Received Date:
26 February 2020 Revised Date:
10 April 2020 Available Online:
01 July 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21702175, 21961037) and the 1000 Youth Talents Plan
Abstract:
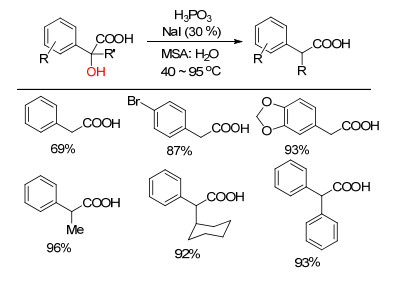
Functional groups are atoms or groups that determine the chemical properties of organic compounds. It often plays the role of guiding group in organic synthesis chemistry. Defunctionalization is the chemical transformation of a substrate with more functional groups into a compound with fewer functional groups, which has positive applications in solving environmental problems, resource shortage and biomass degradation. But due to the bond energy, heating, acid or base are often involved in defunctionalization. In recent years, defunctionalization has been moving toward a greener and more sustainable direction. Metal catalysis provides a new way for defunctionalization. The recent applications of different metal-mediated defunctionalization in organic synthesis and their mechanism are summarized.
Verduyckt, J.; Van Hoof, M.; De Schouwer, F.; Wolberg, M.; Kurttepeli, M.; Eloy, P.; Gaigneaux, E. M.; Bals, S.; Kirschhock, C. E. A.; De Vos, D. E. ACS Catal. 2016, 6, 7303. doi: 10.1021/acscatal.6b02561
Tobisu, M.; Nakamura, R.; Kita, Y.; Chatani, N. J. Am. Chem. Soc. 2009, 131, 3174. doi: 10.1021/ja810142v
[62]
Hooper, J. F.; Young, R. D.; Weller, A. S.; Willis, M. C. Chem.-Eur. J. 2013, 19, 3125. doi: 10.1002/chem.201204056
[63]
Vandekerkhove, A.; Claes, L.; De Schouwer, F.; Van Goethem, C.; Vankelecom, I. F. J.; Lagrain, B.; De Vos, D. E. ACS Sustainable Chem. Eng. 2018, 6, 9218. doi: 10.1021/acssuschemeng.8b01546
[64]
Chatani, N.; Tatamidani, H.; Ie, Y.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc. 2001, 123, 4849. doi: 10.1021/ja0103501
[65]
Tatamidani, H.; Yokota, K.; Kakiuchi, F.; Chatani, N. J. Org. Chem. 2004, 69, 5615. doi: 10.1021/jo0492719
[66]
Mazziotta, A.; Madsen, R. Eur. J. Org. Chem. 2017, 2017, 5417.
Seo, S.; Taylor, J. B.; Greaney, M. F. Chem. Commun. 2012, 48, 8270. doi: 10.1039/c2cc33306f
[87]
Liao, R.-Z.; Chen, S.-L.; Siegbahn, P. E. M. ACS Catal. 2015, 5, 7350. doi: 10.1021/acscatal.5b01502
[88]
Ren, Y.-L.; Tian, M.; Tian, X.-Z.; Wang, Q.; Shang, H.; Wang, J.; Zhang, Z. C. Catal. Commun. 2014, 52, 36. doi: 10.1016/j.catcom.2014.03.036
[89]
(a) Zhang, L.; Koreeda, M. J. Am. Chem. Soc. 2004, 126, 13190.(b) Jordan, P. A.; Miller, S. J. Angew. Chem., Int. Ed. 2012, 51, 2907.
[90]
García, N.; García-García, P.; Fernández-Rodríguez, M. A.; Rubio, R.; Pedrosa, M. R.; Arnáiz, F. J.; Sanz, R. Adv. Synth. Catal. 2012, 354, 321. doi: 10.1002/adsc.201100877
[91]
Sousa, S. C. A.; Fernandes, T. A.; Fernandes, A. C. Eur. J. Org. Chem. 2016, 2016, 3109. doi: 10.1002/ejoc.201600441
[92]
(a) Dupuy, S.; Lazreg, F.; Slawin, A. M. Z.; Cazin, C. S. J.; Nolan, S. P. Chem. Commun. 2011, 47, 5455. (b) Dupuy, S.; Nolan, S. P. Chem.-Eur. J. 2013, 19, 14034.
Diéguez, H. R.; López, A.; Domingo, V.; Arteaga, J. F.; Dobado, J. A.; Herrador, M. M.; Quílez del Moral, J. F.; Barrero, A. F. J. Am. Chem. Soc. 2010, 132, 254. doi: 10.1021/ja906083c



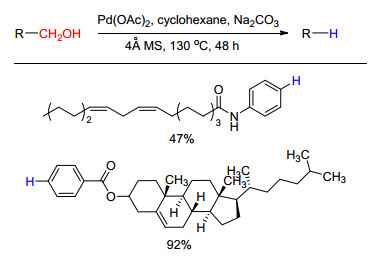
 下载:
下载:
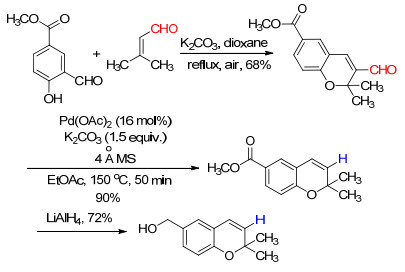

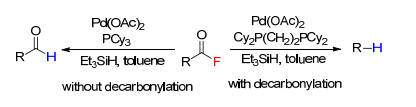
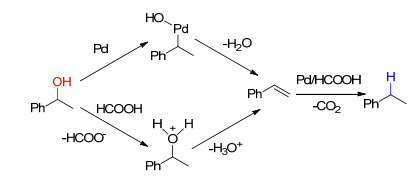




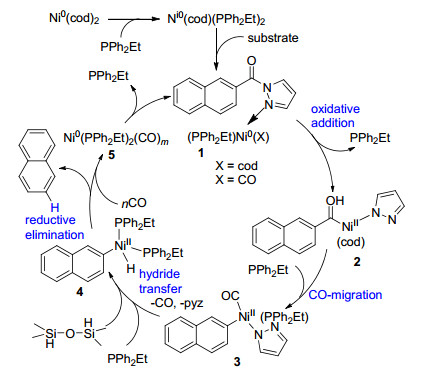

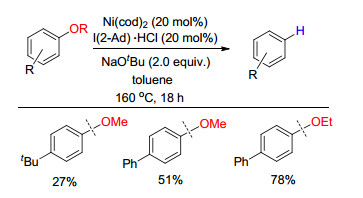
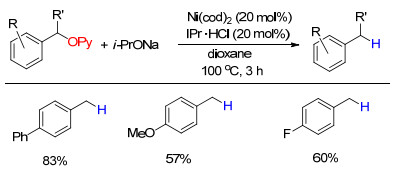
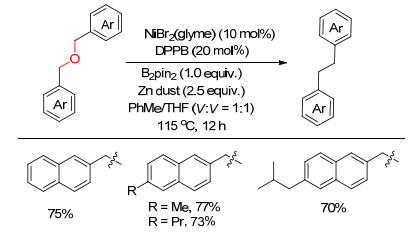







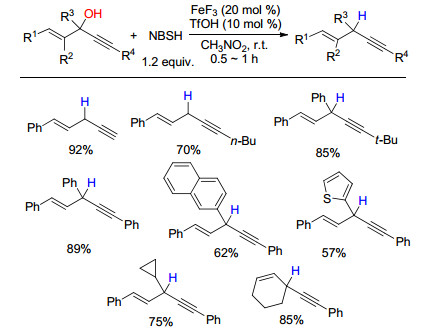
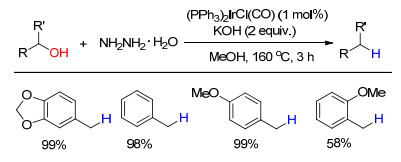
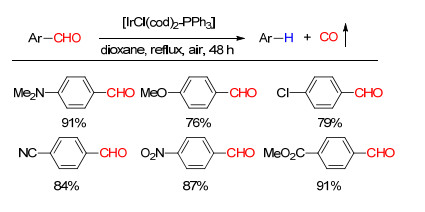

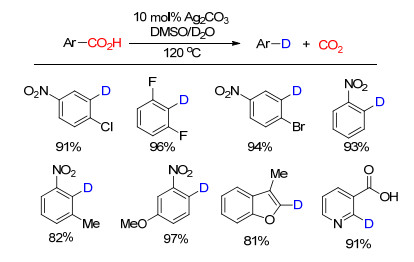
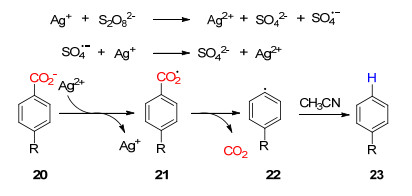





















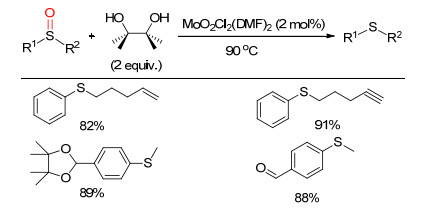
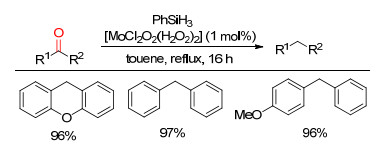

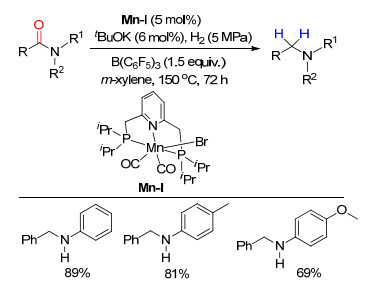















 下载:
下载:


































 下载:
下载:
