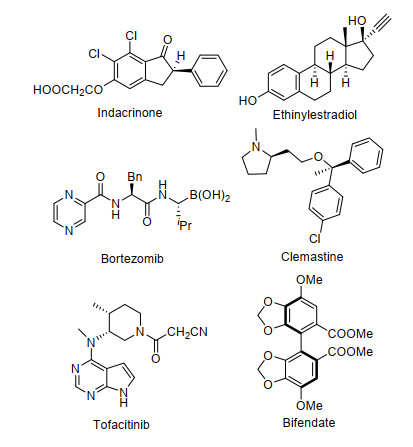
图 1.
一些含有手性结构单元的畅销和常用药物
Figure 1.
Some popular and commonly used drugs with important chiral units

手性药物[1]是指分子结构中含有手性中心或者不对称中心的药物.它包括单一的立体异构体、两个或两个以上立体异构体的混合物.由于受体、酶或离子通道具有高度的立体结构特异性, 所以手性药物的不同立体异构体与靶点的相互作用有所不同, 从而产生不同的药理学活性、代谢过程及生物毒性.包含“反应停”在内的一系列医学事件深刻表明获取光学纯药物以及对其立体异构体的研究在疾病研究和药物发展中尤为重要.因此, 1992年美国食品药品监督管理局(FDA)规定, 新的手性药物上市之前必须分别对左旋体和右旋体进行药效和毒性试验, 否则不允许上市. 2006年1月, 我国国家食品药品监督管理总局(SFDA)也出台了相应的政策法规.
在我们常用的小分子药物中, 叔(季)碳手性中心、甾体、β-芳基胺、三级醇、三级胺的结构单元是广泛存在的(图 1), 例如indacrinone是具有手性叔碳中心结构单元的降血压药物; 用于性激素调节的甾体类药物ethinylestradiol含有五个手性中心; bortezomib是典型的具有手性β-芳基胺结构单元的药物, 其可以用于治疗多发性骨髓瘤和套细胞淋巴瘤; clemastine是目前世界上公认的最好的抗组胺药之一, 其含有手性二芳基三级醇的结构; 具有连续两个手性中心和三级胺结构的药物分子tofacitinib是治疗类风湿关节炎、溃疡性结肠炎、银屑病等多种炎症的实用药物; 具有轴手性的右旋联苯双酯可用于治疗病毒性肝炎和药物性肝损伤引起转氨酶升高症状.
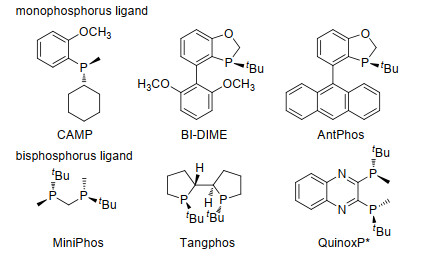
发展高对映选择性、高产率和原子经济性的催化反应对手性药物的高效合成至关重要.目前构建分子的手性中心一般有如下的一些方法:外消旋体的拆分, 使用手性试剂(当量级使用), 手性助剂参与的不对称合成、不对称催化等[2].其中不对称催化, 尤其是过渡金属催化的不对称反应具有底物普适性广、原子经济性强、环境耐受性佳、反应容易放大等特点, 决定了过渡金属催化在手性药物合成中的应用潜力.手性配体的特性, 包括电子效应、立体效应和空间效应等直接影响了催化剂的活性和立体选择性.设计发展具有新型骨架的配体和催化剂是解决过渡金属催化的不对称反应中高效性和选择性的重要途径.近年来, 手性膦配体在不对称催化领域得到了长足的发展, 并广泛应用于手性药物分子的合成中. P-手性膦配体具有显著结构特征, 表现出独特的催化特性和应用前景. 图 2列举了一些代表性的P-手性单膦和双膦配体[3].本文将综述我们研究小组在基于苯并氧杂膦烷刚性骨架的P-手性膦配体的设计和发展、P-手性膦配体促进的高效不对称反应方法学和最终应用于手性药物的高效合成方面的研究思路和实践[4].
手性配体的结构对过渡金属催化的不对称反应的活性和立体/对映选择性有至关重要的影响.苯并氧杂膦烷是具有刚性的结构单元, 基于此类骨架的配体有发展成为优势配体的潜力(图 3).当苯并氧杂膦烷中的磷原子上含有大位阻取代基(叔丁基)时, 所发展的配体往往具有稳定的P-手性.这些光学活性纯的P-手性单膦和双膦配体在过渡金属催化的不对称反应中往往表现出独特的催化特性[5].具体来讲, 单膦配体I具有以下特点(图 3): (1)配体中的苯并氧磷杂五元环的刚性结构使其具有确定的构象, 这保证了在催化反应中催化剂与底物作用时具有确定的优势面和劣势面, 从而显著提高催化反应的活性和立体/对映选择性; (2)磷原子上的供电子取代基使这类配体更加富电子, 同时具有P-手性的配体的手性因素更加靠近过渡金属中心, 因而含有这种配体的催化剂有更高的催化效率和更好的手性诱导; (3)通过改变配体中R1、R2以及下面芳环的结构可以对配体的电性及位阻进行调控, 从而优化配体的催化活性和选择性; (4)单膦配体I的联芳基结构以及叔丁基取代基增大了磷原子周围的位阻, 使得这类配体具有优良的化学稳定性, 增大了操作的简便性和实用性.
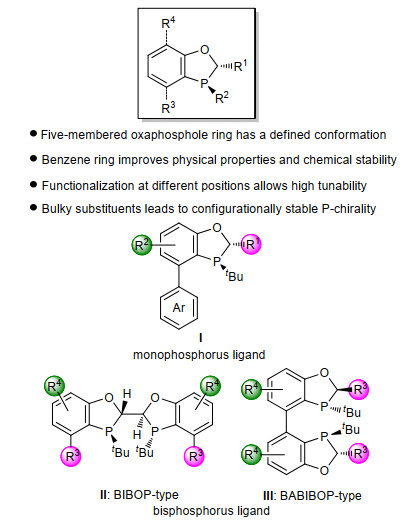
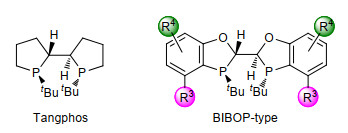
基于苯并氧磷杂五元环刚性骨架的双膦配体有两种类型:具有C2对称性的BIBOP类型配体II和具有C2对称性的联芳基BABIBOP类型配体III. BIBOP类型配体II是基于双膦配体TangPhos发展而来(图 4).和TangPhos相比, 芳环的引入使这一类配体具有更好的空气稳定性.由于能够通过拆分手段获得光学纯的配体骨架, BIBOP类型配体II的合成又更加简便.由于具有联芳基结构, BABIBOP类型配体III有着和BIBOP类型配体II截然不同的空间构型, 因而也表现出和配体II互补的催化活性.值得指出的是, 手性双膦配体II和III同样具有结构和电性可调的特点, 通过改变配体中R3或R4基团可以有效地对配体的催化活性进行优化[5].
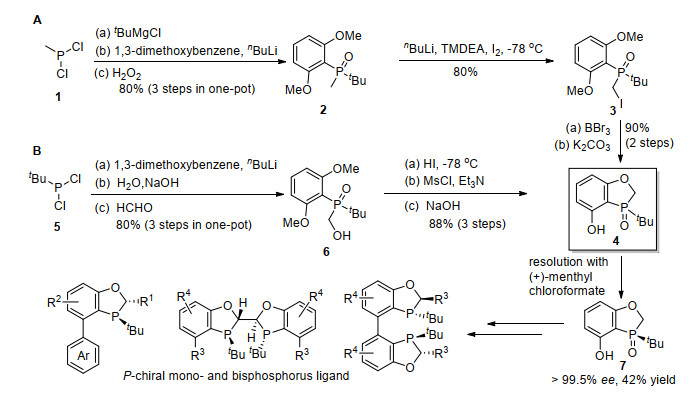
苯并氧杂膦烷的合成路线如Scheme 1所示.最初的路线以甲基二氯膦(1)为起始原料, 其与叔丁基格氏试剂反应, 然后用锂化的1, 3-二甲氧基苯和H2O2连续处理, 可以以80%的收率得到氧化膦2.在nBuLi/I2的条件下, 在2的α位上进行碘化, 以80%的收率合成化合物3.用BBr3对甲氧基进行脱保护, 然后在碱性条件下闭环, 可以以90%的收率得到关键结构单元4.整个过程料, 经过锂化的1, 3-二甲氧基苯、水和甲醛处理, 可以以80%的分离产率得到6.用HI进行甲氧基脱保护, 然后形成甲磺酸酯和闭环得到4.以手性氯甲酸薄荷酯为试剂对4进行拆分, 可以高效地得到光学纯的7, 分离产率为42%.以该手性结构单元为关键合成中间体, 合成了一系列具有P手性的单膦和双膦配体[5].
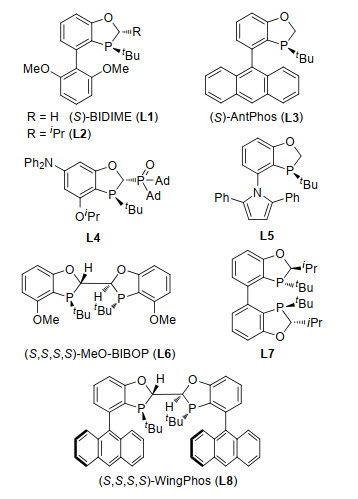
图 5中列出了常用的基于苯并氧杂膦烷结构的P-手性单膦和双膦配体, 其中单膦配体L1~L5在过渡金属钯催化的不对称Suzuki-Miyaura偶联和分子内去芳构环化反应中显示出优异的催化特性.我们利用相应的方法学实现了布洛芬、氟比洛芬、萘普生、雷公藤对醌H和包括棉酚在内的联芳基药物的合成.手性双膦配体L6~L8被应用于Rh催化的不对称氢化和加成反应中, 表现出高立体选择性和反应效率.因此实现了托法替尼、艾司西酞普兰和cipargamin的高效合成.
按照不同的反应类型阐述P-手性膦配体所表现出的独特反应效率和选择性, 以及它们在手性药物合成中的成功应用.
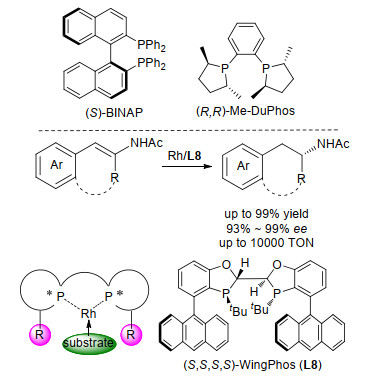
三取代烯酰胺的不对称氢化是一种制备手性氨基酸和β-芳基胺的理想的方法.虽然目前此方面的研究有许多报道, 并且获得了高对映选择性, 但是提升手性催化剂的转化数(TON), 即实现在低催化剂用量条件下的不对称氢化仍然面临巨大的挑战.为了解决这一难题, 我们设计并合成了含有深口袋的手性双膦配体Wing- Phos (L8), 期望可以通过改变金属与配体的配位模式与过渡态结构来提高催化剂的活性和稳定性(图 6).配体L8具有以下几个优势: (1)与目前常用于不对称氢化的BINAP和DuPhos等配体相比, WingPhos具有截然不同的构象, 所形成的金属催化剂也有不同的手性环境, 从而表现出与BINAP和DuPhos等配体互补的催化性能; (2)由于两个蒽基的存在, WingPhos具有深且构象确定的口袋, 可以实现远程手性诱导; (3) WingPhos易于制备且在空气中稳定, 具有工业应用的潜力.研究发展, Rh/L8催化剂可以使三取代β-芳基烯酰胺在温和条件下发生不对称氢化, 得到一系列含有不同官能团的手性β-芳基酰胺化合物.在底物结构中, 除了链状三取代的烯酰胺外, 氢化反应还可以兼容环状三取代的烯酰胺化合物.值得指出的是, 在三取代β-芳基烯酰胺的不对称氢化反应中, Rh/L8催化剂的转化数达到10000, 同时反应的转化率和对映选择性不受影响.这些结果表明WingPhos具有很大的工业应用前景, 可用于许多含有β-芳基胺结构单元的药物分子和天然产物的合成中[6].
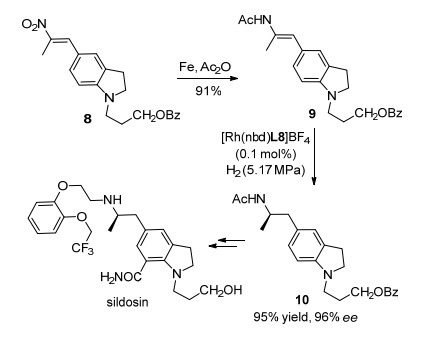
西洛多辛(sildosin)是一种重要的α1肾上腺素受体拮抗剂, 主要用于治疗与良性前列腺增生或肥大相关的泌尿系统疾病.其分子内含有β-芳基手性仲胺的结构可以通过相应烯胺衍生物的不对称氢化合成.如Scheme 2所示, 烯酰胺化合物9可以通过易于制备的硝基苯乙烯衍生物还原得到.在Rh/L8催化剂的存在下, 9经过高对映选择性氢化反应转化为手性酰胺10.参照文献报道的步骤可以将此关键中间体10转化为西洛多辛.
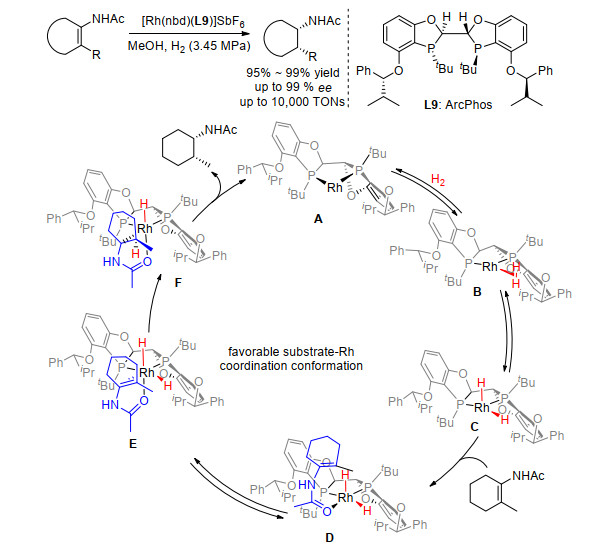
和三取代烯酰胺的不对称氢化相比, 四取代烯酰胺的不对称氢化更具有挑战性.一方面, 取代基的增多导致烯酰胺底物的位阻增大, 降低了氢化反应的活性, 并使得催化剂的用量增加.另一方面, 目前所发展的配体或催化剂往往适用于某种特定结构的底物的氢化, 官能团和底物结构的兼容性较差.为了解决这些难题并发展普适性强的四取代烯酰胺的不对称氢化方法, 认为配体的设计与发展至关重要.在发展了深口袋的Wing-Phos的基础上, 设想通过提高配体整体的富电性来增强配体的催化活性.同时通过引入手性的深口袋来增强催化剂对氢化反应的手性诱导, 继而提高反应的对映选择性.基于富电子的BIBOP型骨架设计合成了一系列新型的含有手性深口袋的双膦配体.在环状四取代烯酰胺的不对称氢化中, 发现使用ArcPhos可以得到优异的产率和对映选择性.经过克级规模试验发现, 催化剂Rh/L9最高的转化数可以达到10000, 具有很好的实用前景[7].通过31P NMR、配体与金属的金属络合物的XRD单晶衍射以及计算化学的辅助手段, 提出了可能的催化循环(图 7).该不对称氢化的反应路径是经过“dihydride”的过程(C), 而对映选择性的决定步骤在于酰胺的双键与Rh金属中心的配位过程(D vs E).反应经能量最有利的过渡态E发生Rh-H对底物双键的插入和后续还原消除给出相应的手性产物.利用这个不对称氢化作为关键步骤, 我们实现了药物分子托法替尼的合成.
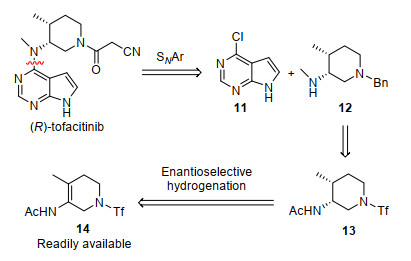
托法替尼是用于治疗类风湿性关节炎、银屑病关节炎和溃疡性关节炎的重要药物[7].其是由辉瑞公司针对免疫抑制作用所开发的一种小分子Janus激酶(JAK)抑制剂. Janus激酶是四个与受体相关的酪氨酸激酶(JAK1-3, TYK2)的家族, 主要参与包括造血和免疫反应等一系列生理过程.托法替尼最近被美国食品药品监督管理局批准用于治疗类风湿性关节炎.由于其显示出一些不利的副作用, 欧洲药品管理局目前尚未批准.托法替尼主要是通过对其外消旋体的手性拆分来获得的[8].目前所报道的两例关于托法替尼的不对称合成路线效率很低[9], 不适于该化合物的大量制备.为了发展一条高效的不对称合成托法替尼的路线, 可以通过四取代烯酰胺的不对称氢化策略来实现托法替尼的高效不对称合成(Scheme 3).具体地, 托法替尼可以通过中间体11和12经过芳环的亲核取代得到.中间体12则可以通过酰胺13经酰基保护基脱除和两次烷基化得到.容易制备的环状烯酰胺14可通过不对称氢化转变为手性酰胺13.
基于对托法替尼的反合成分析, 我们参照已报道的方法很容易地合成了烯酰胺底物14 (Scheme 4).使用Rh/L9催化剂对14进行不对称氢化, 手性酰胺13可以以99%的收率和96% ee顺利得到.这一化合物的绝对构型通过X射线晶体学分析确定.在Li/NH3条件下, 13的三氟甲磺酰基保护基顺利脱除, 接着经苄基引入, 脱乙酰基保护基和甲基化步骤顺利转化为12. 11和12在碳酸钾的条件下进行偶联, 并经后续苄基脱除以及与氰基乙酸的缩合反应得到(R)-托法替尼.值得指出的是, 在所有转化中, 产物的光学纯度没有任何损失.我们最终实现了(R)-托法替尼的克级规模合成.
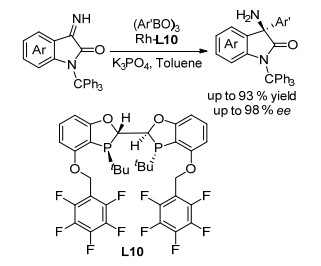
手性α-三级胺结构广泛存在于许多药物分子中.不对称Strecker和Mannich型反应是合成特定类型的手性α-三级胺的重要方法, 但是这两类反应不能用来合成α, α-二芳基三级胺这类重要的手性片段.在早期, 过渡金属催化的烷基锌试剂对酮亚胺的不对称加成成为获得手性α-芳基-三级胺的方法之一, 也为手性α, α-二芳基三级胺的合成提供了思路.相比于金属试剂具有制备繁琐、环境敏感、官能团兼容性不好等缺点, 有机硼试剂往往具有简单易得和稳定性高的明显优势.因此, 芳基硼试剂对亚胺的不对称亲核加成是一种合成手性α, α-二芳基三级胺的便捷方法.但目前报道的方法都要先将亚胺进行保护, 然后发生加成反应, 最后再进行脱保护.通常这些脱保护条件都很剧烈, 官能团容忍性较差.因此, 发展一个芳基硼试剂对无保护亚胺的不对称亲核加成反应具有重要的意义.我们基于BIBOP配体骨架进一步对配体结构进行了优化, 成功发展了配体L10.最终实现了Rh/L10催化的芳基硼酸酐对靛红衍生的N-H亚胺的不对称加成反应(Scheme 5).该方法学的底物普适性好, 可以兼容含有不同取代基和位阻的底物.利用该方法学可以以优秀的收率和对映选择性合成一系列手性3-氨基-3-芳基吲哚酮结构[10].
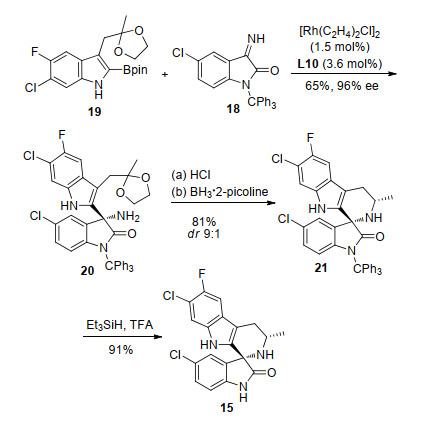
Cipargamin是诺华制药Thierry Diagana等[10]开发的用于治疗疟疾的潜在药物.其具有很好的药代动力学性质, 在啮齿动物模型中有很好的抗疟疗效[11].目前该化合物已经进入临床二期的研究.经过对这个分子的逆合成分析, 我们认为其哌啶环可以通过手性三级胺的分子内还原胺化构建(Scheme 6).而化合物16结构中的手性三级胺基团可以方便地利用芳基硼试剂17对取代的靛红亚胺18的对映选择性加成合成得到.
在实际的合成过程中, 我们利用已知化合物吲哚硼酯19和亚胺18作为起始原料, 使用Rh/L10催化剂成功实现了19对18的不对称加成反应, 以65%的产率和96%的ee值顺利得到手性胺20 (Scheme 7).接着经过脱除缩酮保护基和立体选择性分子内还原胺化, 20顺利转化成螺环结构21, 最后脱除三苯甲基保护基成功地合成cipargamin.
除了对酮亚胺的不对称加成合成手性α-芳基三级胺外, 芳基硼酸或硼酸酐对酮的不对称加成反应则是构建手性叔碳中心的重要方法.基于手性双膦配体WingPhos, 我们也成功实现了芳基硼酸酐对芳基酮的不对称加成反应, 同时利用该方法学实现了手性药物分子艾司西酞普兰(Escitalopram)的首次对映选择性合成[12].
艾司西酞普兰是一种具有高选择性的5-羟色胺再摄取抑制剂.它在治疗中度至重度广泛性焦虑症(GAD)或社交焦虑症(SAD), 恐慌症(伴或不伴恐惧症)以及强迫症(OCD)方面有很好的效果且普遍耐受.工业生产艾司西酞普兰主要依靠外消旋混合物的化学或酶促拆分实现[13], 其总收率和效率都较低.目前艾司西酞普兰的高效不对称合成仍然是一个重大挑战.从合成角度分析, 我们认为艾司西酞普兰分子内的四氢呋喃环可以通过叔醇22经双氰基化-内酯化-内酯还原构建(Scheme 8).手性三级醇22则可以利用芳基硼试剂对芳基酮23的不对称加成得到.于是研究了4-氟苯基硼酸酐对芳基酮23的不对称加成反应(Scheme 9).令人欣慰的是, 使用Rh/L8催化剂, 我们成功地得到光学纯的加成产物22 (>99% ee).经二甲胺参与的取代反应和钯催化的双氰基化-内酯化过程, 22以73%的总收率转化为内酯25.随后25经过DIBAL-H, MsCl/三乙基胺和NaBH4还原等处理得到艾司西酞普兰.
含手性季碳的并环骨架广泛存在于众多具有生物活性的萜类和甾体类天然产物和药物分子中.发展针对这类含手性季碳多环骨架的合成方法具有非常重要的意义.不对称环化是一类高效的构建环系结构骨架的方法, 其中, 通过过渡金属催化的不对称去芳构环化是一种具有吸引力的构建含季碳的多环结构的途径.
我们课题组[14]最近发展了一个不对称去芳构环化反应试图实现一系列萜类、甾体、生物碱和聚酮等具有重要生物活性分子的高效合成.我们提出并设计了一个不对称去芳构环化方法.如图 8所示, 结构26可以经过钯催化的分子内两个芳环之间的不对称去芳构偶联而转化为含有手性季碳中心的并环骨架.实现这一环化反应的关键在于如何解决该反应的活性、区域选择性和对映选择性的问题.依照我们提出的催化循环, 该反应首先经过钯对芳基溴26的氧化加成可能得到手性膦配体配位的芳基钯中间体Ia或Ib.接下来的芳基取代位点决定了反应的化学选择性.由钯中间体Ia出发的芳基取代发生在酚羟基对位(即a位), 形成去芳构化钯中间体IIa, 再经过还原消除得到所需要的含有手性季碳的去芳构环化产物27; 而由钯中间体Ib出发的芳基取代发生在酚羟基的邻位(即b位)得到IIb, 最终得到非手性产物28.从可能的反应产物分析, 尽管平面性分子28在热力学方面较含有手性季碳三环结构27更稳定, 然而27的生成从反应动力学角度来看可能更有利.当L*是一个大位阻单齿膦配体时, Ib构象由于存在可能的配体和酚羟基负离子间作用而变得不稳定, 平衡将向Ia构象的生成倾斜, 促使产物27的形成.同时, 膦配体的手性因素可以对去芳构环化反应进行高效率地诱导, 实现高对映选择性的转化.因此, 我们设想利用大位阻单齿P-手性膦配体来调控该反应的活性和选择性问题.通过研究配体结构和反应活性/选择性的关系, 我们发现大位阻单膦配体如AntPhos可以给出理想的产率和对映选择性.该不对称环化反应表现出很好的官能团和底物结构类型的容忍性.
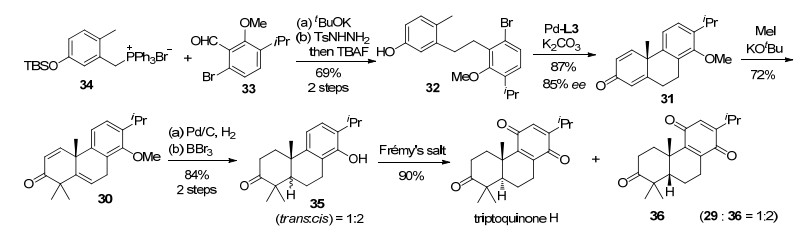
我们利用不对称去芳构环化策略实现了具有全碳手性季碳中心的三环化合物(雷公藤对醌类关键骨架)的高效合成.雷公藤对醌是从中药雷公藤中分离出来的一类重要的天然产物, 用于治疗发烧、发冷、浮肿等症状.雷公藤对醌H作为代表性的雷公藤化合物之一, 具有显著的免疫抑制活性[15].从结构上分析, 雷公藤对醌H属于三环二萜苯醌结构.其对苯醌类结构可以通过30的芳基氧化得到(Scheme 10), 而含有手性季碳的并环结构31可以通过不对称环化反应由芳基溴代物32得到.一个季鏻盐34和醛33之间的Wittig反应可以实现环化底物32的制备.
雷公藤对酮H的合成从醛33和季鏻盐34之间的Wittig反应开始(Scheme 11).随后经双键还原和TBS保护基脱除得到关环前体32.在Pd/L3催化条件下, 32的不对称去芳构环化顺利进行, 我们以87%的收率和85%的ee值得到含有季碳中心的环化产物31.化合物31在tBuOK/MeI的条件下实现了双键迁移和甲基化反应.得到烯酮30之后, 再经过双键氢化和三溴化硼脱甲基得到以反式/顺式异构体的混合物35 (反式:顺式=1:2).用弗雷米盐氧化后, 混合物35成功转化为雷公藤对醌H和36.两种产物可以很方便地通过制备高效液相色谱分离.
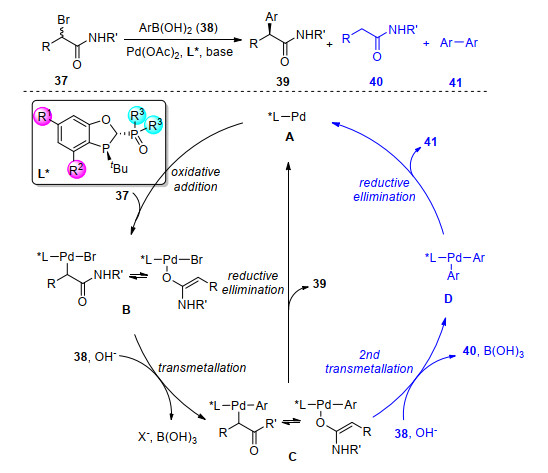
手性的α-芳基羰基化合物在有机合成中是一个非常重要的中间体, 其通过多步转化可以合成诸多具有重要生理活性的天然产物和药物分子.对于这类结构的构建, 通常采用的策略是卤代芳烃与α-羰基金属试剂间的反应, 但该方法仅仅适用于活性较高的卤代芳烃.其他方法如使用当量的金属试剂与预制备的烯醇化合物反应, 则存在许多缺陷, 如部分金属试剂对空气及水分敏感、官能团兼容性差、使用的试剂高毒等.因此, 发展更高效的方法学实现低活性、种类广泛的芳基卤化物和类卤化物或α-卤代羰基化合物参与的偶联反应迫在眉睫.对于前者, 过渡金属催化的α-芳基化反应已经取得了长足的发展.但由于该催化过程需要使用强碱, 反应局限于季碳手性中心的构建, 这大大限制了该反应的适用范围.与之相比, 过渡金属催化α-卤代羰基化合物与有机金属试剂的偶联反应条件更温和, 且此类反应还可以构建羰基α位的叔碳手性中心, 因而具有很大的发展潜力和应用价值.
在过渡金属催化的α-卤代羰基化合物与有机金属试剂, 如芳基硼酸的偶联反应的研究过程中, 我们发现脱卤、β-H消除和异构化等副反应, 这大大影响了这类偶联反应的效率.根据反应的机理, α-卤代羰基化合物经过零价钯物种氧化加成、转金属化后, 形成中间体C(图 9).中间体C可以直接还原消除得到我们所要的交叉偶联产物, 或发生β-H消除以及经过第二次转金属产生脱溴以及联芳基的副产物.为了抑制副反应的发生, 我们提出发展大位阻的配体, 并在该配体中引入具有弱配位能力的辅助基团, 如图 9中的配体L*.我们设想此类大位阻配体的空间环境迫使中间体C到D的过程不易发生, 从而有效地达到抑制二次转金属化的目的.这类“P, P=O”型配体中弱配位点P=O的引人, 不仅便于促进还原消除过程, 同时有效占有了钯中心周围的其他配位位点, 有效地抑制了β-H消除、异构化和还原脱卤等副反应.[16]
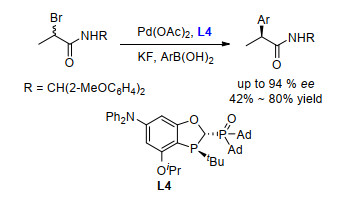
经过对配体结构的优化, 我们发现手性配体L4可以用于钯催化的α-溴代酰胺和芳基硼酸的不对称Suzuki-Miyaura交叉偶联反应中(Scheme 12).利用Pd/ L4催化的偶联反应可以方便地合成一系列手性的α-芳基烷基酰胺类化合物.
手性的芳基丙酸类非甾体药物于20世纪60年代问世.这类药物具有高效的解热、镇痛和抗炎等作用, 其中最有代表性的有布洛芬、萘普生及氟比洛芬等.布洛芬于1961年由英国人Stewart Adams首次发现.它主要用于发热、缓解轻度至中度的疼痛, 包括痛经、偏头痛及类风湿关节炎等.与布洛芬相比, 氟比洛芬的毒性更低并具有出色的耐受性, 其镇痛效果目前是丙酸类非甾体药物中最强的.萘普生是非甾体类抗炎药物, 除了解热、镇痛等功效与布洛芬类似外, 萘普生还可以提高人体免疫力, 并对幽门螺杆菌有较好的抑制作用.此外, 萘普生还具有调节血糖的作用[17].通过钯催化的α-溴代酰胺和芳基硼酸的不对称Suzuki-Miyaura交叉偶联反应, 可以很方便地合成布洛芬、氟比洛芬和萘普生(Scheme 13).以布洛芬的手性合成为例, 芳基硼酸43和外消旋烷基溴代物42的交叉偶联得到高光学活性的手性酰胺44 (90% ee).将酰胺基团的胺基辅基脱除后可以得到布洛芬.相应地, 氟比洛芬和萘普生也可以通过类似的合成方法高效制备.

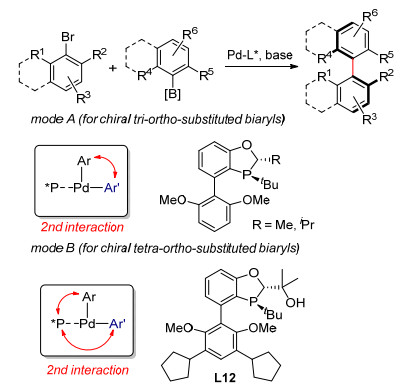
具有轴手性的联芳基结构广泛存在于药物和天然产物分子中, 例如临床上广泛使用的万古霉素类型的环状多肽抗生素和具有包括抗肿瘤等活性的棉酚等.目前所报道的关于具有轴手性的联芳基化合物的合成方法众多且各具特色.在所有构建轴手性联芳基结构的策略中, 不对称偶联无疑是最为简洁且直接的方法.通过不对称偶联合成具有轴手性的联芳基结构, 尤其是高度官能团化的联芳基化合物的挑战在于提高偶联反应的反应活性和对映选择性的高效调控.在发展高立体选择性的交叉偶联反应方面, 我们通过设计新型配体和借助次级相互作用的催化模式成功建立了用于合成邻位三取代[18]和四取代[19]手性联芳基化合物的不对称Suzuki-Miyaura偶联(图 10).基于所发展的偶联反应, 我们实现了棉酚的不对称合成.
棉酚(gossypol)是一个多酚羟基双萘醛类手性药物分子.这个化合物是由Longmore和Marchlewski两位科学家于19世纪末从棉花籽油中首次分离得到[19].棉酚主要存在于棉花的根、茎、叶和种子内, 且在棉籽仁中含量最高.在中国, 棉酚最早是用作男性避孕药来使用.随着对其生物活性研究的深入, 人们发现棉酚及其衍生物具有多种生物活性, 包括抗寄生虫、抗癌、抗病毒以及抗感染等活性.棉酚中的邻位四取代联芳基结构的存在使其沿联萘基碳碳键的旋转具有一定的能垒, 因而该分子具有轴手性.从棉花中提取分离得到的棉酚往往是两个对映体(+)-(S)-棉酚和(-)-(R)-棉酚的混合物.在生物活性方面, (+)-棉酚、(-)-棉酚以及它们的混合物也会有一定的差别, 例如, 棉酚的消旋体具有抗生育活性, 而(+)-棉酚则发现不具备这一活性; (-)-棉酚或者含有较高比例的(-)-棉酚棉籽油具有抑制乳腺脂肪细胞增殖的活性, 因而具有抗乳腺癌和预防肥胖的潜力[31]; 另有研究表明, (-)-棉酚可以在mRNA和蛋白质水平上干预促血管生成因子从癌细胞的释放过程, 因而可以抑制肿瘤血管生成过程.正是由于棉酚两个对映体在生物活性方面的差异, 发展能够得到高光学纯度的单一棉酚对映体的方法至关重要.目前已报道且普遍采用的获得高光学纯度的单一棉酚对映体的方法是化学拆分法.此方法通过棉酚的消旋体和手性拆分试剂(通常是手性氨基酸)作用生成相应的席夫碱, 然后通过高效液相色谱法或重结晶方法获得单一的立体异构体, 再经过拆分试剂的脱除得到高光学纯度的单一棉酚对映体.此方法的局限性在于当量手性拆分试剂的使用, 这无疑增加了整个过程的成本; 同时, 棉酚消旋体的获得也是该方法规模化的一个制约因素.相比较而言, 利用廉价易得的原料, 通过化学合成的方法来得到高光学纯度的单一棉酚对映体更有前景.至今为止, 通过不对称合成的方法得到高光学纯度的单一棉酚对映体目前仅有一例报道, 即Meyers课题组利用手性辅基底物参与的乌尔曼偶联反应为关键步骤实现的棉酚的不对称全合成.手性辅基的使用增加了生产的成本; 同时, 手性辅基的引入和脱除也使得合成路线不够简洁实用.因而发展一种简洁高效地获得高光学纯度的单一棉酚对映体的化学合成方法有着重要的意义.
根据对棉酚的逆合成分析, 我们认为其高度官能团化的联芳基结构可以由前体45通过后期的异丙基化, 脱保护基和甲酰化反应实现(Scheme 14).轴手性联芳基结构则可以利用分子间的不对称Suzuki-Miyaura偶联构建.取代的萘基底物可以有萘醌前体的选择性还原得到.醌类化合物49和二烯48参与的环加成反应可以合成萘醌47.
我们发展了如Scheme 15所示的棉酚的不对称合成路线.首先已知化合物共轭二烯48与苯醌化合物49发生环加成反应, 制得萘醌化合物.将萘醌化合物进行选择性地还原并原位将酚羟基保护得到取代的萘化合物47.在溴化氧化条件下将化合物47转化为萘甲醛51, 同时脱除萘酚羟基上的乙酰基保护基得到51. 51参与的烷基化反应得到偶联前体52.在过渡金属钯催化剂和手性配体BaryPhos (L12)的存在下, 化合物52进行Miyaura硼化反应, 并进一步发生Suzuki-Miyaura偶联反应, 以62%的产率和83% ee制得具有轴手性的联芳基化合物53.通过重结晶可以进一步提高该联芳基化合物的光学纯度(>99% ee).得到53后, 催化氢解和三氟甲磺酰化制得54, 接着芳基异丙基偶联在Pd/L13催化剂存在下顺利进行得到55.最后脱甲基和甲酰化反应制得(-)-棉酚.
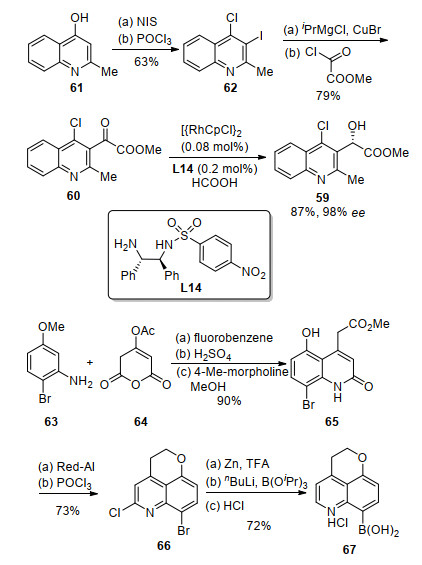
手性联芳基化合物57是基于喹啉结构发展而来的具有别构效应的整合酶抑制剂, 是旨在解决获得性免疫缺陷综合征(艾滋病)治疗过程中药物耐药性问题的一个潜在药物(Scheme 16).除了轴手性联芳基结构外, 这个化合物分子内还含有一个手性仲醇的叔丁醚单元.为了发展一条高效简洁的合成路线, 我们认为其联芳基结构可以通过58和59参与的不对称Suzuki-Miyaura交叉偶联构建[21, 22].通过使用合适的配体, 可以实现对该偶联反应的立体选择性的调控.手性二级醇可以经过相应酮前体60的不对称还原合成.喹啉衍生物61经酰基化反应可以合成得到60.
如Scheme 17所示, 由已知化合物61出发, 经过碘代和氯化反应可以合成62. 62经过金属卤交换后与酰氯反应, 得到酰化产物60.随后在Rh/L14的催化下, 60经过不对称氢化反应合成得到手性醇59.芳基硼酸58的合成是由63和64作为起始原料, 在不饱和酸酐64的存在下, 苯胺63首先发生酰基化反应, 随后在浓硫酸条件下发生关环反应和酯化反应得到酯65. 65在Red-Al条件下被还原为相应的醇.其经过POCl3介导的环化和锌粉还原脱氯转化为芳基溴66.最后在正丁基锂/硼酸三异丙酯条件下, 66转化为相应的硼酸盐酸盐67.
作为合成的关键步骤, Pd/L1催化的58和59的Suzuki-Miyaura交叉偶联反应可以以73%的收率和5:1的dr值得到目标产物68 (Scheme 18).进一步的配体结构优化提高了偶联反应的立体选择性, 即当使用配体L15时, 反应的dr值可以提升至15:1.值得指出的是, 通过重结晶可以将手性醇68的光学纯度提升至99% ee.最后经过改进的Jackson叔丁基化和酯的水解合成得到目标产物57.
综上所述, 发展新型高效的不对称合成方法对手性药物的简洁合成具有重要意义.近年来, 手性配体促进的过渡金属催化的不对称转化在手性药物合成中的应用越来越广泛.本文介绍了基于苯并手性氧杂膦烷结构单元的单、双膦配体的设计理念, 并详细总结了这些具有独特骨架的配体在过渡金属催化的不对称偶联、环化、氢化和加成等反应中的应用.利用所发展的不对称反应方法学, 我们实现了包括托法替尼、cipargamin、艾司西酞普兰、雷公藤对醌H、布洛芬、氟比洛芬、萘普生和棉酚在内的一系列重要手性药物分子的高效不对称合成.可以预见, 未来新型手性配体/催化剂和不对称方法学的不断发展必将进一步促进手性药物的绿色合成.
林国强, 王梅祥, 手性合成和手性药物, 化学工业出版社, 北京, 2008, pp. 1~4.Lin, G. Q.; Wang, M. X. Chiral Synthesis and Chiral Drugs, Chemical Industry Press, Beijing, 2008, pp. 1~4 (in Chinese).
林国强, 孙兴文, 陈耀全, 李月明, 陈新滋, 手性合成——不对称反应及其应用, 科学出版社, 北京, 2013, pp. 36~38.Lin, G. Q.; Sun, X. W.; Chen, Y. Q.; Li, Y. M.; Chen, X. Z. Chiral Synthesis-Asymmetric Reactions and Their Applications, Science Press, Beijing, 2013, pp. 36~38 (in Chinese).
Tang, W; Zhang, X. Chem. Rev. 2003, 103, 3029. doi: 10.1021/cr020049i
汤文军, 中国化学会第十一届全国天然有机化学学术会议论文集, 第一册, 上海, 2019, p. 71.Tang, W. In Proceedings of the 11th National Conference on Natural Organic Chemistry of the Chinese Chemical Society, Volume 1, Shanghai, 2019, p. 71 (in Chinese).
(a) Xu, G.; Senanayake, C. H; Tang, W. Acc. Chem. Res. 2019, 52, 1601.
(b) Wu, T.; Xu, G.; Tang, W. Aldrichim. Acta 2020, 53, 27.
(c) Tang, W.; Li, K. Strem Chem. 2019, XXXI, 1.
Liu, G.; Liu, X.; Cai, Z.; Jiao, G.; Xu, G.; Tang, W. Angew. Chem., Int. Ed. 2013, 52, 42359.
Li, C.; Wan, F; Chen, Y.; Peng, H.; Tang, W.; Yu, S.; McWilliams, H. C.; Mustakis, J.; Samp, L.; Maguire, R. J. Angew. Chem., Int. Ed. 2019, 58, 13573. doi: 10.1002/anie.201908089
Daniella M. S.; Yuka K.; Alejandro V.; Michael W.; Massimo G.; John J. O. Nat. Rev. Drug Discovery 2017, 16, 843. doi: 10.1038/nrd.2017.201
Patil, Y. S.; Bonde, N. L.; Kekan, A. S.; Sathe, D. G.; Das, A. Org. Process Res. Dev. 2014, 18, 1714. doi: 10.1021/op500274j
Zhu, J.; Huang, L.; Dong, W.; Li, N.; Yu, X.; Deng, W.; Tang, W. Angew. Chem., Int. Ed. 2019, 58, 16119. doi: 10.1002/anie.201910008
Rottmann, M.; McNamara, C.; Yeung, B. K. S. Science 2010, 329, 1175. doi: 10.1126/science.1193225
Huang, L.; Zhu, J.; Jiao, G.; Wang, Z.; Yu, X.; Deng, W.; Tang W. Angew. Chem., Int. Ed. 2016, 55, 4527. doi: 10.1002/anie.201600979
Dhillon, S.; Scott, L. J.; Plosker, G. L. CNS Drugs 2006, 20, 763. doi: 10.2165/00023210-200620090-00010
Yang, H.; Tang, W. Chem. Rec. 2020, 20, 23. doi: 10.1002/tcr.201900003
Cao, Z.; Du, K.; Liu, J. H.; Tang, W. Tetrahedron 2016, 72, 1782. doi: 10.1016/j.tet.2016.02.043
Li, B.; Li, T.; Aliyu, M. A.; Li, Z.; Tang, W. Angew. Chem., Int. Ed. 2019, 58, 11355. doi: 10.1002/anie.201905174
Dodou, K.; Anderson, R. J.; Lough, W. J.; Small, D. A. P.; Shelley, M. D.; Groundwater, P. W. Bioorg. Med. Chem. 2005, 13, 4228. doi: 10.1016/j.bmc.2005.04.026
(a) Xu, G.; Fu, W.; Liu, G.; Senanayake, C. H.; Tang, W. J. Am. Chem. Soc. 2014, 136, 570.
(b) Xu, G.; Zhao, Q.; Tang, W. Chin. J. Org. Chem. 2014, 34, 1919 (in Chinese).
(徐广庆, 赵庆, 汤文军, 有机化学, 2014, 34, 1919.)
Yang, H.; Sun, J.; Gu, W.; Tang, W. J. Am. Chem. Soc. 2020, 142, 8036. doi: 10.1021/jacs.0c02686
Lu, Y.; Dong, C.; Huang, J.; Zhou, H.; Wang, W. Future Med. Chem. 2017, 9, 1243. doi: 10.4155/fmc-2017-0046
Fandrick, K. R.; Li, W.; Zhang, Y.; Tang, W.; Gao, J.; Rodriguez, S.; Patel, N. D.; Reeves, D. C.; Wu, J.-P.; Sanyal, S.; Gonnella, N.; Qu, B.; Haddad, N.; Lorenz, J. C.; Sidhu, K.; Wang, J.; Ma, S.; Grinberg, N.; Lee, H.; Tsantrizos, Y.; Poupart, M.-A.; Busacca, C. A.; Yee, N. K.; Lu, B. Z.; Senanayake, C. H. Angew. Chem., Int. Ed. 2015, 54, 7144. doi: 10.1002/anie.201501575
Haddad, N.; Mangunuru, H. P. R.; Fandrick, K. R.; Qu, B.; Sieber, J. D.; Rodriguez, S.; Desrosiers, J. N.; Patel, N. D.; Lee, H.; Kurouski, D.; Grinberg, N.; Yee, N. K.; Song, J. J.; Senanayake, C. H. Adv. Synth. Catal. 2016, 358, 3522. doi: 10.1002/adsc.201600889

图 1 一些含有手性结构单元的畅销和常用药物
Figure 1 Some popular and commonly used drugs with important chiral units

图 2 一些典型的P-手性的单膦和双膦配体
Figure 2 Selected examples of typical P-chiral mono- and bis- phosphine ligands

图 3 基于苯并氧杂膦烷结构的配体类型
Figure 3 Ligand structures based on chiral benzooxaphosphane skeleton

图 7 四取代烯酰胺的不对称氢化:催化循环
Figure 7 Proposed catalytic cycle for asymmetric hydrogenation of tetra-substituted enamides

图式 5 Rh催化的芳基硼酸酐对无保护亚胺的不对称亲核加成
Scheme 5 Asymmetric nucleophilic addition of arylboronic anhydride to unprotected imine

图 9 Pd催化的烷基溴代物参与的Suzuki-Miyaura偶联反应
Figure 9 Pd-catalyzed Suzuki-Miyaura coupling using alkyl bromides

图式 13 布洛芬、氟比洛芬和萘普生的合成路线
Scheme 13 Asymmetric synthesis of ibuprofen, flurbiprofen, and naproxen

图 10 基于配体和催化模式设计的不对称Suzuki-Miyaura偶联反应
Figure 10 Asymmetric Suzuki-Miyaura coupling based on design of ligand and catalysis mode

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:










 下载:
下载:




















 下载:
下载: