌催化剂并研究了它们的立体选择性和反应活性.
2010年, Collins课题组[33]报道了几个具有C1对称的NHC配体钌催化剂(Ru-20~Ru-23), 由于催化剂的顺式(顺式是指与钌卡宾位于同一侧的NHC配体上的烷基)和反式异构体难以分离, 所以这几个催化剂是以混合物的形式使用.研究结果表明, 对称三烯66的RCM反应具有一定的对映选择性, 但是反应的立体选择性并不高(ee<52%)(表 16).
下载:
导出CSV
 |
|||
| Entry | Cat. | Conversion/% | ee/% |
| 1 | Ru-20 | 95 | 8 |
| 2 | Ru-21 | 95 | 38 |
| 3 | Ru-22 | 90 | 52 |
| 4 | Ru-23 | 86 | 50 |
同年, Hoveyda课题组[21]为了提高催化剂的立体选择性, 研究了具有两个不同N-取代基的NHC催化剂.结果发现这些催化剂在催化对称三烯发生RCM时立体选择性有所提高, 产物烯烃的ee值可以达到66% (Scheme 14).
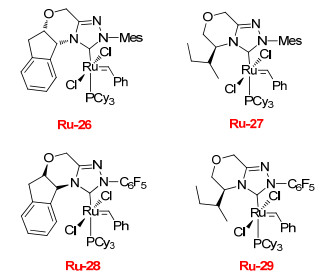
2013年, Grela课题组[34]报道了一组以1, 2, 4-三唑为骨架的钌催化剂(Ru-26~Ru-30)及其在不对称关环复分解反应(ARCM)中的应用研究(图 2).研究结果表明, 与传统催化剂相比, 这种具有1, 2, 4-三唑骨架的钌催化剂催化效果并不理想(表 17).
 下载:
导出CSV
下载:
导出CSV
 |
|||
| Entry | Catalyst | Conv./% | ee/% |
| 1 | Ru-26 | >95 | 26 |
| 2 | Ru-27 | 64 | 4 |
| 3 | Ru-28 | 59 | 9 |
| 4 | Ru-29 | 85 | 8 |
2014年和2016年, Grisi课题组[35-36]先后报道了在NHC配体骨架上同时含有烷基和苯基取代的不对称钌催化剂(Ru-30~Ru-33)及其反应活性和立体选择性.研究结果表明, 这种具有不对称取代的NHC骨架在增强催化剂的立体选择性方面有着积极作用, 经过烯烃复分解反应得到了中等对映选择性的闭环产物.相同催化剂用量条件下, N原子上取代基的变化对催化剂的选择性没有明显影响, 同时还发现在反应体系中加入NaI可以提高其立体选择性(表 18).
下载:
导出CSV
 |
|||||
| Entry | Ru-X (mol%) | Solvent | Additive | Conversion/% | ee/% |
| 1 | Ru-30 (2.5) | DCM | — | >98 | 18 |
| 2 | Ru-30 (4.0) | THF | NaI | >95 | 53 |
| 3 | Ru-31 (2.5) | DCM | — | >98 | 33 |
| 4 | Ru-31 (4.0) | THF | NaI | >98 | 50 |
| 5 | Ru-32 (2.5) | DCM | — | >98 | 19 |
| 6 | Ru-32 (4.0) | THF | NaI | >95 | 52 |
| 7 | Ru-33 (2.5) | DCM | — | >98 | 33 |
| 8 | Ru-33 (4.0) | THF | NaI | >98 | 47 |
2014年, Grubbs课题组[37]考察了钌催化的含有硅醚或酰胺的末端三烯烃发生的RCM的立体选择性.结果表明, 在Ru-2作用下反应得到了中等以上的收率和较好的对映体选择性(表 19).另外研究还发现, 当产物的环稳定性不高时, 及时除去产物中的乙烯可以有效地抑制反应向逆方向进行.
下载:
导出CSV
 |
||||
| Entry | 68 | 69 | Yield/% | ee/% |
| 1 |  |
 |
65 | 69 |
| 2 |  |
 |
29 | 67 |
| 3 |  |
 |
95 | 54 |
| 4 |  |
 |
72 | 47 |
| 5 |  |
 |
90 | 57 |
2011年, Blechert课题组[38]报道了一种含有喹啉骨架NHC配体的手性钌催化剂(Ru-34和Ru-35), 并研究了此类催化剂在烯烃开环复分解反应中的立体选择性, 结果发现由于喹啉环的存在, 限制了N-芳基的自由旋转, 将其固定在适当的位置, 从而增强了用于形成钌环丁烷的手性口袋, 因此获得了具有较好对映选择性的手性二烯产物, ee可达到98%, 转化率也高达99%(表 20).
下载:
导出CSV
| Substrate | Product | Ru-X | Conv./% (E:Z) | ee/% E (Z) |
 |
 |
 |
>99 (1:2) | 79 (87) |
 |
 |
 |
>99 (2:1) | 82 (82) |
 |
 |
Ru-34 Ru-35 |
96 (2:1) >99 (2:1) |
98 (92) 95 (91) |
2012年, Hoveyda课题组[6-7]研究了NHC配体骨架上同时含有两个芳氧基取代的钌催化剂(Ru-36)在AROCM中的立体选择性.结果发现当使用烯醇醚或硫醇醚作为偶联配偶体时, 这些催化剂经过AROCM得到立体专一的二烯取代的四氢吡喃产物, 而且这些产物中Z-异构体占绝对优势(表 21).
下载:
导出CSV
 |
|||||
| Entry | Product | R | Yield/% | Z:E | ee/% |
| 1 | 78a | OnBu | 80 | 95:5 | 96 |
| 2 | 78b | OCy | 64 | 98:2 | 96 |
| 3 | 78c | OPMP | 67 | 95:5 | 94 |
| 4 | 78d | OCH2CF3 | 65 | 94:6 | 92 |
| 5 | 78e | O(CH2)2Cl | 63 | 95:5 | 96 |
| 6 | 78f | SPh | 67 | 91:9 | 92 |
2013年, 基于Hoveyda课题组研究结果, Grubbs课题组[8]在降冰片烯与末端烯烃的AROCM中使用Ru-2作为催化剂, 立体选择性地得到Z-构型二烯烃产物(表 22).同时发现当降冰片烯衍生物进行AROCM时, 结构中含醚键的降冰片烯衍生物也具有良好的适用性, 即使是具有刚性的降冰片烯衍生物也可以得到较好的立体选择性产物.
下载:
导出CSV
 |
||||
| Entry | Substrate | Yield/% | Z:E | ee/% |
| 1 |  |
64 | 95:5 | 93 |
| 2 |  |
58 | 98:2 | 75 |
| 3 |  |
55 | 76:24 | >98 |
| 4 |  |
56 | 15:85 | 94 |
| 5 |  |
40 | 70:30 | 95 |
2014年, Grubbs课题组[39]继续研究了环丁烯衍生物与末端烯烃在催化剂Ru-2作用下发生AROCM反应, 结果表明, 反应可以选择性得到Z-构型二烯烃产物.由于环丁烯衍生物的四元环具有较大的环张力和较弱的空间位阻, 环丁烯衍生物可以与各种烷基烯烃发生AROCM, 而降冰片烯只可以与甲基烯烃发生反应.此类反应可以制备各种反式1, 2-二醇类化合物(表 23).
下载:
导出CSV
 |
|||||
| Entry | R | R' | Yield/% | Z:E | ee/% of Z (E) |
| 1 | H | Bz | 67 | 75:25 | 91 (67) |
| 2 | Bz | H | 69 | 75:25 | 96 (82) |
| 3 | Bn | Ac | 79 | 85:15 | 95 |
| 4 | Bn | CH2C(O)Me | 65 | 90:10 | 92 (84) |
从文献报道看来, 在ACM领域的研究进展一直比ARCM和AROCM慢得多.根据反应的过程和机理来推断, 不对称CM可能是烯烃不对称复分解转化中难度最大的, 催化剂不仅要控制反应的立体化学, 还要控制过程中烯烃的对映体面方向选择性. 2006年Grubbs课题组[40]报道第一个ACM例子(表 24), 在催化剂Ru-37的催化下, cis-1, 4-二乙酰基丁烯与几种对称二烯烃化合物发生CM, 产物的收率和立体选择性都不高(<54% yield, <52% ee).
下载:
导出CSV
 |
||||
| Entry | Substrate | Product | ee/% | Yield/% |
| 1 |  |
 |
52 | 54 |
| 2 |  |
 |
40 | 17 |
| 3 |  |
 |
37 | 48 |
| 4 |  |
 |
4 | 23 |
2014年, Grubbs课题组[37]再次尝试了催化剂Ru-2在cis-1, 4-二乙酰基丁烯与对称二烯烃化合物发生CM时的立体选择性, 研究结果发现立体选择性仍然不理想(Scheme 15).
近年来, 研究者发现了许多烯烃复分解反应的催化剂, 用于合成特定立体构型烯烃.这种立体选择性的合成方法在复杂天然分子的合成中得到了广泛的应用[41-45].随着应用范围的扩展, 又推动了新型催化剂的发展.
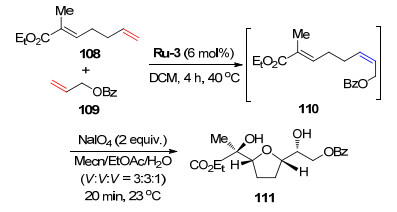
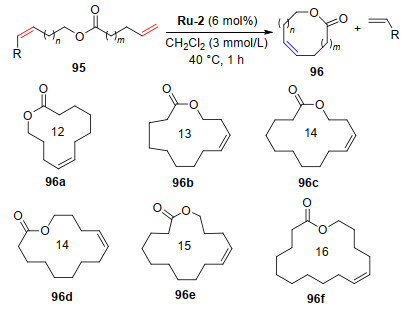
大环类化合物是天然产物中重要的一类物质, 这类分子在医药和香料行业中起着举足轻重的作用, 麝香酮就是一个典型的例子. 2013年, Grubbs课题组[4]使用催化剂Ru-2巧妙地合成了一系列目前被广泛应用于香水制备行业的麝香酮类似物(Scheme 16).
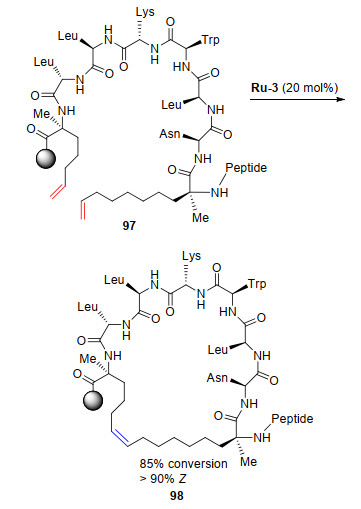
烯烃复分解反应在生物学领域的应用是一个新兴领域, 通过复分解反应可以将氨基酸与侧链烯烃结合到肽和蛋白质中, 但是控制产物氨基酸中烯烃几何构型是一个关键难点[47-51]. 2014年, Grubbs课题组[52]研究了催化剂Ru-2和Ru-3在多种肽的烯烃复分解反应中构型选择性.研究发现在这类钌催化的复分解反应中得到了较好的产率和很高的Z-构型产物. 2015年[53], Grubbs课题组使用催化剂Ru-3催化的RCM巧妙地合成了“钉合”肽(Scheme 17), 获得了较高的转化率和优异的Z-构型产物选择性.
2014年, Grubbs课题组[39]借助不对称开环复分解反应, 使用Ru-8催化合成了1, 2-反二醇.随后他们利用催化剂Ru-8采用两步反应简便地合成了南方松甲虫雄性诱导信息素[54]中的一个主要成分(Scheme 18).
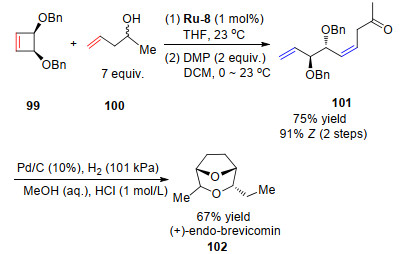
近年来, 钌催化的立体选择性烯烃复分解反应在有机合成反应中的应用越来越广泛.自从日本Shin-Etsu化学制品公司研制开发了合成信息素灭虫法, 信息素代替农药作为第二代杀虫剂受到了很多关注[55-57].
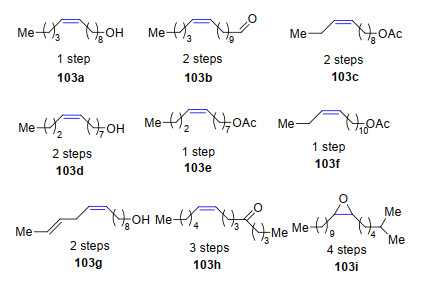
2013年, Grubbs课题组[58]利用烯烃不对称复分解反应合成了9种信息素(103a~103i)[59], 这些信息素结构中的Z-构型双键就是依靠催化剂Ru-1的立体选择性催化来实现的(Scheme 19).
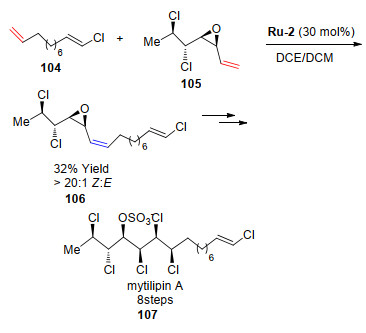
Mytilipin A是从亚得里亚海贻贝中分离出的氯磺基脂质家族的成员[60-62], 其结构引起了合成领域的广泛关注. 2013年, Vanderwal课题组[63]利用催化剂Ru-2实现了Mytilipin A关键中间体(106)的合成, 仅仅需要8步反应即可高对映选择性地得到Mytilipin A, 收率也高于早期报道的方法(Scheme 20).
2015年, Grubbs课题组[64]研究了烯烃复分解与双键氧化的串联反应.首先使用催化剂Ru-3催化烯烃复分解反应, 立体选择性地得到某一构型的烯烃, 然后继续发生双键的氧化反应, 立体选择性地得到反式二醇产物.诸多天然产物中含有四氢呋喃反式二醇类化合物. 2016年, Grubbs课题组[65]利用烯烃复分解-环氧化反应串联, 巧妙地合成了2, 5-二取代四氢呋喃反式二醇(Scheme 21).
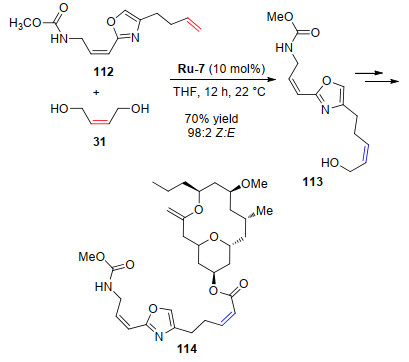
(+)-Neopeltolide是一种大环海洋天然产物[66-67], 具有抗肿瘤细胞系增殖活性, 其制备方法引起了人们的极大兴趣[68-74]. 2015年, Hoveyda课题组[75]报道了这个化合物迄今为止最短的合成路线, 文献报道的方法最长包括28个步骤, 而该方法只需要11步.他们采用四种不同的Z-选择性烯烃复分解反应步骤来构建这个分子, 其中一步采用钌催化剂Ru-7, 该步骤得到了高选择性Z-构型的醇(113), 进一步反应得到了(+)-Neopeltolide (Scheme 22).
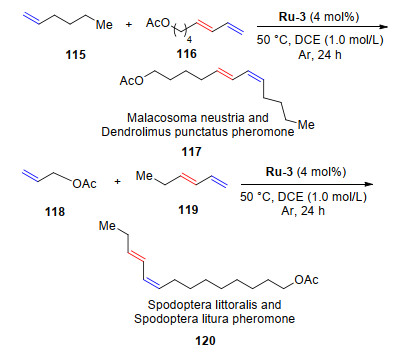
2016年, Grubbs课题组[9]报道了末端烯烃与3E-1, 3-二烯烃在催化剂Ru-3的催化下, 可以高选择性地得到Z, E-共轭的二烯烃类化合物.利用此性质, 该课题组使用经济、安全、温和的反应条件, 采用两步反应完成了原本需要六步才能实现的毛虫(天幕毛虫和马尾松毛虫)生物信息素和夜蛾(灰翅夜蛾和斜纹夜蛾)生物信息素的合成(Scheme 23).
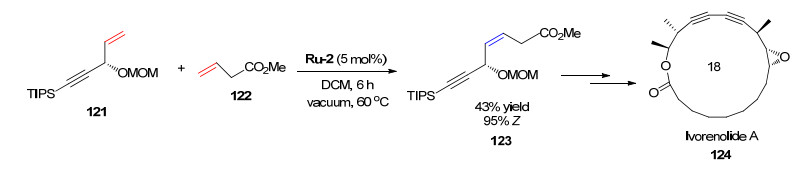
Z-选择性烯烃复分解反应在大环内酯类化合物的合成中也表现出明显的优势[76-77]. Ivorenolide A是一种从Khaya ivorensis的树皮中分离出来的免疫抑制剂. 2016年, Collins课题组[76]在Ivorenolide A的全合成工作中, 利用烯烃复分解反应实现了关键中间体123的合成, 并最终以14步反应简便地合成了Ivorenolide A (Scheme 24).
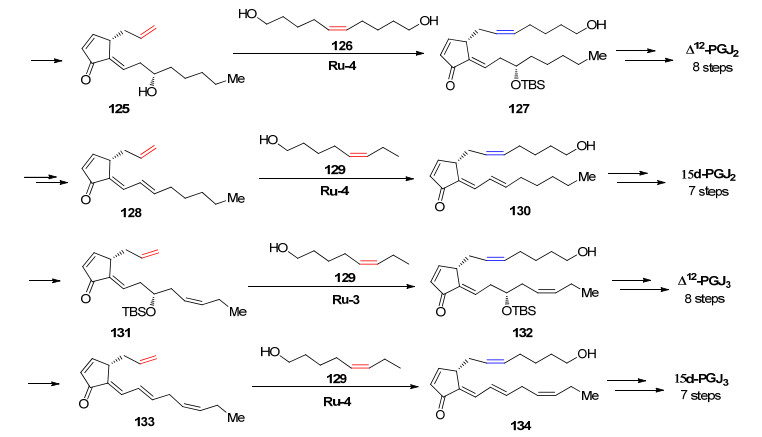
前列腺素是一种存在于哺乳动物组织中的天然分子, 这些化合物表现出广泛的生物学功能和医学应用价值.各种前列腺素的合成对前列腺癌的治疗有着深远影响.最近的研究发现Δ12-前列腺素J家族的四个化合物(Scheme 25)分子中具有独特的共轭二烯酮结构, 并表现出抗癌活性. 2019年, Grubbs课题组[78]利用Ru-4催化的烯烃复分解反应立体选择性地合成了这四个化合物, 此路线非常简便, 最长路线只有7~8步.
烯烃复分解反应为碳碳双键的构建特别是特定构型烯烃的合成提供了新方法.在过去十年里, 人们对烯烃络合钌杂环丁烷在立体选择性烯烃复分解反应中的作用进行了深入广泛的探索, 并取得了较大进展.一系列高活性和高选择性的复分解反应催化剂被成功开发, 并将其应用到药物发现和材料制备的过程中, 不但极大地优化了合成路线, 大大提高了生产效率, 而且制备工艺安全、简便、经济、环保.尽管如此, 该领域仍存在较多值得深入研究的问题, 在立体选择性合成手性分子方面仍存在很大的扩展空间.随着人们对烯烃复分解反应研究的不断深入, 更多高活性、高立体选择性的钌催化剂将继续被开发出来, 并被应用到医药、食品、化工和生物技术产业等相关领域.
Jean-Louis, H. P.; Chauvin, Y. Makromol. Chem. 1971, 141, 161. doi: 10.1002/macp.1971.021410112
Keitz, B. K.; Endo, K.; Patel, P. R.; Herbert, M. B.; Grubbs, R. H. J. Am. Chem. Soc. 2012, 134, 693. doi: 10.1021/ja210225e
Marx, V. M.; Herbert, M. B.; Keitz, B. K.; Grubbs, R. H. J. Am. Chem. Soc. 2013, 135, 94. doi: 10.1021/ja311241q
Rosebrugh, L. E.; Herbert, M. B.; Marx, V. M.; Keitz, B. K.; Grubbs, R. H. J. Am. Chem. Soc. 2013, 135, 1276. doi: 10.1021/ja311916m
Ahmed, T. S.; Grubbs, R. H. Angew. Chem.. Int. Ed. 2017, 56, 11213. doi: 10.1002/anie.201704670
Khan, R. K. M.; Zhugralin, A. R.; Torker, S.; O'Brien, R. V.; Lombardi, P. J.; Hoveyda, A. H. J. Am. Chem. Soc. 2012, 134, 12438. doi: 10.1021/ja3056722
Khan, R. K. M.; O'Brien, R. V.; Torker, S.; Li, B.; Hoveyda, A. H. J. Am. Chem. Soc. 2012, 134, 12774. doi: 10.1021/ja304827a
Hartung, J.; Grubbs, R. H. J. Am. Chem. Soc. 2013, 135, 10183. doi: 10.1021/ja4046422
Luo, S. X.; Cannon, J. S.; Taylor, B. L. H.; Engle, K. M.; Houk, K. N.; Grubbs, R. H. J. Am. Chem. Soc. 2016, 138, 14039. doi: 10.1021/jacs.6b08387
Keitz, B. K.; Endo, K.; Herbert, M. B.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 9686. doi: 10.1021/ja203488e
Endo, K.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8525. doi: 10.1021/ja202818v
Bronner, S. M.; Herbert, M. B.; Patel, P. R.; Marx, V. M.; Grubbs, R. H. Chem. Sci. 2014, 5, 4091. doi: 10.1039/C4SC01541J
Cannon, J. S.; Grubbs, R. H. Angew. Chem.. Int. Ed. 2013, 52, 9001. doi: 10.1002/anie.201302724
Quigley, B. L.; Grubbs, R. H. Chem. Sci. 2014, 5, 501. doi: 10.1039/C3SC52806E
Herbert, M. B.; Grubbs, R. H. Angew. Chem.. Int. Ed. 2015, 54, 2. doi: 10.1002/anie.201410932
Koh, M. J.; Khan, R. K. M.; Torker, S.; Yu, M.; Mikus, M. S.; Hoveyda, A. H. Nature 2015, 517, 181. doi: 10.1038/nature14061
Tiede, S.; Berger, A.; Schlesiger, D.; Rost, D.; Lühl, A.; Blechert, S. Angew. Chem.. Int. Ed. 2010, 49, 3972. doi: 10.1002/anie.201000940
Miyazaki, H.; Herbert, M. B.; Liu, P.; Dong, X.; Xu, X.; Keitz, B. K.; Ung, T.; Mkrtumyan, G.; Houk, K. N.; Grubbs, R. H. J. Am. Chem. Soc. 2013, 135, 5848. doi: 10.1021/ja4010267
Johns, A. M.; Ahmed, T. S.; Jackson, B. W.; Grubbs, R. H.; Pederson, R. L. Org. Lett. 2016, 18, 772. doi: 10.1021/acs.orglett.6b00031
Montgomery, T. P.; Ahmed, T. S.; Grubbs, R. H. Angew. Chem.. Int. Ed. 2017, 56, 11024. doi: 10.1002/anie.201704686
Khan, R. K. M.; Torker, S.; Hoveyda, A. H. J. Am. Chem. Soc. 2013, 135, 10258. doi: 10.1021/ja404208a
Ahmed, T. S.; Grubbs, R. H. J. Am. Chem. Soc. 2017, 139, 1532. doi: 10.1021/jacs.6b11330
Ahmed, T. S.; Grubbs, R. H. J. Am. Chem. Soc. 2017, 139, 1532. doi: 10.1021/jacs.6b11330
Xu, C. F.; Shen, X.; Hoveyda, A. H. J. Am. Chem. Soc. 2017, 139, 10919. doi: 10.1021/jacs.7b06552
Xu, C. F.; Liu, Z. X.; Torker, S.; Shen, X.; Xu, D. M.; Hoveyda, A. H. J. Am. Chem. Soc. 2017, 139, 15640. doi: 10.1021/jacs.7b10010
Ahmed, T. S.; Montgomery, T. P.; Grubbs, R. H. Chem. Sci. 2018, 9, 3580. doi: 10.1039/C8SC00435H
Torker, S.; Khan, R. K. M.; Hoveyda, A. H. J. Am. Chem. Soc. 2014, 136, 3439. doi: 10.1021/ja410606b
Khan, R. K. M.; Torker, S.; Hoveyda, A. H. J. Am. Chem. Soc. 2014, 136, 14337. doi: 10.1021/ja505961z
Mikus, M. S.; Torker, S.; Xu, C.; Li, B.; Hoveyda, A. H. Organometallics 2016, 35, 3878. doi: 10.1021/acs.organomet.6b00773
Montgomery, T. P.; Grandner, J. M.; Houk, K. N.; Grubbs, R. H. Organometallics 2017, 36, 3940. doi: 10.1021/acs.organomet.7b00555
Dumas, A.; Müller, D. S.; Curbet, I.; Toupet, L.; Rouen, M.; Baslé, O.; Mauduit, M. Organometallics 2018, 37, 829. doi: 10.1021/acs.organomet.7b00836
Seiders, T. J.; Ward, D. W.; Grubbs, R. H. Org. Lett. 2001, 3, 3225. doi: 10.1021/ol0165692
Stenne, B.; Timperio, J.; Savoie, J.; Dudding, T.; Collins, S. K. Org. Lett. 2010, 12, 2032. doi: 10.1021/ol100511d
Gawin, R.; Pieczykolan, M.; Malinska, M.; Wozniak, K.; Grela, K. Synlett 2013, 24, 1250. doi: 10.1055/s-0033-1338877
Paradiso, V.; Bertolasi, V.; Grisi, F. Organometallics 2014, 33, 5932. doi: 10.1021/om500731k
Paradiso, V.; Bertolasi, V.; Costabile, C.; Grisi, F. Dalton Trans. 2016, 45, 561. doi: 10.1039/C5DT03758A
Hartung, J.; Dornan, P. K.; Grubbs, R. H. J. Am. Chem. Soc. 2014, 136, 13029. doi: 10.1021/ja506611k
Kannenberg, A.; Rost, D.; Eibauer, S.; Tiede, S.; Blechert, S. Angew. Chem.. Int. Ed. 2011, 50, 3299. doi: 10.1002/anie.201007673
Hartung, J.; Grubbs, R. H. Angew. Chem.. Int. Ed. 2014, 53, 3885. doi: 10.1002/anie.201310767
Berlin, J. M.; Goldberg, S. D.; Grubbs, R. H. Angew. Chem.. Int. Ed. 2006, 45, 7591. doi: 10.1002/anie.200602469
Werrel, S.; Walker, J. C. L.; Donohoe, T. J. Tetrahedron Lett. 2015, 56, 5261. doi: 10.1016/j.tetlet.2015.07.008
Montgomery, T. P.; Johns, A. M.; Grubbs, R. H. Catalysts 2017, 7, 87. doi: 10.3390/catal7030087
Ogba, O. M.; Warner, N. C.; O'Leary, D. J.; Grubbs, R. H. Chem. Soc. Rev. 2018, 47, 4510. doi: 10.1039/C8CS00027A
Potukuchi, H. K.; Colomer, I.; Donohoe, T. J. Adv. Heterocycl. Chem. 2016, 120, 43. doi: 10.1016/bs.aihch.2016.04.006
Hoveyda, A. H.; Malcolmson, S. J.; Meek, S. J.; Zhugralin, A. R. Angew. Chem.. Int. Ed. 2010, 49, 34. doi: 10.1002/anie.200904491
Donohoe, T. J.; Jones, C. R.; Barbosa, L. C. A. J. Am. Chem. Soc. 2011, 133, 16418. doi: 10.1021/ja207835w
Song, W.; Wang, Y.; Qu, J.; Lin, Q. J. Am. Chem. Soc. 2008, 130, 9654. doi: 10.1021/ja803598e
Van Hest, J. C. M.; Kiick, K. L.; Tirrell, D. A. J. Am. Chem. Soc. 2000, 122, 1282. doi: 10.1021/ja992749j
Bernardes, G. J. L.; Chalker, J. M.; Errey, J. C.; Davis, B. G. J. Am. Chem. Soc. 2008, 130, 5052. doi: 10.1021/ja800800p
Lin, Y. A.; Boutureira, O.; Lercher, L.; Bhushan, B.; Paton, R. S.; Davis, B. G. J. Am. Chem. Soc. 2013, 135, 12156. doi: 10.1021/ja403191g
Zhu, Y.; van der Donk, W. A. Org. Lett. 2001, 3, 1189. doi: 10.1021/ol015648a
Mangold, S. L.; O'Leary, D. J.; Grubbs, R. H. J. Am. Chem. Soc. 2014, 136, 12469. doi: 10.1021/ja507166g
Mangold, S. L.; Grubbs, R. H. Chem. Sci. 2015, 6, 4561. doi: 10.1039/C5SC01507C
Silverstein, R. M.; Brownlee, R. G.; Bellas, T. E.; Wood, D. L.; Browne, L. E. Science 1968, 159, 889. doi: 10.1126/science.159.3817.889
Henrick, C. A. Tetrahedron 1977, 33, 1845. doi: 10.1016/0040-4020(77)80372-4
Ortiz, A.; Quesada, A.; Sanchez, A. J. Chem. Ecol. 2004, 30, 991. doi: 10.1023/B:JOEC.0000028463.43564.40
Howse, P. E.; Stevens, I. D. R.; Jones, O. T. Insect Pheromones and their Use in Pest Management, Springer, Dordrecht, Netherlands, 1998.
Herbert, M. B.; Marx, V. M.; Pederson, R. L.; Grubbs, R. H. Angew. Chem.. Int. Ed. 2013, 52, 310. doi: 10.1002/anie.201206079
Ando, T.; Inomata, S.; Yamamoto, M. In Topics in Current Chemistry, Ed.: Schulz, S., Springer, Berlin/Heidelberg, 2004, pp. 51~96.
Ciminiello, P.; Fattorusso, E.; Forino, M.; Di Rosa, M.; Ianaro, A.; Poletti, R. J. Org. Chem. 2001, 66, 578. doi: 10.1021/jo001437s
Ciminiello, P.; Dell'Aversano, C.; Fattorusso, E.; Forino, M.; Magno, S.; Di Rosa, M.; Ianaro, A.; Poletti, R. J. Am. Chem. Soc. 2002, 124, 13114. doi: 10.1021/ja0207347
Ciminiello, P.; Dell'Aversano, C.; Fattorusso, E.; Forino, M.; Magno, S.; Di Meglio, P.; Ianaro, A.; Poletti, R. Tetrahedron 2004, 60, 7093. doi: 10.1016/j.tet.2003.12.072
Chung, W.; Carlson, J. S.; Bedke, D. K.; Vanderwal, C. D. Angew. Chem.. Int. Ed. 2013, 52, 10052. doi: 10.1002/anie.201304565
Dornan, P. K.; Wickens, Z. K.; Grubbs, R. H. Angew. Chem.. Int. Ed. 2015, 54, 7134. doi: 10.1002/anie.201501505
Dornan, P. K.; Lee, D.; Grubbs, R. H. J. Am. Chem. Soc. 2016, 138, 6372. doi: 10.1021/jacs.6b02653
Yu, M.; Schrock, R. R.; Hoveyda, A. H. Angew. Chem.. Int. Ed. 2014, 53, 1. doi: 10.1002/anie.201310509
Wright, A. E.; Botelho, J. C.; Guzmán, E.; Harmody, D.; Linley, P.; McCarthy, P. J.; Pitts, T. P.; Pomponi, S. A.; Reed, J. K. J. Nat. Prod. 2007, 70, 412. doi: 10.1021/np060597h
Youngsaye, W.; Lowe, J. T.; Pohlki, F.; Ralifo, P.; Panek, J. S. Angew. Chem.. Int. Ed. 2007, 46, 9211. doi: 10.1002/anie.200704122
Custar, D. W.; Zabawa, T. P.; Scheidt, K. A. J. Am. Chem. Soc. 2008, 130, 804. doi: 10.1021/ja710080q
Woo, S. K.; Kwon, M. S.; Lee, E. Angew. Chem.. Int. Ed. 2008, 47, 3242. doi: 10.1002/anie.200800386
Fuwa, H.; Naito, S.; Goto, T.; Sasaki, M. Angew. Chem.. Int. Ed. 2008, 47, 4737. doi: 10.1002/anie.200801399
Paterson, I.; Miller, N. A. Chem. Commun. 2008, 4708.
Guinchard, X.; Roulland, E. Org. Lett. 2009, 11, 4700. doi: 10.1021/ol902047z
Fuwa, H.; Saito, A.; Sasaki, M. A. Angew. Chem.. Int. Ed. 2010, 49, 3041. doi: 10.1002/anie.201000624
Yu, M.; Schrock, R. R.; Hoveyda, A. H. Angew. Chem.. Int. Ed. 2015, 54, 215. doi: 10.1002/anie.201409120
De Léséleuc, M.; Godin, É.; Parisien-Collette, S.; Lévesque, A.; Collins, S. K. J. Org. Chem. 2016, 81, 6750. doi: 10.1021/acs.joc.6b01500
Zhang, B.; Wang, Y.; Yang, S. P.; Zhou, Y.; Wu, W. B.; Tang, W.; Zuo, J. P.; Li, Y.; Yue, J. M. I. J. Am. Chem. Soc. 2012, 134, 20605. doi: 10.1021/ja310482z
Li, J. M.; Ahmed, T. S.; Xu, C.; Stoltz, B. M.; Grubbs, R. H. J. Am. Chem. Soc. 2019, 141, 154. doi: 10.1021/jacs.8b12816

图式 2 钌催化的烯烃复分解反应立体选择性机理
Scheme 2 Stereoselectivity mechanism of ruthenium-catalyzed olefin metathesis

图式 4 双环氧杂环烯烃与烯醇醚或苯基乙烯基硫醚发生的ROCM
Scheme 4 ROCM of oxabicyclic alkenes and enol ethers or phenyl vinyl sulfide

图式 10 顺式1, 4-二乙酰基-2-丁烯与顺式-4-辛烯发生的CM
Scheme 10 Cross metathesis of cis-1, 4-diacetoxy-2-butene with cis-4-octene

图式 11 顺-1, 4-二乙酰基-2-丁烯和顺-4-辛烯发生的CM
Scheme 11 CM of cis-1, 4-diacetoxy-2-butene with cis-4-octene

图式 12 利用构型保持的烯烃复分解反应合成Z或E烯丙基醇
Scheme 12 Synthesis of Z- or E-allylic alcohols by stereoretentive olefin metathesis

图式 14 手性NHC配体催化不对称RCM中立体选择性
Scheme 14 Stereoselectivity of unsymmetrical RCM catalyzed by chiral NHC ligands

图 2 含1, 2, 4-三唑为骨架的钌催化剂
Figure 2 Various cyclometalated catalyst with 1, 2, 3-triazole skeleton

图式 18 利用ROCM合成昆虫信息素(+)-endo-brevicomin
Scheme 18 Synthesis of (+)-endo-brevicomin employing RO- CM

图式 19 Grubbs课题组利用烯烃复分解反应合成的9种昆虫信息素
Scheme 19 Nine insect pheromones synthesized by Grubbs and coworkers employing OM Bonds formed by OM are highlighted

图式 20 借助Z-选择性CM构建mytilipin A全合成的关键中间体
Scheme 20 Synthesis of key intermediates of mytilipin A employing Z-selective CM

图式 21 利用Z-选择性复分解-环氧化反应串联合成反式二醇类化合物
Scheme 21 Synthesis of trans-diols employing Z-selective CM/oxidative reaction

图式 22 利用钌催化的Z-选择性CM构建(+)-neopeltolide全合成的关键中间体
Scheme 22 Employment of ruthenium-catalyzed Z-selective CM to join fragments of (+)-neopeltolide

图式 23 通过Z-选择性CM合成昆虫信息素
Scheme 23 Synthesis of insect pheromones via Z-selective cross-metathesis

图式 24 利用Z-选择性CM合成Ivorenalide A
Scheme 24 Formal synthesis of ivorenalide A utilizing Z-selective CM

图式 25 借助构型保持的CM简便地合成Δ12-前列腺素J家族的四个化合物
Scheme 25 Concise syntheses of Δ12-prostaglandin J natural products employing stereoretentive CM
表 1 催化剂Ru-2和Ru-3在大环RCM中的立体选择性
Table 1. Stereoselectivity of macrocyclic ring-closing metathesis employing Ru-2 and Ru-3
|
|
 下载: 导出CSV
下载: 导出CSV
表 3 降冰片烯二醇与末端烯烃发生的ROCM
Table 3. ROCM of norbornene diols with terminal olefins
|
||||
| Entry | R | Ru-6/mol% | Yield/% | Z/% |
| 1 | Ph | 1 | 92 | 97 |
| 2 | m-FC6H4 | 1 | 93 | 96 |
| 3 | (CH2)2OTBS | 5 | 68 | >98 |
| 4 | (CH2)2C(O)NHPh | 5 | 65 | >98 |
| 5 | (CH2)7Me | 5 | 58 | >98 |
| 6 | |
5 | 93 | 93 |
| 7 | |
5 | 97 | >98 |
| 8 | |
2 | 84 | 91 |
| 9 | |
5 | 80 | >98 |
| 10 | OnBu | 2 | 95 | >98 |
| 11 | SEt | 5 | 80 | 92 |
下载: 导出CSV
表 4 Ru-8催化的末端烯烃自身CM
Table 4. CM of terminal olefins catalyzed using Ru-8
|
||||
| Entry | R | Ru-8/mol% | Yield/% | Z/% |
| 1 | CH2Ph | 0.10 | >95 | >95 |
| 2 | CH2Ph | 0.01 | 74 | >95 |
| 3 | (CH2)3OH | 0.10 | >95 | >95 |
| 4 | (CH2)8CO2Me | 0.10 | 77 | 95 |
下载: 导出CSV
表 5 钌催化剂对交叉烯烃复分解反应的效果比较
Table 5. Comparison of CM using ruthenium catalysts
|
||||||
| Entry | R | R1 | R2 | Cat. (mol%) | Yield/% | Z/% |
| 1 | CH2Ph | CH2OAc | CH2OAc | Ru-2 (1.0) | 58 | 91 |
| 2 | (CH2)Me | H | H | Ru-2 (0.5) | 70 | 91 |
| 3 | (CH2)3Me | (CH2)7OAc | H | Ru-2 (0.5) | 67 | 91 |
| 4 | (CH2)Me | H | H | Ru-3 (0.5) | 71 | >95 |
| 5 | (CH2)3Me | (CH2)7OAc | H | Ru-3 (0.1) | 60 | >95 |
下载: 导出CSV
表 6 Ru-3催化的末端烯烃与非共轭二烯烃CM
Table 6. CM of terminal olefins and nonconjugated diolefins using Ru-3
|
|||
| Entry | R | Yield/% | Z/% |
| 1 | CH2Ph | 63 | > 95 |
| 2 | (CH2)8CHO | 70 | > 95 |
| 3 | (CH2)7CO2Me | 82 | > 95 |
| 4 | CH2NHPh | 68 | > 95 |
| 5 | CH2OCO2Me | 79 | > 95 |
下载: 导出CSV
表 8 顺-2-丁烯-1, 4-二醇与端烯发生的CM
Table 8. CM scope of cis-2-butene-1, 4-diol with terminal olefins
|
||||
| Entry | R | Time/h | Yield/% | Z/% |
| 1 | (CH2)9Me | 4 | 72 | 96 |
| 2 | (CH2)2OTBS | 4 | 65 | 93 |
| 3 | (CH2)2OPNP | 4 | 74 | 96 |
| 4 | (CH2)2OnBu | 12 | 57 | 91 |
| 5 | (CH2)2CO2Bn | 4 | 80 | 98 |
| 6 | (CH2)4Phth | 4 | 64 | 98 |
| 7 | (CH2)8CHO | 4 | 80 | 94 |
| 8 | (CH2)3CO2H | 4 | 70 | 96 |
| 9 | Cy | 4 | 59 | 98 |
| 10 | |
4 | 73 | 98 |
| 11 | |
8 | 66 | 95 |
| 12 | |
8 | 63 | 92 |
| 13 | |
8 | 56 | 96 |
| 14 | |
8 | 54 | 87 |
下载: 导出CSV
表 9 降冰片烯衍生物与苯乙烯发生的ROCM
Table 9. ROCM of various norbornene substrates with styrene
|
||||
| Entry | Substrate | E:Z | Coversion/% | ee/% |
| 1 | |
24:1 13:1 |
>98 >98 |
82 90 |
| 2 | |
21:1 23:1 |
>98 >98 |
82 90 |
| 3 | |
19:1 | 61 | 86 |
| 4 | |
>30:1 | >98 | 76 |
| 5 | |
21:1 | >98 | 70 |
下载: 导出CSV
表 10 E-异构体富集的大环烯烃复分解反应
Table 10. Reaction of E-isomer enriched macrocycles ethenolysis
|
|||
| Entry | Substrate | Initial E/% | Final E/% |
| 1 | 19 | 55 | > 95 |
| 2 | 20 | 80 | > 95 |
| 3 | 21 | 80 | > 95 |
下载: 导出CSV
表 11 E-异构体富集的链状烯烃复分解反应
Table 11. Reaction of E-isomer enriched chain olefin
|
||||
| Entry | R | n | Initial E/% | Final E/% |
| 1 | Me | 3 | 52 | 90 |
| 2 | OAc | 7 | 78 | > 95 |
| 3 | OH | 4 | 68 | 90 |
| 4 | CO2Me | 6 | 80 | > 95 |
| 5 | NHPh | 3 | 80 | > 95 |
| 6 | C(O)Me | 2 | 72 | > 95 |
下载: 导出CSV
表 12 1-癸烯与反式-4-辛烯的交叉复分解反应
Table 12. CM of 1-decene and trans-4-octene
|
||||
| Entry | Ru-X | Conv./% | Yield/% of 46 | E/Z |
| 1 | Ru-4 | 92 | 4 | 88/12 |
| 2 | Ru-9 | 37 | 7 | > 99/1 |
| 3 | Ru-10 | 53 | 29 | > 99/1 |
| 4 | Ru-11 | 55 | 31 | > 99/1 |
下载: 导出CSV
表 13 油酸甲酯发生的自身复分解反应
Table 13. Self-metathesis of methyl oleate
|
||||
| Entry | Ru (mol%) | 47 | 48/% (Z/E) | 49/% (Z/E) |
| 1 | Ru-4 (0.01) | >99% Z | 24 (>99/1) | 24 (>99/1) |
| 2 | Ru-9 (0.50) | >99% Z | 10 (>99/1) | 10 (>99/1) |
| 3 | Ru-9 (7.50) | >97% E | 24 (4/96) | 24 (4/96) |
下载: 导出CSV
表 14 钌催化的自身复分解反应
Table 14. Self-metathesis catalyzed by ruthenium catalysts
|
||||
| Entry | 50 | Ru (mol%) | 51/% (E/Z) | 52/% (E/Z) |
| 1 | E-50 | Ru-12 (1.00) | 25 (>99/1) | 25 (>99/1) |
| 2 | E-50 | Ru-13 (1.00) | 6 (>99/1) | 6 (>99/1) |
| 3 | Z-50 | Ru-12 (0.10) | 25 (<1/99) | 25 (<1/99) |
| 4 | Z-50 | Ru-13 (0.05) | 25 (<1/99) | 25 (<1/99) |
下载: 导出CSV
表 15 烯烃复分解反应合成大环化合物
Table 15. Synthesis of macrocyclic lacones employing olefin metathesis
|
|
下载: 导出CSV
表 16 利用手性NHC配体催化的不对称RCM
Table 16. RCM catalyzed by chiral NHC ligands
|
|||
| Entry | Cat. | Conversion/% | ee/% |
| 1 | Ru-20 | 95 | 8 |
| 2 | Ru-21 | 95 | 38 |
| 3 | Ru-22 | 90 | 52 |
| 4 | Ru-23 | 86 | 50 |
下载: 导出CSV
表 17 1, 2, 4-三唑为骨架的钌催化剂在不对称RCM中的立体选择性
Table 17. Stereoselectivity of catalysts with 1, 2, 4-azole skeleton in ARCM
|
|||
| Entry | Catalyst | Conv./% | ee/% |
| 1 | Ru-26 | >95 | 26 |
| 2 | Ru-27 | 64 | 4 |
| 3 | Ru-28 | 59 | 9 |
| 4 | Ru-29 | 85 | 8 |
下载: 导出CSV
表 18 不同取代基团的NHC对RCM立体选择性的影响
Table 18. Effects of the N-alkyl groups on the stereoselectivity of RCM
|
|||||
| Entry | Ru-X (mol%) | Solvent | Additive | Conversion/% | ee/% |
| 1 | Ru-30 (2.5) | DCM | — | >98 | 18 |
| 2 | Ru-30 (4.0) | THF | NaI | >95 | 53 |
| 3 | Ru-31 (2.5) | DCM | — | >98 | 33 |
| 4 | Ru-31 (4.0) | THF | NaI | >98 | 50 |
| 5 | Ru-32 (2.5) | DCM | — | >98 | 19 |
| 6 | Ru-32 (4.0) | THF | NaI | >95 | 52 |
| 7 | Ru-33 (2.5) | DCM | — | >98 | 33 |
| 8 | Ru-33 (4.0) | THF | NaI | >98 | 47 |
下载: 导出CSV
表 19 含有硅醚或酰胺的末端三烯烃发生的RCM的立体选择性
Table 19. Stereoselectivity of terminal trienes containing silyl ethers or amides in RCM
|
||||
| Entry | 68 | 69 | Yield/% | ee/% |
| 1 | |
|
65 | 69 |
| 2 | |
|
29 | 67 |
| 3 | |
|
95 | 54 |
| 4 | |
|
72 | 47 |
| 5 | |
|
90 | 57 |
下载: 导出CSV
表 20 含喹啉骨架的钌催化剂在烯烃开环复分解反应中的立体选择性
Table 20. Stereoselectivity of catalyst with Quinoline skeleton in ROCM
| Substrate | Product | Ru-X | Conv./% (E:Z) | ee/% E (Z) |
|
|
|
>99 (1:2) | 79 (87) |
|
|
|
>99 (2:1) | 82 (82) |
|
|
Ru-34 Ru-35 |
96 (2:1) >99 (2:1) |
98 (92) 95 (91) |
下载: 导出CSV
表 21 钌催化剂Ru-36催化的开环复分解反应
Table 21. ROCM employing Ru-36
|
|||||
| Entry | Product | R | Yield/% | Z:E | ee/% |
| 1 | 78a | OnBu | 80 | 95:5 | 96 |
| 2 | 78b | OCy | 64 | 98:2 | 96 |
| 3 | 78c | OPMP | 67 | 95:5 | 94 |
| 4 | 78d | OCH2CF3 | 65 | 94:6 | 92 |
| 5 | 78e | O(CH2)2Cl | 63 | 95:5 | 96 |
| 6 | 78f | SPh | 67 | 91:9 | 92 |
下载: 导出CSV
表 22 Ru-2催化的降冰片烯与末端烯烃的开环复分解反应
Table 22. ROCM of norbornene and terminal olefins catalyzed by Ru-2
|
||||
| Entry | Substrate | Yield/% | Z:E | ee/% |
| 1 | |
64 | 95:5 | 93 |
| 2 | |
58 | 98:2 | 75 |
| 3 | |
55 | 76:24 | >98 |
| 4 | |
56 | 15:85 | 94 |
| 5 | |
40 | 70:30 | 95 |
下载: 导出CSV
表 23 环丁烯衍生物与末端烯烃的AROCM
Table 23. AROCM of cyclobutenes and terminal olefins
|
|||||
| Entry | R | R' | Yield/% | Z:E | ee/% of Z (E) |
| 1 | H | Bz | 67 | 75:25 | 91 (67) |
| 2 | Bz | H | 69 | 75:25 | 96 (82) |
| 3 | Bn | Ac | 79 | 85:15 | 95 |
| 4 | Bn | CH2C(O)Me | 65 | 90:10 | 92 (84) |
下载: 导出CSV
表 24 cis-1, 4-二乙酰基丁烯与对称二烯烃发生的CM
Table 24. CM of cis-1, 4-diacetoxy-2-butene and symmetrical diolefins
|
||||
| Entry | Substrate | Product | ee/% | Yield/% |
| 1 | |
|
52 | 54 |
| 2 | |
|
40 | 17 |
| 3 | |
|
37 | 48 |
| 4 | |
|
4 | 23 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们














 下载:
下载: